
Figure 1.
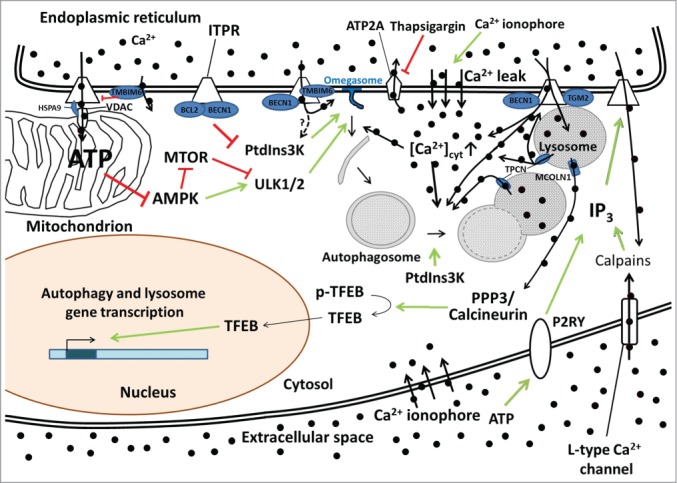
The various possible mechanisms of Ca2+-ITPR-mediated control of autophagy. Constitutive ITPR-mediated Ca2+ release into mitochondria inhibits a proximal step in the autophagy pathway by fueling mitochondrial energetics and ATP production and limiting AMPK activity. The ER Ca2+-leak channel TMBIM6 can impede ATP production by lowering the steady-state ER Ca2+ concentration and thus reduce the amount of Ca2+ available for transfer into the mitochondria. ITPRs can also function as scaffolding molecules, thereby suppressing autophagy independently of their Ca2+-release activity by promoting the interaction of BCL2 with BECN1 and thus preventing the formation of the active class III phosphatidylinositol 3-kinase (PtdIns3K) complex. ITPR-mediated Ca2+ release can also be enhanced by BECN1 and TMBIM6 and dampened by TGM2, thereby influencing omegasome formation (possibly through PtdIns3K activation) and autophagosome maturation/trafficking. ITPR-mediated Ca2+ release can also influence the lysosomal Ca2+ concentration and lysosomal Ca2+ release through TPCNs, likely influencing lysosomal fusion events, or through MCOLN1, influencing autophagic and lysosomal gene transcription through a pathway involving PPP3/calcineurin and TFEB. TPCNs reciprocally also influence ITPRs via a Ca2+-induced Ca2+-release mechanism. Autophagosome synthesis, maturation and fusion are also affected by Ca2+-mobilizing agents such as thapsigargin (that inhibits the ER Ca2+ pump ATP2A) and Ca2+ ionophores that increase the cytosolic Ca2+ concentration ([Ca2+]cyt). IP3 production and the subsequent IP3-mediated Ca2+ release can also be regulated by a feedback loop involving calpain activation by L-type Ca2+ channel-mediated Ca2+ entry or the activation of P2RY (purinergic receptor, G-protein coupled). The black circles represent Ca2+ ions, with thick black arrows indicating the direction of the Ca2+ fluxes. Green arrows indicate stimulatory effects, red lines inhibitory ones.