

Abstract
Objective:
The genetic risk architecture of Alzheimer disease (AD) is complex with single pathogenic mutations leading to early-onset AD, while both rare and common genetic susceptibility variants contribute to the more widespread late-onset AD (LOAD); we sought to discover novel genes contributing to LOAD risk.
Methods:
Whole-exome sequencing and genome-wide genotyping were performed on 11 affected individuals in an extended family with an apparent autosomal dominant pattern of LOAD. Variants of interest were then evaluated in a large cohort of LOAD cases and aged controls.
Results:
We detected a single rare, nonsynonymous variant shared in all 11 LOAD individuals, a missense change in the tetratricopeptide repeat domain 3 (TTC3) gene. The missense variant, rs377155188 (p.S1038C), is predicted to be damaging. Affecteds-only multipoint linkage analysis demonstrated that this region of TTC3 has a LOD score of 2.66 in this family.
Conclusion:
The TTC3 p.S1038C substitution may represent a segregating, rare LOAD risk variant. Previous studies have shown that TTC3 expression is consistently reduced in LOAD patients and negatively correlated with AD neuropathology and that TTC3 is a regulator of Akt signaling, a key pathway disrupted in LOAD. This study demonstrates how utilizing whole-exome sequencing in a large, multigenerational family with a high incidence of LOAD could reveal a novel candidate gene.
Alzheimer disease (AD) is the most common form of dementia in the elderly, accounting for up to 75% of all dementia patients.1 Mutations in the amyloid precursor protein (APP), presenilin 1 (PSEN1), and presenilin 2 (PSEN2) genes have been shown to cause autosomal dominant, early-onset AD.2–4 Late-onset AD (LOAD) is etiologically more complex and presents with intermediate to high heritability (58%–79%).5 The apolipoprotein E (APOE) gene contributes significantly to LOAD risk; the APOE ε4 allele confers an increased risk to LOAD, while the rare APOE ε2 allele is protective.6–9 In addition to the APOE gene, genome-wide association studies identified 19 further loci that confer low individual, but reproducible risk to LOAD.10 Moreover, rare variants in TREM2 have been shown to contribute to LOAD risk.11 It seems likely that additional, yet unknown, rare variants with intermediate to large effects may contribute to the complex genetic architecture of LOAD. We identified a large, multiplex LOAD family with an autosomal dominant pattern of inheritance and negative for pathogenic variants in known AD genes. We performed whole-exome sequencing (WES) in 11 affected family members and prioritized rare variants that segregated with disease. A single alteration in tetratricopeptide repeat domain 3 (TTC3) was identified in all 11 LOAD individuals. Our study shows that WES in a single extended family with a high LOAD burden can lead to the identification of rare genetic variants that may contribute to risk for AD.
METHODS
Standard protocol approvals, registrations, and patient consents.
All individuals ascertained in this study provided written informed consent prior to their participation. If the study participant was not competent to provide consent, then written consent was obtained from the immediate next of kin or his or her legal representative. All participants were ascertained using the protocol approved by the appropriate institutional review board. Oversight of the study falls under the University of Miami (IRB #20070307).
Collection and characterization of an extended LOAD family.
We identified a large family (no. 1229) that was consistent with an autosomal dominant inheritance pattern of LOAD. Family 1229 has 15 affected individuals, 3 of whom have a neuropathologically confirmed diagnosis of LOAD. All affected individuals meet the standard National Institute of Neurological and Communicative Disorders and Stroke and Alzheimer Disease and Related Disorders Association (NINCDS-ADRDA) criteria for LOAD with a disease onset of 65 years of age or older.12 Besides direct neurologic examinations, a battery of neuropsychiatric testing was performed to monitor preclinical symptoms of LOAD and comorbidities (Parkinsonism, depression) that are known to influence the course of preclinical AD. The family was evaluated to determine APOE ε genotypes and screened for high-risk variants in APP, PSEN1, and PSEN2.
Genome-wide genotyping and linkage analysis.
