

Abstract
An aberrant interaction between hematopoietic stem cells and mesenchymal stromal cells has been linked to disease and shown to contribute to the pathophysiology of hematologic malignancies in murine models. Juvenile myelomonocytic leukemia is an aggressive malignant disease affecting young infants. Here we investigated the impact of juvenile myelomonocytic leukemia on mesenchymal stromal cells. Mesenchymal stromal cells were expanded from bone marrow samples of patients at diagnosis (n=9) and after hematopoietic stem cell transplantation (n=7; from 5 patients) and from healthy children (n=10). Cells were characterized by phenotyping, differentiation, gene expression analysis (of controls and samples obtained at diagnosis) and in vitro functional studies assessing immunomodulation and hematopoietic support. Mesenchymal stromal cells from patients did not differ from controls in differentiation capacity nor did they differ in their capacity to support in vitro hematopoiesis. Deep-SAGE sequencing revealed differential mRNA expression in patient-derived samples, including genes encoding proteins involved in immunomodulation and cell-cell interaction. Selected gene expression normalized during remission after successful hematopoietic stem cell transplantation. Whereas natural killer cell activation and peripheral blood mononuclear cell proliferation were not differentially affected, the suppressive effect on monocyte to dendritic cell differentiation was increased by mesenchymal stromal cells obtained at diagnosis, but not at time of remission. This study shows that active juvenile myelomonocytic leukemia affects the immune response-related gene expression and function of mesenchymal stromal cells. In contrast, the differential gene expression of hematopoiesis-related genes could not be supported by functional data. Decreased immune surveillance might contribute to the therapy resistance and progression in juvenile myelomonocytic leukemia.
Introduction
The bone marrow (BM) niche represents the supportive environment for hematopoietic stem cells (HSC).1,2 Mesenchymal stromal cells (MSCs), being precursors to osteoblasts, adipocytes and chondrocytes and a cellular constituent of the niche, are crucial for maintenance of quiescent HSC.3 MSCs, or differentiated subpopulations of these cells, are used in vitro as a model for the BM microenvironment. Soluble factors as well as direct cell-to-cell contact have been described to play a role in normal MSC-HSC interaction.4,5
Hematopoietic malignancies such as leukemia originate in the BM. Although leukemic blast cells can be detected throughout the body during disease, the leukemic stem cells are thought to remain in the BM, and more specifically in the hematopoietic stem cell niche.6 It is widely accepted that malignant cells have a negative impact on the normal hematopoiesis causing anemia and thrombocytopenia. However, the effect of the malignant cells on the BM microenvironment has not been studied extensively.
Recent studies in mice have demonstrated that myeloid neoplasms affect the normal niche structure.7–9 These alterations contribute potentially to the formation of the leukemic niche in which leukemic stem cells are difficult to target by conventional chemotherapy or irradiation.10 Studies describing MSC characteristics in human myeloproliferative neoplasms are mostly limited to adult patients, demonstrating conflicting results with regard to genetic abnormalities, gene expression and MSC function.11–14
Juvenile myelomonocytic leukemia (JMML) is an aggressive leukemia occurring in young children, predominantly in infants between birth and four years of age. Patients usually present with hepatosplenomegaly, fever and monocytosis.15 Monosomy 7 is the most common karyotype abnormality detected in 25% of cases, and numerous leukemogenic mutations have been identified mainly involving the RAS-RAF-ERK pathway, e.g. PTPN11, K-RAS and c-CBL.16,17 Hematopoietic stem cell transplantation (HSCT) is the standard first-line treatment. Unfortunately, the 1-year relapse rate ranges between 30% and 50%.18,19
We hypothesize that the aggressive and therapy-resistant characteristics of JMML may point towards an altered BM microenvironment based on previous experimental data suggesting support of a neoplasm by the stromal compartment.8,9 In the current study, we aimed to identify stromal factors involved in the support of JMML. MSCs of patients with JMML were obtained at diagnosis and after HSCT, and expanded from BM. These patient-derived MSCs were compared to healthy pediatric donor-derived MSCs by investigating their capacity for immunomodulation and hematopoietic support and their gene expression profiles.
Methods
Patients
Children referred to our center for HSCT were included in this study according to a protocol approved by the institutional review board (P08.001). BM of 9 consecutive children with JMML was collected prior to treatment initiation. In addition, BM after HSCT was collected from 5 of these 9 children. The patients were classified following previously described criteria. BM samples were sent to the EWOG-MDS reference center in Freiburg, Germany, for mutation analysis. BM samples of pediatric HSCT donors (n=10) were used as control group (HC). Informed consent was obtained from all parents. This study was conducted according to the Declaration of Helsinki. Details of the methods used are available in the Online Supplementary Appendix.21
Mesenchymal stromal cell expansion and characterization
Mesenchymal stromal cell were expanded from fresh BM and characterized as previously described.22 Monosomy 7 by FISH and chimerism by variable number of cytosine adenine (CA) repeat analysis were determined in expanded MSC before and after HSCT, respectively.23,24 MSC gene expression and function was investigated using MSCs obtained at passage 2–3 and 3–5, respectively.
