

Abstract
ULK1 and ATG13 assemble with RB1CC1/FIP200 and ATG101 to form a macroautophagy (hereafter autophagy) induction (ULK1) complex in higher eukaryotes. The yeast counterpart, the Atg1 complex, is comprised of Atg1 and Atg13 (ULK1 and ATG13 homologs), Atg17 (a proposed functional homolog of RB1CC1), and either the Atg101 subunit (in Schizosaccharomyces pombe) or the Atg29-Atg31 heterodimer (in Saccharomyces cerevisiae). With mutual exclusivity of, and no detectable homology between, the Atg29-Atg31 dimer and Atg101, knowledge about the roles of these proteins in autophagy induction is an important piece in the puzzle of understanding the molecular mechanism of autophagy initiation. A recent study reporting the structure of the S. pombe homolog Atg101 bound to the Atg13HORMA domain is a notable contribution to this knowledge (see the punctum in this issue of the journal).
Keywords: Atg protein, autophagy, stress, structure, vacuole, yeast
Suzuki et al., purified Atg101 and the HORMA domain of Atg13 from S. pombe (Sp) and solved the crystal structure of the SpAtg101-SpAtg13HORMA complex.1 They found that SpAtg101 adopts the HORMA domain fold, as does the N terminus of SpAtg13. Overall, the SpAtg101-SpAtg13HORMA heterodimer resembles the asymmetric homodimer consisting of the open (O) and closed (C) conformations of the Mad2 protein, a well-studied HORMA domain family member. Mad2 is a remarkable protein in that it is able to switch between open (autoinhibited) and closed (active) conformations. The switch from O-Mad2 to the C-Mad2 occurs via an intriguing mechanism in which the N and C termini traverse from one side of the stable core (comprised of 3 α helices and 3 β sheets) of the protein to another. The resulting active C-Mad2 conformation exposes an interacting interface formed by the stable β6 strand and the traversing β6-β8 loop (named the safety belt). Thereby, the flexible safety belt recognizes a target (Fig. 1A). The structure of the SpAtg101-SpAtg13HORMA complex (Fig. 1B) reveals that SpAtg101 adopts an O-Mad2-like conformation that, in contrast to O-Mad2, appears to be locked and incapable of switching. The suggested reason for this lock is that the αC-β5 loop in Atg101 is shorter than the corresponding loop in O-Mad2. A mutation that shortens the αC-β5 loop in O-Mad2 also locks the open conformation.2 Another distinct feature in the O-Mad2-like SpAtg101 is a longer β4-β5 loop that is enriched in conserved Trp and Phe residues; denoted as the WF finger. It remains unclear how or if the WF finger positions and interplays with the disordered C terminus of 21 residues that is missing in the crystal structure. Interestingly, this highly mobile C terminus that follows the β6 strand in SpAtg101 maps onto the safety belt and 2 β strands in Mad2.
Figure 1.