Genotype data were obtained for 9 LOAD-affected individuals of family 1229 using the Human OmniExpress BeadChip array (Illumina, San Diego, CA). Genotype quality control comprised the exclusion of single-nucleotide variants (SNVs) with call rates below 98%. The B allele frequency plots and the log R ratio plots were used to search for potential copy number variants surrounding our top variant. These data were analyzed with MERLIN to determine an affecteds-only multipoint linkage LOD score with LOAD in family 1229 assuming an autosomal dominant model.13 Two-point age-dependent penetrance linkage was also performed for the variant of interest using an age curve as previously described.14
Whole-exome sequencing.
WES was performed in all 11 affected individuals of family 1229. WES used 3 μg of DNA from each sample. Exome capture and sequence library construction were performed on a Sciclone G3 NGS Workstation (Caliper Life Sciences, Hopkinton, MA) using the SureSelect Human All Exon 50 Mb Kit (Agilent Technologies, Santa Clara, CA) and the Paired-End Multiplexed Sequencing library kit (Illumina) for sequence library preparation. Two exome sample libraries were sequenced per lane on a HiSeq 2000 Sequencing System (Illumina) in paired-end 2 × 100 base pair runs. In addition, WES of 373 non-AD samples of European ancestry were also run and analyzed at the John P. Hussman Institute for Human Genomics in order to get minor allele frequency (MAF) estimates and control for WES artifacts.
SNV and insertion-deletion calling and annotation.
Sequencing data were processed using the Illumina RTA base calling pipeline v1.8. Alignment to the human reference genome (hg19) was executed with the Burrows-Wheeler Aligner and variant calling performed with the Genome Analysis Toolkit (GATK).15,16 GATK parameters for variant quality control included duplicate sequence read removal, base quality score recalibration (QUAL >50), and variant quality score recalibration (VQSLOD >0).17 SNVs passing quality measurements were annotated with SeattleSeq (http://snp.gs.washington.edu/SeattleSeqAnnotation137/). We define rare SNVs and insertions-deletions (indels) as those with a MAF <1% in dbSNP139 (http://www.ncbi.nlm.nih.gov/snp/), 1000 Genomes Project populations, the 5,379 European and African-American samples from the NHLBI GO Exome Sequencing Project (ESP), Seattle, WA (http://evs.gs.washington.edu/EVS/), and 373 Caucasian exomes processed at the John P. Hussman Institute for Human Genomics.18,19 Variants were evaluated for phylogenetic conservation in mammals with the Genomic Evolutionary Rate Profiling score and PhastCons.20,21 A Genomic Evolutionary Rate Profiling score >3 and a PhastCons score >0.7 were considered phylogenetically conserved. A variant's predicted deleteriousness to protein function was assessed by the Likelihood Ratio Test, the Mutation Assessor, Mutation Taster, PolyPhen-2 (HumDiv), and PROVEAN/SIFT programs using the default score cutoffs for deleteriousness.22–26
SNV validation by Sanger sequencing.
Ten rare SNVs identified by WES that passed quality control and were shared in >80% (≥9/11) of affected individuals of family 1229 were evaluated by Sanger sequencing in the 11 affected and 5 unaffected individuals of this family.
Evaluating the TTC3 variant in a LOAD cohort.
The TTC3 variant was evaluated using a custom TaqMan genotyping assay (Life Technologies, Carlsbad, CA). A large cohort of 6,669 LOAD cases and 5,585 controls (≥60 years) of European ancestry from the Alzheimer's Disease Genetics Consortium (ADGC) cohort were used. Power calculations were performed with the Genetic Power Calculator.27
RESULTS
Clinical and neuropathologic characterization.
DNA samples were collected from family 1229 from 11 affected and 5 unaffected members of at least 65 years old (figure 1, table e-1 at Neurology.org/ng). Clinically, each of the 11 affected individuals presented with late-onset dementia (range: 70–85 years old). The initial course of disease followed a typical AD pattern, with early forgetfulness and progressive worsening in areas of cognitive function such as short-term memory, orientation, executive function as reflected in activities of daily living, and language retrieval (word finding).
Figure 1. Pedigree of family 1229.