Functional assays
The effect of MSCs on proliferation of peripheral blood mononuclear cells (PBMCs) was investigated in co-cultures stimulated with phytohemagglutinin (PHA) by measuring 3H-thymidine incorporation.
The suppressive effect of MSC on NK-cell activation was determined by stimulation of purified NK cells with IL-2 for five days. NK-cell activation was measured by flow cytometry investigating DNAM-1, NKp30 and NKp44 expression.
To evaluate the effect of MSCs on antigen-presenting cells, monocytes were isolated from PBMCs and cultured with growth factors for five or seven days to differentiate towards immature dendritic cells (DC) or mature DC, respectively. Cells were phenotyped for the expression of CD14 and CD1a after co-culture.
Short-term co-culture assays with MSCs and hematopoietic progenitor cells (HPC) were performed to determine the supportive capacity of MSCs for HPCs maintenance and differentiation. Therefore, HPCs were isolated from healthy transplant donors using CD34 positive selection. Proliferation (day 7) and differentiation (days 7 and 14) were assessed using 3H-thymidine incorporation and flow cytometry, respectively.
To determine a direct effect of MSCs on HPC differentiation into colony forming units (CFU), MSCs were added to freshly purified HPCs in methylcellulose containing growth factors and cultured for 14 days (CFU-assay).
Gene expression
Total RNA was isolated from MSCs and mRNA was profiled using Deep-SAGE (serial analysis of gene expression) sequencing using Illumina technology.25 Data were mapped against the UCSC-hg19 reference genome using Bowtie, with permission of one mismatch and only retaining unique mappings.
Expression of genes of interest was validated using independent biological samples by RT-qPCR after generation of cDNA using the listed primers (Online Supplementary Table S1), as previously described.25 Expression levels were calculated relative to the average expression of two housekeeping genes.
Statistical analysis
Mann-Whitney and Wilcoxon matched-pairs signed-rank tests were performed to compare different groups in functional assays and in the validation of mRNA expression. Differential gene expression analysis was performed in R, using EdgeR, Globaltests and Limma data analysis packages.26–28 Correction for multiple testing was performed according to Benjamini and Hochberg.29 STRING software was used for analysis of protein interactions.30 Adjusted P-values <0.05 were considered statistically significant.
Results
Patients
Mesenchymal stromal cells were successfully expanded from all 9 patients with JMML (Table 1) at diagnosis and 10 HC. In addition, 7 BM samples collected after HSCT, derived from 5 of the 9 JMML patients included, were used for MSC expansion (Table 1). These MSCs were of patient origin, as tested by CA-repeat analysis. Median age at diagnosis was 2.1 years (range 0.5–3.5), whereas the age of HC ranged between 1.1 and 16.4 years (median 7.4). Children with different genetic mutations were included as shown in Table 1. Monosomy 7, present at diagnosis in 2 children, was not detected in the MSCs at diagnosis.
Table 1.
Patients’ characteristics.
Mesenchymal expansion and characterization
All expanded MSC populations fulfilled the criteria proposed by Horowitz et al.,31 i.e. differentiation towards adipocytes and osteoblasts, a characteristic phenotype (CD73+, CD90+, CD105+, HLA-DR− and no expression of lineage markers) and adherence to plastic. Representative examples of differentiation of MSCs prior to and after HSCT are shown in Figure 1A and B. Besides these criteria, MSCs have been described to suppress PHA-induced PBMC proliferation. MSCs derived from patients and controls did not differ in their immunosuppressive capacity of PHA-induced PBMC proliferation (Figure 1C).
Figure 1.
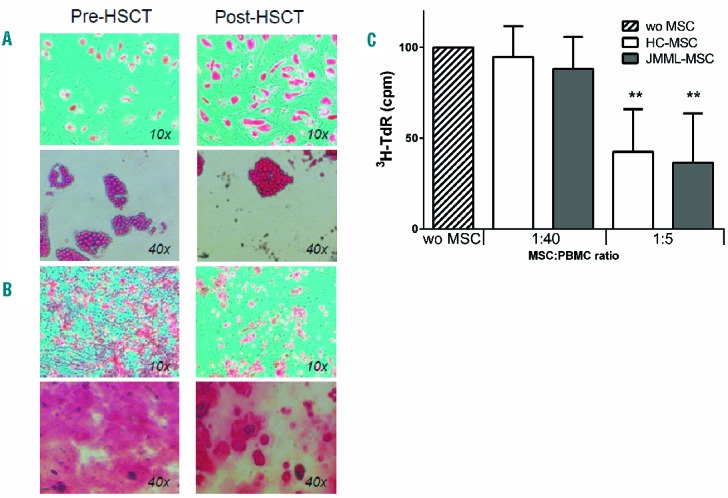
Expansion and characterization of mesenchymal stromal cells (MSC) from juvenile myelomonocytic leukemia (JMML) patients at diagnosis and post-hematopoietic stem cell transplantation (HSCT) and healthy controls. MSCs were expanded and differentiation capacity towards adipocytes (A) and osteoblasts (B) was evaluated. MSCs were cultured with differentiation factors for three weeks and Oil Red O and Alizarine Red was used to stain fat and calcium deposition, respectively (examples are representative for MSCs of 9 JMML patients at diagnosis and 5 JMML patients after HSCT). (C). Healthy control (HC)-MSCs and JMML-MSCs showed a comparable dose-dependent suppressive effect on PBMC proliferation after PHA stimulation. Boxes indicate the mean proliferation at the indicated PBMC:MSC ratio relative to the proliferation induced without (wo) MSCs. Error bars represent standard deviation. Experiments were performed with n=7 JMML-MSCs and n=4 HC-MSCs. Statistics were performed using Mann-Whitney tests: ** P<0.01.