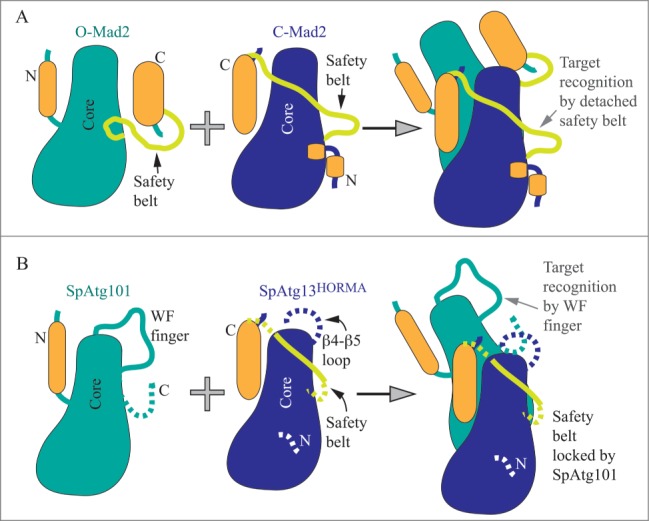
Schematic models of HORMA domain architectures. (A) Open (O-Mad2) autoinhibited and closed (C-Mad2) active conformation of the Mad2 protein that together form the asymmetric homodimer. The structurally stable core (in teal or blue) consists of αA, αB, and αC helices and β4, β5, and β6 strands. During the conformational change from O-Mad2 to C-Mad2, the N and C termini (in orange; consisting mostly of β strands) traverse from one side of the core to another. O-Mad2 stabilizes C-Mad2, in which the safety belt is detached. The target (a ligand) is recognized by forming the intermolecular β sheet with β6 of C-Mad2 that also uses its safety belt (light green) to tighten binding with the ligand. (B) The O-Mad2-like SpAtg101 interacts with the C-Mad2-like SpAtg13HORMA to form a heterodimer. The O-Mad2-like conformation of SpAtg101 is locked (by the short αC-β5 loop within the core). The WF finger acts in target recognition. The function of the missing (likely disordered) C terminus is unknown. The C-Mad2-like conformation of SpAtg13HORMA is very unstable and requires Atg101 for stabilization, which at the same time locks the safety belt and disables its function. The purpose of the β4-β5 loop in SpAtg13HORMA is not known. Dashed lines represent very flexible domains missing in the crystal structure.
In comparison to the O-Mad2-like conformation of SpAtg101, the HORMA domain of SpAtg13 adopts the locked C-Mad2-like conformation, similar to the structure of the Lachancea thermotolerans Atg13HORMA domain reported previously.3 L. thermotolerans is a budding yeast as opposed to S. pombe. A comparison of the topology of the Atg13HORMA domain from budding and fission yeast reveals that the budding yeast Atg13HORMA domain is stabilized by locking its conformation via an additional 3-stranded β sheet (comprised of the β4″, β4,″ and β8″ strands), named the cap. This locking substructure is not seen in the SpAtg13HORMA, and the sequence alignment presented in the paper1 suggests that it is also absent in ATG13 from higher eukaryotes. The absent cap together with numerous missing residues in the crystal structure of the SpAtg13HORMA suggests that this architecture alone is very unstable. So, what is responsible for the stability to the Atg13HORMA from S. pombe? Suzuki et al. show that it is SpAtg101 interacting with SpAtg13HORMA. The interacting surface in the SpAtg101-SpAtg13HORMA heterodimer is analogous to that in the Mad2 asymmetric homodimer, and consists of αA, αC, β3, and the αA-β2 loop in SpAtg101 and of αC, β8′, and the αA-αB loop in SpAtg13HORMA. An intermolecular β sheet formed by the interacting loops of SpAtg101 and SpAtg13HORMA strengthen this interaction. The contacts observed in the structure were validated by mutational analysis.
Probing the functional importance of the heterodimer showed that without binding to ATG13HORMA, ATG101 cannot incorporate into the ULK1 complex, albeit the assembly of other subunits in the complex is independent of the presence of ATG101. Besides stabilization of ATG13HORMA, the ATG101-ATG13 interaction also stabilizes the ULK1 subunit in the complex. Moreover, the impairment in this interaction leads to accumulation of SQSTM1/p62, an autophagy receptor protein, and to a decrease in autophagic flux, measured as the bafilomycin A1-dependent increase in LC3-II. Both of these autophagic readouts are also sensitive to mutation in the WF finger of ATG101, despite the results showing no involvement of the WF finger in the ATG101-ATG13 interaction. Therefore, the authors tested whether the mutations in the ATG101-ATG13 interface and in the WF finger affect the recruitment of other autophagy proteins, in particular LC3, WIPI1, and ZFYVE1/DFCP1. Indeed, the ATG101-ATG13 interaction interface and the WF finger were found to be important for the recruitment of these downstream factors to the autophagosome-formation site in mouse embryonic fibroblasts. These data were interpreted to indicate that the WF finger, a unique feature in ATG101 positioned near its β6 strand, is needed for autophagy, as is the interaction of ATG101 with ATG13HORMA. The authors proposed that the WF finger along with the β6 strand in ATG101 acts in target recognition. Suzuki et al. conclude that ATG101 has at least 2 functions in autophagy, which are the stabilization of the ATG13HORMA architecture and the recruitment of downstream factors to the autophagosome-formation site.