TTC3 p.S1038C (rs377155188) is the only rare, nonsynonymous variant detected by whole-exome sequencing (WES) shared by all 11 LOAD individuals. The age beneath each individual is either the age at onset for individuals with LOAD or the age at examination for unaffected family members. The genotypes are the wild type (C/C) or the alteration (C/G) that causes the TTC3 missense alteration.
Patients were assessed for features suggestive of alternate dementing pathologies, such as dementia with Lewy bodies, frontotemporal dementia, and vascular dementia. Abnormal features suggestive of a frontotemporal etiology such as frontal release signs, early changes in personality, behavior, or language changes were not present in any family members. A positive snout reflex was noted approximately 9 years into the disease course in one individual (0122), but no other individual exhibited frontal release signs or symptoms. The late appearance of a single frontal release sign is not suggestive of a diagnosis of frontotemporal dementia. A second individual (9006) exhibited mild parkinsonian features (masked facies, hunching, micrographia, poor balance) when examined at age 84, ten years into the disease course. The later onset of comorbid parkinsonian features is a known entity in AD. Four affected individuals underwent autopsy with subsequent neuropathologic results (table e-1). All individuals demonstrated classic Braak and Braak pathology, ranging in stage from III to V.
Complete segregation of the rare TTC3 p.S1038C substitution with LOAD.
WES in 11 LOAD members of family 1229 resulted in an average coverage of 83% (±0.04% SD) of the human exome sequence at a minimal depth of 20 reads per base pair. On average, 11,227 (±305 SD) exonic SNVs were called per exome, for a total of 32,784 unique SNVs across all 11 affected family members. The majority of disease-causing variants identified to date are nonsynonymous, protein-altering SNVs including nonsense, missense, and splice site variants as well as small insertion-deletion polymorphisms (insertions/deletions). Of all exonic variants detected in family 1229, about half (16,363 SNVs) were found to be nonsynonymous.
Family 1229 is characterized by a suggestive autosomal dominant pattern of LOAD inheritance (figure 1). We therefore hypothesized that a single (or a few) highly penetrant disease variant, with considerable individual risk, could explain the high burden of LOAD observed in this family. Statistical power estimates for the detection of LOAD variants suggest that published genome-wide association studies would likely have identified loci that harbor variants with aMAF >1% in the general population.7 Thus, we further filtered for rare variants (MAF <1%), which reduced the candidate variants to 2,897 SNVs. Among these rare and nonsynonymous variants, only a single variant, rs377155188, was identified in all 11 affected family members; this variant is in the TTC3 gene and encodes for a serine to cysteine substitution at amino acid position 1038 (figure 1; table 1).
Table 1.
Top rare LOAD candidate variants identified in family 1229
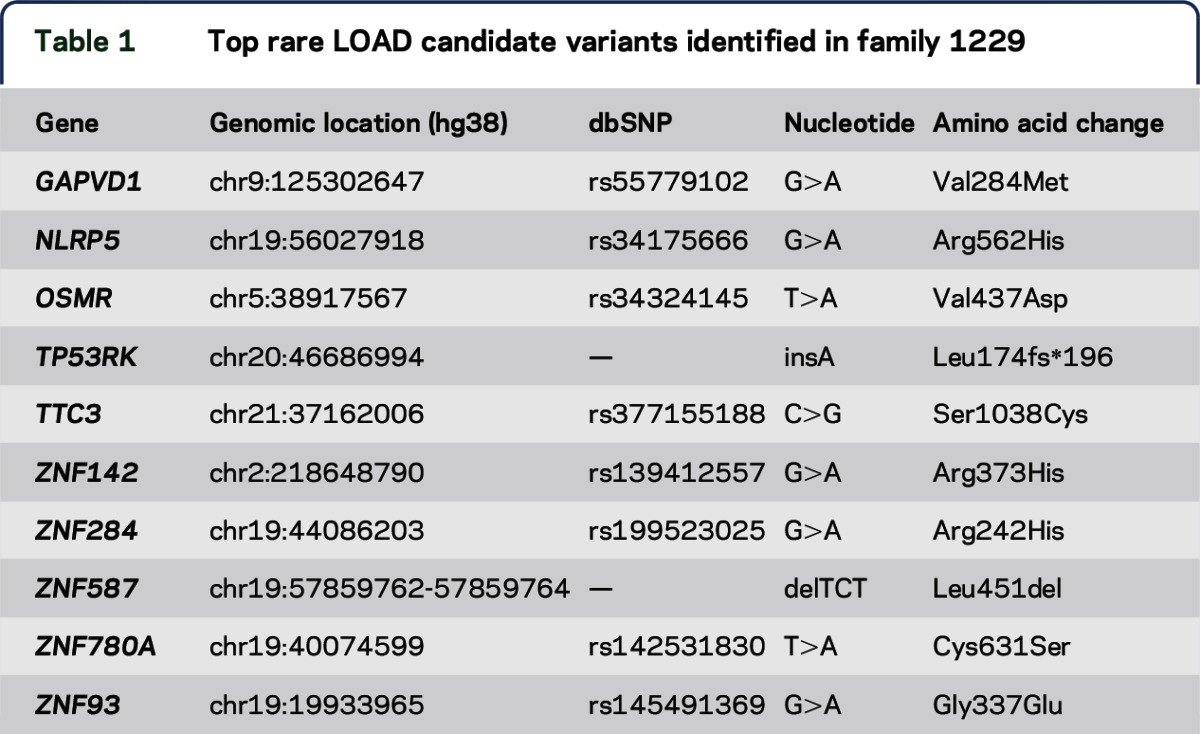