Hematopoietic support
Mesenchymal stromal cells have been described to play an important role in the tight regulation of hematopoiesis in the BM. We investigated the capacity of JMML-MSCs derived at diagnosis to support hematopoiesis in vitro. HPC proliferation induced by SCF and Flt3-L was enhanced in the presence of MSCs and dependent on the HPC : MSC ratio (Figure 2A). JMML-MSCs and HC-MSCs did not differ in this respect. Despite increased HPC proliferation, the percentage of CD34+ cells after 14 days of culture was significantly better maintained in the presence of MSCs (Figure 2B). Cells that lost CD34 expression expressed lineage markers such as CD14. The frequency of CD14+ cells amongst CD45+ lineage-committed cells was significantly higher after 7–14 days of culture of CD34+ cells with MSCs (Figure 2C). The supportive effect by MSCs was comparable between MSCs of JMML patient and MSCs of HC.
Figure 2.
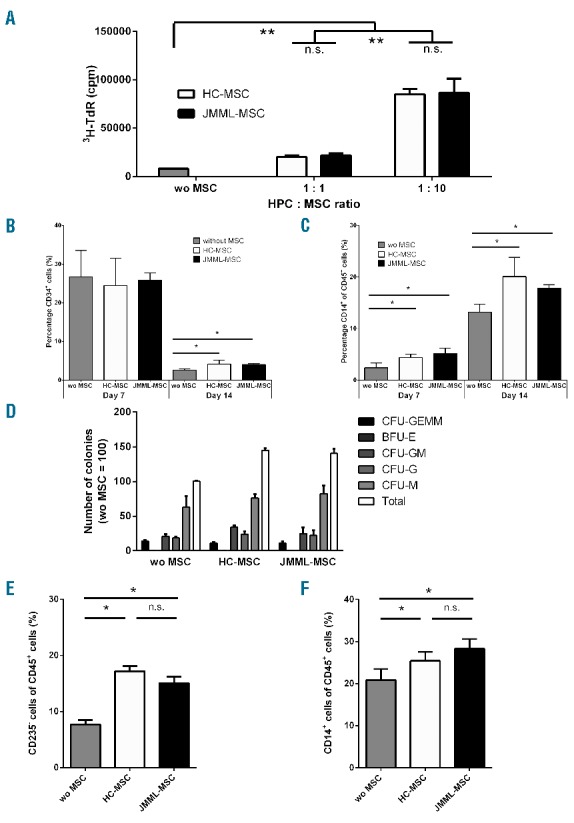
Juvenile myelomonocytic leukemia mesenchymal stromal cells (JMML-MSC) support the proliferation and differentiation of HPCs in vitro. (A). Both in the presence of JMML patient and healthy control (HC) derived MSCs the proliferation of CD34+ cells (HPCs) was increased after seven days of culture. MSCs alone did not show 3H-thymidine incorporation (data not shown). (B). HPCs lost the expression of CD34 after 14 days of culture; however, in the presence of MSCs (HPC : MSC ratio 1:5) the decline in CD34 expression was diminished. (C). HPCs acquired lineage markers, e.g. CD14, but no differences were seen between JMML-MSCs and HC-MSCs. (D). A significantly (P<0.05) increased number of colonies was seen in CFU-assays in the presence of MSCs (HPC : MSC ratio 1:60). (E and F). Cells harvested after colony formation contained increased percentages of CD235a negative and CD14 positive cells within the CD45+ cell population. Boxes indicate the mean, and error bars represent standard deviation. Experiments were performed with n=4 JMML-MSCs and n=2 HC-MSCs. Statistics were performed using Mann-Whitney tests: *P<0.05; **P<0.01; n.s.: non-significant.
To evaluate the effect of MSCs on functional differentiation of HPCs, MSCs were added to semi-solid cultures of isolated HPCs in methylcellulose containing growth factors. Both the total number of CFU and the different types of colonies increased in the presence of MSCs (Figure 2D). The percentage of CD45+CD235a− cells was increased after culture with MSCs suggesting support of differentiation and proliferation of myeloid cells at the cost of CD45−CD235a+ erythroblasts in this culture condition (Figure 2E). Indeed, also in this culture configuration the percentage of monocytes (CD14+ cells) was significantly increased in the presence of both HC as well as JMML-MSCs (Figure 2F). Also in these differentiation assays, no difference was observed between JMML-MSCs and HC-MSCs.
