
Fig. 3.
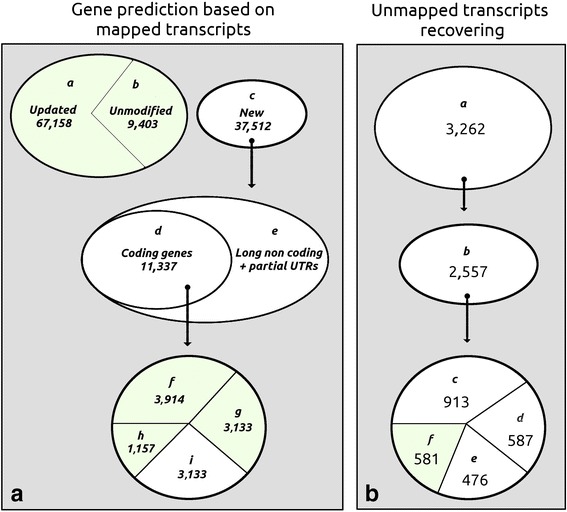
Gene prediction statistics. Statistics inferred using the mapped transcripts are shown in (a), while statistics inferred using the unmapped contigs are evidenced in (b). The light green areas indicates the reliable transcripts selected as result of this analysis. b the areas (a) and (b) represent the updated and unmodified gene predictions referred to the genome annotation data. The new gene prediction stressed in (c) can be classified into coding genes (d) and unreliable information such as long non-coding genes and partial UTRs (e). About 62.2 % of coding genes (d) consisting of (f) and (g) were significantly similar to a sequence stored into the non-redundant database; while a percentage of 37.8 % of (d) consisting of (g) and (h) had significant long ORF predictions. (i) represents the number of unreliable coding genes discarded, while (a), (b), (f), (g) and (h) represent the gene predictions considered in the gene expression analysis. b the area (a) represents the number of transcripts (3262) that mapped less than 10 % of their sequence length onto the reference genome. (b) 2557 out of 3262 transcripts evidenced a significant coding potential, (c) 913 transcripts had ORFs open at 5′-end; (d) 587 transcripts resulted in ORFs open at both ends; (e) 476 transcripts resulted in ORFs open at 3′-end, and (f) 581 resulted complete ORFs