We evaluated the remaining rare, nonsynonymous variants. None were shared in 10 of 11 affected family members, but 9 variants were shared in 9 affected family members (table 1). All 10 variants in at least 9 LOAD family members were confirmed by traditional Sanger sequencing (table 1).
The TTC3 variant, rs377155188, is extremely rare in European Americans (MAF = 0.00012) and absent in African Americans according to the NHLBI ESP.16 The p.S1038C substitution in the TTC3 protein is predicted to be damaging by 5 distinct algorithms. Furthermore, it is located in a highly conserved site.
The TTC3 heterozygous genotypes identified through WES were validated by Sanger sequencing in all 11 affected individuals. Wild-type genotypes were obtained for 4 out of 5 unaffected family members ≥65 years old (figure 1). Individual no. 9018 was the only aged and unaffected heterozygous carrier of TTC3 rs377155188. However, this individual did not receive a formal cognitive examination as did the 3 other unaffected family members. This individual's negative affection status of LOAD has been based on family history data after the individual's death at age 76. This person's age at death is only 1 year above the average age of LOAD onset in this family (table e-1); thus, given the absence of a formal examination, this carrier may still have been presymptomatic at age 76.
We then evaluated the prevalence of rs377155188 by genotyping 12,254 unrelated individuals (6,669 LOAD cases and 5,585 controls) of European ancestry from the ADGC. Over all individuals, rs377155188 showed a low frequency (MAF = 0.020%), as has been reported by the ESP for Americans of European descent (MAF = 0.012%).16 We observed 5 heterozygous individuals in total. Four were LOAD cases, all of whom carried the APOE ε3/ε4 genotype. Only a single cognitively normal control carried the TTC3 variant (0.018%). This individual was last examined at age 84 and also carried a protective APOE ε2/ε3 genotype. These genotypes resulted in an allelic association odds ratio of 3.35, which did not reach statistical significance (Fisher exact test, p = 0.248). Power calculations show that we are underpowered to detect a significant effect in our cohort (49.6% power to achieve significance at α = 0.05; assuming a disease frequency of 0.10), given the extremely rare allele frequency of the rs377155188 variant in our cohort and the general population.27
Linkage analysis in LOAD family 1229.
We used genome-wide common SNP genotyping data available for 9 LOAD-affected members of family 1229 to calculate the affecteds-only multipoint linkage score under an autosomal dominant model of inheritance. Two regions were identified with a LOD score greater than 2: chr19:48.89–51.07 Mb (LOD = 2.49) and chr21:38.08–40.12 Mb (LOD = 2.66). Of the top variants of interest, only the one in TTC3 on chromosome 21q22.13 fell within a linkage region. The genotyping data were also used to determine whether a copy number was located on chromosome 21 surrounding the top variant of interest, but none was found (figure e-1). In addition, age-dependent penetrance analysis of the TTC3 variant confirmed the affecteds-only results (LOD = 2.70).