Gene expression analysis
Because the functional tests executed so far did not reveal differences between JMML- and HC-MSCs, we explored possible differences in mRNA expression profiles by performing Deep-SAGE sequencing, a next generation sequencing-based approach with higher sensitivity than traditional microarrays (de novo JMML n=8; HC n=8).32 The median number of obtained reads that fulfilled quality control criteria was 15.9×106 reads (range 11.4×106–30.6×106). A median of 65.6% of all reads aligned uniquely to the reference genome (range 59.3%–68.4%). The percentage of the aligned reads mapping to an annotated exon was 84.5% (range: 74.7%–86.3%).
The differentially expressed genes (n=162; P<0.001) are listed in Online Supplementary Table S2. A heat-map of the top 100 differentially expressed genes shows clustering of the JMML- and HC-derived MSCs (Online Supplementary Figure S1). After correction for multiple testing, in total 43 genes were differentially expressed between JMML and HC MSCs [criterion: false discovery rate (FDR) < 0.05; see Online Supplementary Table S2)]. The top 6 genes (Dickkopf-1 (DKK1), ectonucleotide pyrophosphatase/phosphodiesterase 2 (ENPP2), DEAD (Asp-Glu-Ala-Asp) box helicase 17 (DDX17), monooxygenase DBH-like 1 (MOXD1), Adenomatosis Polyposis Coli Down-Regulated 1-Like (APCDD1L) and tumor necrosis factor alpha-induced protein 2 (TNFAIP2)), that were expressed at intermediate to high levels (> 100 reads per million sequenced reads, in both patients and controls) were selected for RT-qPCR validation (Figure 3).
Figure 3.
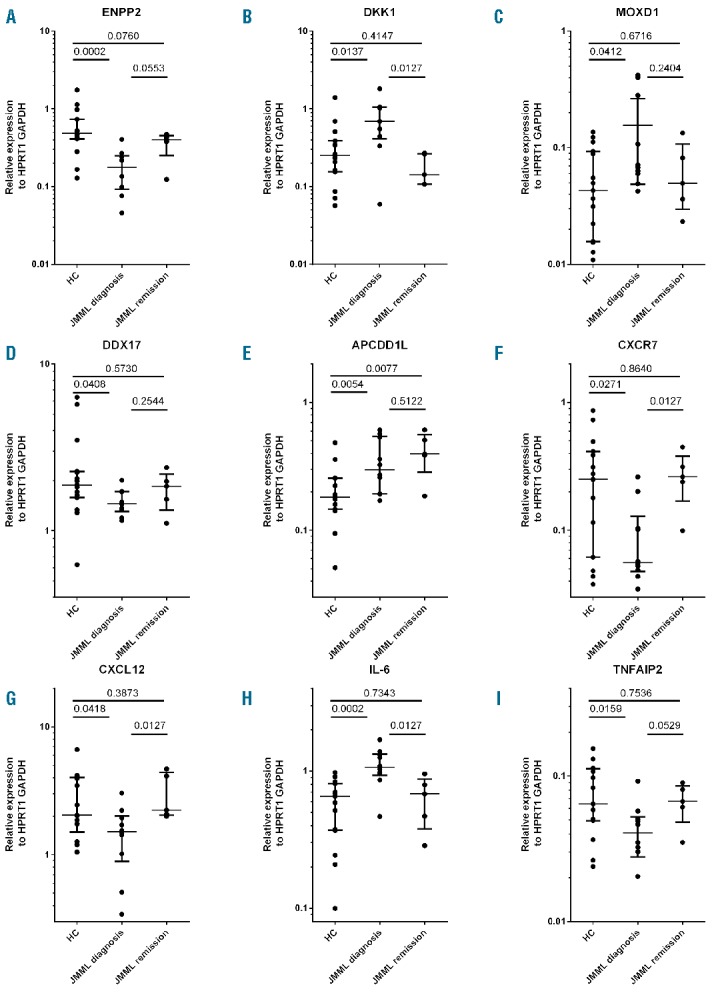
Gene expression of differentially expressed genes in mesenchymal stromal cells of juvenile myelomonyctic leukemic (MSC-JMML) patients at time of diagnosis and after hematopoietic stem cell transplantation (HSCT). Genes identified by Deep-SAGE sequencing were validated by RT-qPCR in JMML-MSCs expanded from bone marrow samples obtained prior to (n=9) and after (n=6) HSCT at time of remission and in MSCs derived from healthy donors (HC, n=10). Expression levels were expressed relative to that of the average of the housekeeping genes HPRT1 and GADPH. The results are indicated as median (horizontal line) and interquartile range (whiskers) on a logarithmic scale. Statistics were performed using Mann-Whitney tests.