The novelty of this study is that it offers an explanation for structural and mechanistic differences between the ULK1 and Atg1 complex. A key revelation is that the C-Mad2-like conformation of ATG13HORMA, unlike C-Mad2, appears to be unable to utilize the function of the flexible safety belt, because the safety belt is locked, and the locking mechanism appears to be different between fission and budding yeast. In fission yeast, Atg101 is the lock that prevents the safety belt in the C-Mad2-like Atg13HORMA from recognizing a target. The WF finger and the β6 strand in the O-Mad2-like conformation of Atg101 serve as a target recognition site instead (Fig. 1B). In budding yeast, the safety belt in the C-Mad2-like Atg13HORMA is proposed to be locked and prevented from target recognition by the cap, the substructure not seen in C-Mad2 or the S. pombe C-Mad2-like Atg13HORMA. Thus, the C-Mad2-like Atg13HORMA in budding yeast engages an intramolecular lock, whereas the fission yeast C-Mad2-like Atg13HORMA uses an intermolecular lock, Atg101, that also provides a recognition site (the WF finger and β6 strand). How the C-Mad2-like Atg13HORMA in budding yeast recognizes a target remains to be discovered. The Atg29-Atg31 heterodimer, which is mutually exclusive with Atg101, does not contain a homologous WF finger, suggesting that the mechanism of downstream factor recruitment in budding yeast will rely on a somewhat different principle. An intriguing question also remains as to why the evolution to higher eukaryotes favored an intermolecular locking mechanism over the intramolecular one in the Atg13HORMA domain. Finding why and how the inherent instability of the C-Mad2-like SpAtg13HORMA in the absence of Atg101 is beneficial can be a clue in solving the mechanism of autophagy regulation. Another interesting structural point is how the mechanism of the autophagy pathway modifies the mobility of the Atg101 and Atg13 HORMA domains compared to Mad2 (Fig. 1). Mad2 employs mobility via movement of the N and C termini from one side of the core to the other and by detachment of the safety belt. In contrast, the autophagic HORMA domains of SpAtg101 and SpAtg13 do not traverse their N and C termini around the core. Instead, the O-Mad2-like SpAtg101 engages the WF finger and somehow moves the very flexible C terminus of 21 residues. The C-Mad2-like SpAtg13HORMA with overall unstable (mobile) architecture carries a long disordered β4-β5 loop of approximately 25 residues, which interestingly maps on the WF finger in the O-Mad2-like SpAtg101 or on the β4'and β4″ strands of the cap (in budding yeast Atg13HORMA), and which is missing in the crystal structure. The functionality of this just described distribution of plasticity in Atg101 and Atg13HORMA and the purpose of the missing domains for autophagy regulation remain to be elucidated.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by NIH grant GM053396 to DJK and the Protein Folding Diseases FastForward Initiative, University of Michigan.
References
- 1.Suzuki H, Kaizuka T, Mizushima N, Noda NN. Structure of the Atg101-Atg13 complex reveals essential roles of Atg101 in autophagy initiation. Nat Struc Mol Biol 2015; 22:572-580; PMID: 26030876; http://dx.doi.org/ 10.1038/nsmb.3036 [DOI] [PubMed] [Google Scholar]
- 2.Mapelli M, Massimiliano L, Santaguida S, Musacchio A. The Mad2 conformational dimer: structure and implications for the spindle assembly checkpoint. Cell 2007; 131:730-743; PMID: 18022367; http://dx.doi.org/ 10.1016/j.cell.2007.08.049 [DOI] [PubMed] [Google Scholar]
- 3.Jao CC, Ragusa MJ, Stanley RE, Hurley JH. A HORMA domain in Atg13 mediates PI 3-kinase recruitment in autophagy. Proc Nat Acad Sci USA 2013; 110:5486-5491; PMID: 23509291; http://dx.doi.org/ 10.1073/pnas.1220306110 [DOI] [PMC free article] [PubMed] [Google Scholar]