DISCUSSION
We performed WES in an extended, multigenerational family with an autosomal dominant inheritance pattern of LOAD. Disease segregation analyses identified the TTC3 p.S1038C substitution as the only rare, nonsynonymous variant completely shared among all LOAD family members. It reached a suggestive multipoint linkage LOD score of 2.66. Moreover, it was absent in 3 aged family members with confirmed normal cognition (figure 1). A fourth aged family member carried the variant in TTC3, but died at age 76, very close to the average age at onset for the 11 AD relatives with the variant, 75.7 years old. Therefore, it is possible that if this individual had lived longer, he may have developed AD. Thus, the TTC3 variant may, like APOE, contribute to LOAD risk; however, it is unlike APP, PSEN1, and PSEN2 and may not be sufficient to initiate AD pathogenesis.
LOAD is a common, complex, and age-related disease. Additional factors, such as the aggregation of several genetic risk variants, might contribute to LOAD. Individual 122 presented with the TTC3 risk allele and an APOE ε3/3 genotype, demonstrating that the presence of an APOE ε4 is not required to confer AD risk in this family (figure 1; table e-1). In addition, the family has 9 ε3/4 AD carriers and a single ε4/4 affected individual. However, the high abundance of APOE ε3/4 genotypes in family 1229 may not be sufficient to explain the apparent autosomal dominant inheritance pattern of LOAD. The average age of LOAD onset among affected family members was 75.7 years. A study representative of the US population determined the prevalence of LOAD at ≥70 years of age to be 9.7% (95% confidence interval: 7.6%–11.9%).28 The LOAD risk of APOE ε3/4 carriers is about 3-fold higher than that found in ε3/3 carriers, while ε4/4 carriers have a 15-fold higher risk of AD.4 We would expect ∼4 phenocopies attributable to the APOE ε4 allele in family 1229 by assuming that the single ε4/4 carrier and 30% of the 9 ε3/4 carriers suffered from LOAD due to their APOE genotype. Thus, it is highly unlikely that APOE ε3/4 accounts for the heavy burden of AD in this family.
TTC3 is an appealing LOAD candidate gene because of its involvement in pathways implicated in LOAD; functional studies in neuronal cell lines have shown that TTC3 plays an important role in ubiquitination, Akt signaling, negative cell cycle control, and inhibition of neuronal differentiation.29–31 In addition, the TTC3 protein carries conserved domains predicted to be involved in microtubule and actin-related processes of the cytoskeleton, apoptosis, cellular stress response, and mitochondrial function.32–36 Therefore, TTC3 might act in multiple LOAD-relevant pathways. Moreover, these processes are intertwined with each other with respect to normal neuronal and synaptic functioning and Aβ and protein tau pathology.32,37–39 The identification of additional AD families with rare, nonsynonymous variants potentially contributing to disease would strengthen TTC3 as an AD candidate gene.
During human embryogenesis, TTC3 becomes predominantly expressed in neuronal tissue with high expression in cortical layers of the brain.40,e1 Human postmortem brain microarray data demonstrate that cortical TTC3 expression is consistently lower in LOAD cases compared to controls without dementia.e2,e3 Moreover, TTC3 expression is negatively correlated with LOAD neuropathology markers Braak stage and frontal atrophy in the prefrontal cortex of LOAD cases. This pattern suggests that TTC3 activity might protect against LOAD; thus, variants that reduce TTC3 expression could contribute to LOAD risk. The p.S1038C substitution may alter the TTC3 protein's conformation and thereby its function via the formation of bisulfide bonds with other cysteine residues, for instance in the short Cys-His–rich motif of the C-terminal H2 ring finger domain of the protein,e4,e5 a domain required for ubiquitin ligase activity.30 TTC3 is an E3 ubiquitin ligase that acts as a specific terminal regulator of Akt signaling, a core intracellular pathway for cell survival.30 Akt/mTOR (mammalian target of rapamycin) signaling is a major cellular senescence pathway implicated in several age-related human diseases. Moreover, a large body of evidence attributes Akt/mTOR signaling as a key neuronal pathway that is disregulated in LOAD.e6 Therapeutic approaches designed to normalize the Akt/mTOR axis protected against AD pathology and cognitive decline in animal models.e7,e10 Some of these therapies modifying this pathway in LOAD patients have been successful.e11 Therefore, genetic variants that damage TTC3 protein function could contribute to LOAD susceptibility by reducing TTC3's ability to counteract activated Akt, thereby facilitating an increase in activity of the Akt/mTOR axis. Further molecular studies that decipher the potential consequences of the alteration reported here are required to fully understand how the genetic alteration may contribute to AD pathogenesis.