In addition, 3 out of the 162 differentially expressed genes were previously reported to be involved in MSC function and were validated using RT-qPCR (CXCL12, CXCR7 and IL-6, Figure 3G, F and H, respectively). CXCL12 (Figure 3G), previously reported to be of importance in HSC-MSC interaction and mobilization of HSCs, was found to be significantly decreased in JMML-MSCs.3 Whereas the commonly involved receptor CXCR4 was not differentially expressed, expression of the alternative receptor CXCR7 was significantly decreased in JMML-MSCs (Figure 3F). String analysis of the top differentially expressed genes (data not shown) revealed that many of the genes up-regulated in JMML-MSCs are associated with the IL-1 superfamily [(IL-1β, IL-6 (Figure 3H), PTHLH, CLU, ATF3, PENK, RGS3 and RGS16)]. DKK1 expression (Figure 3B), related with osteolysis, was also increased. In contrast, expression of genes in the leptin pathway was decreased (LEP, LEPR, KISS1, SLC25A27, RXRA and CBLB). Results of gene ontology (P<0.001 after Holms correction and at least 100 co-variates) are listed in the Online Supplementary Table S3. Pathways related to immune responses and protein ubiquination, involved in cell regulation of cellular interaction,33 were predominantly affected.
Normalization of gene expression in JMML-MSCs after HSCT
Genes of interest, identified by Deep-SAGE, were studied in patient samples after HSCT. BM obtained at remission was available for MSC expansion of 4 patients (5 samples in total). RT-qPCR was used to study gene expression in these samples. ENPP2, DDX17, CXCR7, CXCL12 and TNFAIP2 expression was decreased in JMML-MSCs at diagnosis. However, expression was restored to the level of HC-MSC in samples after HSCT (Figure 3A, D, F, G and I). DKK1, MOXD1 and IL-6 expression was increased in JMML-MSCs at diagnosis, but normalized in JMML-MSCs post-HSCT (Figure 3B, C, and H). APCDD1L (Figure 3E), a paralog of the WNT inhibitor APCDD1,34 was the only gene studied, of which the abnormal (increased) expression at diagnosis remained unchanged after HSCT. BM at time of relapse post HSCT was only available in 2 patients and, therefore, these data were not included in the gene expression analysis.
Immunomodulation
Differential expression of IL-6 and other genes in the IL-1 superfamily suggests a differential effect of JMML patient-derived MSCs on the innate immune system. Escape from NK-cell surveillance is an important survival mechanism in tumorigenesis. However, HC-MSCs and JMML-MSCs derived from BM obtained at diagnosis suppressed NK-cell activation to a similar extent (Figure 4A).
Figure 4.
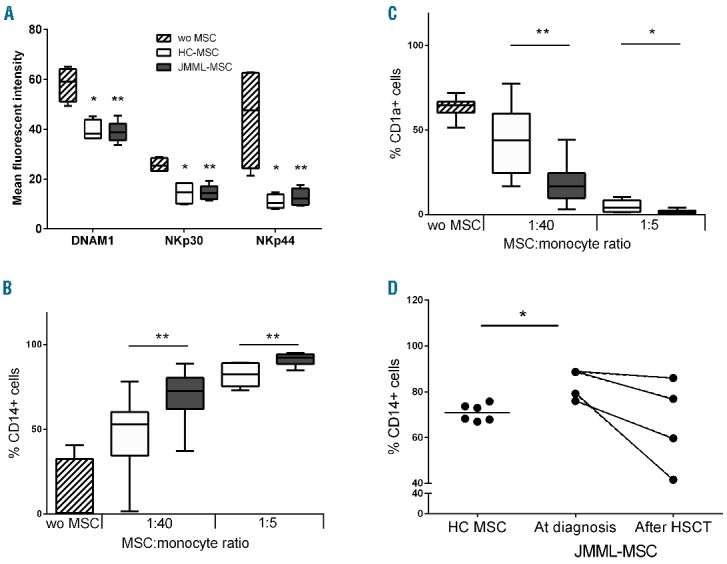
Mesenchymal stromal cells of juvenile myelomonyctic leukemic (JMML-MSCs) expanded from bone marrow at diagnosis have an increased suppressive effect on monocyte to immature dendritic cell differentiation but not on NK-cell activation. (A). NK-cell activation induced by IL-2 (30 IU/mL), measured by mean fluorescence intensity (MFI) of staining for DNAM-1, NKp30 and NKp44 expression on day 5, was suppressed in the presence of MSCs of healthy controls (HC) and JMML patients. MSC: NK cell ratio 1:5 (JMML-MSCs n=4 and HC-MSCs n=4). BC. An increased suppressive effect of JMML-MSCs was observed on the differentiation of monocytes (CD14+ cells, (B) to immature dendritic cells (CD1a+ cells), (C) during five days of stimulation with IL-4 and GM-CSF (JMML-MSCs n=6, gray boxes, and HC-MSCs n=6, white boxes). (D) JMML-MSCs derived from bone marrow (BM) after hematopoietic stem cell transplantation (HSCT) showed a trend towards a lower suppressive effect on the monocyte to immature DC differentiation than JMML-MSCs derived from BM obtained at diagnosis. Paired samples of JMML-MSCs at diagnosis and after HSCT (n=2) and HC-MSCs (n=3) were investigated using monocytes from 2 donors as target populations (MSC : monocyte ratio 1:40). Results in panels A–C are depicted as mean (horizontal line) with interquartile distance (boxes) and range (whiskers). Statistics were performed using Mann-Whitney tests in panels A–D. *P<0.05; **P<0.01; wo: without MSC.