Therefore, this study demonstrates how WES of single, extended families with a high burden of LOAD can lead to the identification of rare, predicted damaging genetic variants with potentially considerable risk to LOAD.
Supplementary Material
ACKNOWLEDGMENT
The authors are grateful to the many patients with Alzheimer disease and their families for their participation, without whom this work would not be possible.
GLOSSARY
- AD
Alzheimer disease
- ADGC
Alzheimer's Disease Genetics Consortium
- APOE
apolipoprotein E
- APP
amyloid precursor protein
- ESP
Exome Sequencing Project
- FTD
frontotemporal dementia
- LOAD
late-onset AD
- MAF
minor allele frequency
- PSEN
presenilin
- SNV
single-nucleotide variant
- TTC3
tetratricopeptide repeat domain 3
- WES
whole-exome sequencing
Footnotes
Supplemental data at Neurology.org/ng
AUTHOR CONTRIBUTIONS
M.A.K., S.Z., L.A.F., R.P.M., J.L.H., E.R.M., G.W.B., J.R.G., G.D.S., and M.A.P.V., conceived and designed the experiments. M.A.K., H.N.C., K.L.H.N., B.W.K., E.R.M., G.W.B., J.R.G., and M.A.P.V., analyzed the exome sequencing data. M.A.K., K.L.H.N., B.W.K., and M.A.P.V., performed the linkage and statistical analysis. S.R., and P.L.W., performed the exome sequencing and Sanger sequencing validation. L.A.F., J.L.H., and G.D.S., collected ADGC samples and performed genotyping and analysis. J.M.V., M.L.C., and R.M.C., collected the samples, made diagnoses, and interpreted the clinical data from the AD families. M.A.K., and H.N.C., wrote the manuscript. B.W.K., L.A.F., J.R.G., and M.A.P.V., edited the manuscript. The authors jointly discussed the experimental results throughout the duration of the study. All authors read and approved the final manuscript.
STUDY FUNDING
This research was supported by grants from the NIH (1R01AG027944, UO1AG032984), the Alzheimer's Association (IIRG09133827), and the BrightFocus Foundation (A2011048).
DISCLOSURE
Dr. Kohli reports no disclosures. Dr. Cukier has received research support from the Brain & Behavior Research Foundation, the Hussman Foundation, and the Alzheimer's Association. Ms. Hamilton-Nelson has received research support from NIH. Ms. Rolati, Dr. Kunkle, Ms. Whitehead, and Dr. Züchner report no disclosures. Dr. Farrer has served on the editorial boards of the American Journal of Alzheimer's Disease & Other Dementias and Clinical Genetics; holds a patent (pending) for use of PLXNA4 as a drug target and biomarker for Alzheimer disease; has been a consultant for Novartis Pharmaceuticals, Gerson Lerman, Guidepoint Global, and Finnegan & Associates, LLP; and has received research support from NIH, the Fidelity Foundation, and the Thome Memorial Foundation. Dr. Martin serves on the editorial board of Frontiers in Statistical Genetics and Methodology and holds a patent for Test for Linkage and Association in General Pedigrees: The Pedigree Disequilibrium Test. Dr. Beecham has received funding from NIH and the Department of Defense. Dr. Haines has received travel funding/honoraria form the American Society of Human Genetics; First United Methodist Church in Schenectady, NY; BCN/Pitt Neurogenetics Meeting, and Novartis Vision Research day; receives publishing royalties for Current Protocols in Human Genetics (John Wiley & Sons); has received research support from the NIH/NIA Consortium for Alzheimer's Sequence Analysis, and NIH/NEI; and receives royalty payments for the use of APOE in clinical diagnostics, Athena diagnostics. Dr. Vance has served on an NIH grant's internal review for the University of Alaska, Fairbanks (received honoraria); has served on the editorial boards of the American Journal of Neurodegenerative Diseases and Neurology: Genetics; holds patents for Method of detecting Charcot-Marie-Tooth disease type 2A, TRPC6 involved in glomerulonephritis, and Methods for identifying an individual at increased risk of developing coronary artery disease; has received research support from NIH/NINDS; and receives royalty payments from Duke University for CMT2A with Athena. Dr. Cuccaro has served on the