The suppressive effect of MSCs on monocyte to dendritic cell (DC) differentiation has been described to be IL-6 dependent.35 Therefore, we analyzed the monocyte to DC differentiation in the absence and presence of MSCs at different MSC : monocyte ratios. The addition of both sources of MSCs resulted in decreased differentiation to immature DCs as measured by a lower percentage of cells expressing the DC marker CD1a on day 5 of co-culture; these cells remained CD14+ (Figure 4B and C). JMML MSCs had a significantly stronger suppressive effect on monocyte to immature DC differentiation than HC MSCs. This was supported by the observation that CD163 expression on CD14+ cells, a marker associated with anti-inflammatory monocytes and macrophages, was higher after co-culture with JMML-MSCs (MFI: 49.3±7.4 SEM) compared to co-culture with HC-MSCs (MFI: 26.6±4.7 SEM; P=0.03) (data not shown).
The differential suppressive effect of JMML MSCs compared to HC MSCs disappeared after two additional days in culture with growth factors (IFN-γ and CD40-L) to support the differentiation of immature DCs to mature DCs (data not shown).
Gene expression, i.e. IL-6, in MSCs obtained after HSCT was comparable to HC. Therefore, the effect of JMML-MSCs expanded from BM after HSCT on monocyte to immature DC differentiation was studied. The percentage of CD14+ cells in co-cultures of monocytes with JMML-MSCs derived from BM obtained after HSCT during remission was variable and did not differ from from that in co-cultures with HC-MSCs. In addition, MSC derived during remission showed a trend to a lower percentage of CD14+ cells compared to the percentage observed in co-cultures with JMML-MSC at diagnosis (Figure 4D).
Discussion
A genetic mutation in the RAS pathway is identified in 90% of JMML patients.17 However, the etiology of therapy resistance and the high relapse rate after HSCT remains unknown. The aggressive nature of the disease in combination with the high burden of malignant cells may lead to a disturbance of the BM microenvironment.
As a model for the BM microenvironment, we used BM derived MSCs. In our study, MSCs of JMML patients had a comparable differentiation capacity compared to MSCs of HC. In contrast to previously reported studies in adult MDS, adipocyte and osteoblast differentiation was not adversely affected.36 This is in line with the unaffected differentiation capacity of MSCs derived from BM of children with MDS.37 However, gene expression analysis revealed increased DKK1 expression in JMML-derived MSCs. DKK1, related with osteolysis and WNT signaling, has been described to be up-regulated in MSCs derived from patients with multiple myeloma.38 Identification of this protein, also expressed by multiple myeloma cells,39 led to therapeutic strategies using antibodies or vaccination against DKK1 with a beneficial effect in murine models.40,41
The control group differed from the patients regarding age at BM collection. However, data were validated using selected controls aged 1–4 years (n=4). We did not observe an age-dependent effect on gene expression in our patient or control MSCs.
In this study, the suppressive effect of JMML-MSCs on differentiation from monocytes to immature DCs was significantly stronger compared to MSCs of healthy controls. The increased IL-6 expression by JMML-MSCs suggests a causal relationship because IL-6 has been described to be essential in the suppressive mechanism.35 Previously, pediatric MDS-derived MSCs showed an even stronger increase in IL-6 expression, whereas no effect on monocyte to DC differentiation was observed.37 Inhibition of differentiation towards professional antigen-presenting cells might contribute to the escape of JMML cells from the immune system and might explain the usually progressive course of this disease. NK cells are involved in the innate defense against malignant transformed cells. Whereas IL-6 production has been reported to suppress NK-cell cytotoxicity against neuroblastoma and lymphoblastoid cells, facilitating tumor escape,42,43 the suppressive effect of MSCs on NK-cell activation was comparable between JMML-MSCs and HC-MSCs. In previous studies, we demonstrated a correlation between the suppressive effect of MSCs on NK-cell activation marker expression, i.e. DNAM-1, NKp30 and NKp44, and cytotoxicity.22
Juvenile myelomonocytic leukemia clinically presents with anemia and thrombocytopenia in combination with monocytosis and leukocytosis. Previously, the misbalance in hematopoiesis in patients with hematologic malignancies was suggested to be caused by factors excreted by malignant cells.44 In addition, disturbed support of hematopoiesis by MSCs might contribute to dysplasia in these patients. CXCL12 expression was decreased in JMML-MSCs, as was also previously shown in studies on MSC from adult CML and pediatric ALL patients.8,45 The CXCL12-CXCR4 interaction is an important mechanism in hematopoietic support and decreased CXCL12 expression has been linked to dysplasia in mice and adult chronic myeloid leukemia.8,9 Our in vitro hematopoiesis experiments did not reveal differences in support of proliferation and differentiation of HPCs between JMML- and control-derived MSCs. Both JMML and healthy control MSCs have a strong supportive effect on hematopoiesis involving multiple pathways, e.g. CXCL12, G-CSF and SCF. Therefore, aberrant expression of one of these molecules could be compensated by other pathways in our in vitro experiments. In this study, we used in vitro and polyclonal expanded MSCs, and, as a consequence, differences in vivo between HC- and JMML-derived MSCs might be lost during culture. However, Zhang et al. demonstrated sustained decreased expression of CXCL12 in vivo and in in vitro expanded MSCs.8 Further in vivo characterization of the MSCs in mice models modeling the hematopoietic niche with human MSCs, such as previously used in e.g. multiple myeloma, might help to identify more subtle differences.46
The different gene expression profile observed in JMML-MSCs versus HC-MSCs is more likely the consequence than the cause of the disease, with JMML cells interacting with the stromal compartment. This conclusion is supported by the observation that after allogeneic HSCT, in which hematopoietic progenitor cells become of donor origin, gene expression profiles in MSCs normalized in paired samples analysis. Of note, the MSCs after HSCT remain of patient origin, suggesting that the aberrant MSC gene expression pattern before HSCT is induced by JMML cells.
In conclusion, our data demonstrate an effect of JMML cells on MSCs during active disease. In vitro hematopoietic support and maintenance of CD34+ HPCs by JMML-MSCs were not affected despite differential gene expression. However, immunosuppression was affected via a bigger decrease in monocyte to immature DC differentiation by JMML-MSCs.
Acknowledgments
The authors would like to acknowledge the medical, nursing and associated non-medical personnel of our referring centers and the Pediatric Stem Cell Transplantation Unit of the Leiden University Medical Center for the excellent clinical care offered to the patients included in this study. The study would not have been possible without the collaboration within the Dutch Children Oncology Group. We are grateful for the support from the Sequencing Analysis Support Core, in particular Dr. H. Mei, and the Leiden Genomic Technology Center of the Leiden University Medical Center, in particular Dr. H. Buermans. We thank Dr. J. Wijnen for the chimerism analysis and Dr. W. Kroes for the chromosome analysis. Dr. M. van Pel advised us on the various hematopoietic assays.
Footnotes
Funding
This study was supported by a grant from KIKA, Dutch Children Cancer-Free Foundation (Grant 38).
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Mendez-Ferrer S, Michurina TV, Ferraro F, et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 2010;466(7308):829–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morikawa S, Mabuchi Y, Kubota Y, et al. Prospective identification, isolation, and systemic transplantation of multipotent mesenchymal stem cells in murine bone marrow. J Exp Med 2009;206(11):2483–2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sugiyama T, Kohara H, Noda M, et al. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity 2006;25(6):977–988. [DOI] [PubMed] [Google Scholar]
- 4.Greenbaum A, Hsu YM, Day RB, et al. CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature 2013;495(7440):227–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang X, Hisha H, Mizokami T, et al. Mouse mesenchymal stem cells can support human hematopoiesis both in vitro and in vivo: the crucial role of neural cell adhesion molecule. Haematologica 2010;95(6):884–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ayala F, Dewar R, Kieran M, et al. Contribution of bone microenvironment to leukemogenesis and leukemia progression. Leukemia 2009;23(12):2233–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schepers K, Pietras EM, Reynaud D, et al. Myeloproliferative Neoplasia Remodels the Endosteal Bone Marrow Niche into a Self-Reinforcing Leukemic Niche. Cell Stem Cell 2013;13(3):285–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang B, Ho YW, Huang Q, et al. Altered microenvironmental regulation of leukemic and normal stem cells in chronic myelogenous leukemia. Cancer Cell 2012;21(4): 577–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arranz L, Sanchez-Aguilera A, Martin-Perez D, et al. Neuropathy of haematopoietic stem cell niche is essential for myeloproliferative neoplasms. Nature 2014;512 (7512):78–81. [DOI] [PubMed] [Google Scholar]
- 10.Kurtova AV, Balakrishnan K, Chen R, et al. Diverse marrow stromal cells protect CLL cells from spontaneous and drug-induced apoptosis: development of a reliable and reproducible system to assess stromal cell adhesion-mediated drug resistance. Blood 2009;114(20):4441–4450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao ZG, Xu W, Yu HP, et al. Functional characteristics of mesenchymal stem cells derived from bone marrow of patients with myelodysplastic syndromes. Cancer Lett 2012;317(2):136–143. [DOI] [PubMed] [Google Scholar]
- 12.Raaijmakers MH. Myelodysplastic syndromes: revisiting the role of the bone marrow microenvironment in disease pathogenesis. Int J Hematol 2012;95(1):17–25. [DOI] [PubMed] [Google Scholar]
- 13.Klaus M, Stavroulaki E, Kastrinaki MC, et al. Reserves, functional, immunoregulatory, and cytogenetic properties of bone marrow mesenchymal stem cells in patients with myelodysplastic syndromes. Stem Cells Dev 2010;19(7):1043–1054. [DOI] [PubMed] [Google Scholar]
- 14.Flores-Figueroa E, Montesinos JJ, Flores-Guzman P, et al. Functional analysis of myelodysplastic syndromes-derived mesenchymal stem cells. Leuk Res 2008; 32(9):1407–1416. [DOI] [PubMed] [Google Scholar]
- 15.Niemeyer CM, Arico M, Basso G, et al. Chronic myelomonocytic leukemia in childhood: a retrospective analysis of 110 cases. European Working Group on Myelodysplastic Syndromes in Childhood (EWOG-MDS). Blood 1997;89(10):3534–3543. [PubMed] [Google Scholar]