editorial board of Child Psychiatry & Human Development. Dr. Gilbert has received research support from the Alzheimer Association. Dr. Schellenberg has served on the Alzheimer's Association Veterans Affairs Cooperative Study Unit Executive Committee, the Autism Genome Project, the National Alliance for Autism Research Medical Advisory Board, the Society of Progressive Supranuclear Palsy Scientific Advisory Board, and the Alzheimer Research Consortium Executive Board—Autism Genome Project Peebler PSP Research Foundation; has received travel/speaker honoraria from the Alzheimer's Disease Center Directors Meeting, the CurePSP International Research Symposium, the Robert Katzman Memorial Lecture, the Colorado 25th Annual Southern California Alzheimer's Disease Research Conference, the University of California Institute for Memory Impairment and Neurological Disorders, Biomarkers in Neuropsychiatric Disorders, NIH, Novartis Institutes for Biomedical Research, Inc., McKnight Brain Institute, Keep Memory Alive Event Center, Lou Ruvo Center for Brain Health, Accelerated Medicines Program Alzheimer's Disease F2F Target Discovery and Preclinical Validation Project Meeting, and the American Association of Neuropathologists; has served on the editorial boards of the Journal of Neural Transmission, Alzheimer's Research, American Journal of Alzheimer's Disease & Other Dementias, Neurodegenerative Diseases, Current Alzheimer Research, and Pathology and Laboratory Medicine International; is an employee of the University of Pennsylvania; has been a consultant for Finnigan Law Firm; and has received research support from Integregen, NIA/NIH, and CurePSP CBD Solutions. Dr. Carney and Dr. Pericak-Vance report no disclosures. Go to Neurology.org/ng for full disclosure forms.
REFERENCES
- 1.Qiu C, Kivipelto M, von Strauss E. Epidemiology of Alzheimer's disease: occurrence, determinants, and strategies toward intervention. Dialogues Clin Neurosci 2009;11:111–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goate A, Chartier-Harlin MC, Mullan M, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature 1991;349:704–706. [DOI] [PubMed] [Google Scholar]
- 3.Sherrington R, Rogaev EI, Liang Y, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature 1995;375:754–760. [DOI] [PubMed] [Google Scholar]
- 4.Rogaev EI, Sherrington R, Rogaeva EA, et al. Familial Alzheimer's disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer's disease type 3 gene. Nature 1995;376:775–778. [DOI] [PubMed] [Google Scholar]
- 5.Gatz M, Reynolds CA, Fratiglioni L, et al. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry 2006;63:168–174. [DOI] [PubMed] [Google Scholar]
- 6.Pericak-Vance MA, Bebout JL, Gaskell PC, Jr, et al. Linkage studies in familial Alzheimer disease: evidence for chromosome 19 linkage. Am J Hum Genet 1991;48:1034–1050. [PMC free article] [PubMed] [Google Scholar]
- 7.Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 1993;261:921–923. [DOI] [PubMed] [Google Scholar]
- 8.Pericak-Vance MA, Haines JL. Genetic susceptibility to Alzheimer disease. Trends Genet 1995;11:504–508. [DOI] [PubMed] [Google Scholar]
- 9.Farrer LA, Cupples LA, Haines JL, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 1997;278:1349–1356. [PubMed] [Google Scholar]
- 10.Lambert JC, Ibrahim-Verbaas CA, Harold D, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet 2013;45:1452–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jonsson T, Stefansson H, Steinberg S, et al. Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med 2013;368:107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology 1984;34:939–944. [DOI] [PubMed] [Google Scholar]
- 13.Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin–rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 2002;30:97–101. [DOI] [PubMed] [Google Scholar]
- 14.Pericak-Vance MA, Bass MP, Yamaoka LH, et al. Complete genomic screen in late-onset familial Alzheimer disease. Evidence for a new locus on chromosome 12. JAMA 1997;278:1237–1241. [PubMed] [Google Scholar]
- 15.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010;20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011;43:491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.1000 Genomes Project Consortium, Abecasis GR, Altshuler D, et al. A map of human genome variation from population-scale sequencing. Nature 2010;467:1061–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tennessen JA, Bigham AW, O'Connor TD, et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science 2012;337:64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cooper GM, Stone EA, Asimenos G, et al. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res 2005;15:901–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Siepel A, Bejerano G, Pedersen JS, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 2005;15:1034–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chun S, Fay JC. Identification of deleterious mutations within three human genomes. Genome Res 2009;19:1553–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res 2011;39:e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods 2010;7:575–576. [DOI] [PubMed] [Google Scholar]
- 25.Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet 2013. Chapter 7: Unit 7.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and indels. PLoS One 2012;7:e46688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Purcell S, Cherny SS, Sham PC. Genetic Power Calculator: design of linkage and association genetic mapping studies of complex traits. Bioinformatics 2003;19:149–150. [DOI] [PubMed] [Google Scholar]
- 28.Plassman BL, Langa KM, Fisher GG, et al. Prevalence of dementia in the United States: the aging, demographics, and memory study. Neuroepidemiology 2007;29:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim SY, Lee JH, Huh JW, et al. Cigarette smoke induces Akt protein degradation by the ubiquitin-proteasome system. J Biol Chem 2011;286:31932–31943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Suizu F, Hiramuki Y, Okumura F, et al. The E3 ligase TTC3 facilitates ubiquitination and degradation of phosphorylated Akt. Dev Cell 2009;17:800–810. [DOI] [PubMed] [Google Scholar]
- 31.Berto G, Camera P, Fusco C, et al. The Down syndrome critical region protein TTC3 inhibits neuronal differentiation via RhoA and citron kinase. J Cell Sci 2007;120:1859–1867. [DOI] [PubMed] [Google Scholar]
- 32.Bamburg JR, Bloom GS. Cytoskeletal pathologies of Alzheimer disease. Cell Motil Cytoskeleton 2009;66:635–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eki T, Abe M, Naitou M, et al. Cloning and characterization of novel gene, DCRR1, expressed from Down's syndrome critical region of human chromosome 21q22.2. DNA Seq 1997;7:153–164. [DOI] [PubMed] [Google Scholar]
- 34.Behl C. Apoptosis and Alzheimer's disease. J Neural Transm 2000;107:1325–1344. [DOI] [PubMed] [Google Scholar]
- 35.Hirai K, Aliev G, Nunomura A, et al. Mitochondrial abnormalities in Alzheimer's disease. J Neurosci 2001;21:3017–3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chaturvedi RK, Flint Beal M. Mitochondrial diseases of the brain. Free Radic Biol Med 2013;63:1–29. [DOI] [PubMed] [Google Scholar]
- 37.Arendt T, Bruckner MK. Linking cell-cycle dysfunction in Alzheimer's disease to a failure of synaptic plasticity. Biochim Biophys Acta 2007;1772:413–421. [DOI] [PubMed] [Google Scholar]
- 38.Bhaskar K, Miller M, Chludzinski A, Herrup K, Zagorski M, Lamb BT. The PI3K-Akt-mTOR pathway regulates Abeta oligomer induced neuronal cell cycle events. Mol Neurodegener 2009;4:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Caccamo A, Magrì A, Medina DX, et al. mTOR regulates tau phosphorylation and degradation: implications for Alzheimer's disease and other tauopathies. Aging Cell 2013;12:370–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ohira M, Ootsuyama A, Suzuki E, et al. Identification of a novel human gene containing the tetratricopeptide repeat domain from the Down syndrome region of chromosome 21. DNA Res 1996;3:9–16. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.