- 16.Niemeyer CM, Kratz CP. Paediatric myelodysplastic syndromes and juvenile myelomonocytic leukaemia: molecular classification and treatment options. Br J Haematol 2008;140(6):610–624. [DOI] [PubMed] [Google Scholar]
- 17.de Vries AC, Zwaan CM, van den Heuvel-Eibrink MM. Molecular basis of juvenile myelomonocytic leukemia. Haematologica 2010;95(2):179–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Locatelli F, Nollke P, Zecca M, et al. Hematopoietic stem cell transplantation (HSCT) in children with juvenile myelomonocytic leukemia (JMML): results of the EWOG-MDS/EBMT trial. Blood 2005;105(1):410–419. [DOI] [PubMed] [Google Scholar]
- 19.Locatelli F, Niemeyer CM. How I treat juvenile myelomonocytic leukemia (JMML). Blood 2015;125(7):1083–1090. [DOI] [PubMed] [Google Scholar]
- 20.Loh ML. Recent advances in the pathogenesis and treatment of juvenile myelomonocytic leukaemia. Br J Haematol 2011; 152(6):677–687. [DOI] [PubMed] [Google Scholar]
- 21.World Medical Association. World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA 2013;310(20):2191–2194. [DOI] [PubMed] [Google Scholar]
- 22.Calkoen FG, Brinkman DM, Vervat C, et al. Mesenchymal stromal cells isolated from children with systemic juvenile idiopathic arthritis suppress innate and adaptive immune responses. Cytotherapy 2013;15(3):280–291. [DOI] [PubMed] [Google Scholar]
- 23.Lankester AC, Bierings MB, van Wering ER, et al. Preemptive alloimmune intervention in high-risk pediatric acute lymphoblastic leukemia patients guided by minimal residual disease level before stem cell transplantation. Leukemia 2010;24(8):1462–1469. [DOI] [PubMed] [Google Scholar]
- 24.Bronkhorst IH, Maat W, Jordanova ES, et al. Effect of heterogeneous distribution of monosomy 3 on prognosis in uveal melanoma. Arch Pathol Lab Med 2011;135 (8):1042–1047. [DOI] [PubMed] [Google Scholar]
- 25.Mastrokolias A, den Dunnen JT, van Ommen GB, et al. Increased sensitivity of next generation sequencing-based expression profiling after globin reduction in human blood RNA. BMC Genomics 2012;13:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010;26(1): 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.R: A language and environment for statistical computing. Vienna, Austria: 2012. [Google Scholar]
- 28.Smyth G. Limma: linear models for microarray data. In: Gentleman R.VCSDRIWH, editor. Bioinformatics and Computational Biology Solutions using R and Bioconductor. New York, NY, USA: Springer, 2005:397–420. [Google Scholar]
- 29.Hochberg Y, Benjamini Y. More powerful procedures for multiple significance testing. Stat Med 1990;9(7):811–818. [DOI] [PubMed] [Google Scholar]
- 30.Franceschini A, Szklarczyk D, Frankild S, et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res 2013; 41(Database issue):D808–D815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Horwitz EM, Le Blanc K, Dominici M, et al. Clarification of the nomenclature for MSC: The International Society for Cellular Therapy position statement. Cytotherapy 2005;7(5):393–395. [DOI] [PubMed] [Google Scholar]
- 32.‘t Hoen PA, Ariyurek Y, Thygesen HH, et al. Deep sequencing-based expression analysis shows major advances in robustness, resolution and inter-lab portability over five microarray platforms. Nucleic Acids Res 2008;36(21):e141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hammond-Martel I, Yu H, Affar eB. Roles of ubiquitin signaling in transcription regulation. Cell Signal 2012;24(2):410–421. [DOI] [PubMed] [Google Scholar]
- 34.Shimomura Y, Agalliu D, Vonica A, et al. APCDD1 is a novel Wnt inhibitor mutated in hereditary hypotrichosis simplex. Nature 2010;464(7291):1043–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Melief SM, Geutskens SB, Fibbe WE, et al. Multipotent stromal cells skew monocytes towards an anti-inflammatory interleukin-10-producing phenotype by production of interleukin-6. Haematologica 2013;98(6): 888–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao Y, Wu D, Fei C, et al. Downregulation of Dicer1 promotes cellular senescence and decreases the differentiation and stem cell-supporting capacities of mesenchymal stromal cells in patients with myelodysplastic syndrome. Haematologica 2015;100(2): 194–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Calkoen FG, Vervat C, van PM, et al. Despite differential gene expression profiles pediatric MDS derived mesenchymal stromal cells display functionality in vitro. Stem Cell Res 2015;14(2):198–210. [DOI] [PubMed] [Google Scholar]
- 38.Corre J, Mahtouk K, Attal M, et al. Bone marrow mesenchymal stem cells are abnormal in multiple myeloma. Leukemia 2007;21(5):1079–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qian J, Xie J, Hong S, et al. Dickkopf-1 (DKK1) is a widely expressed and potent tumor-associated antigen in multiple myeloma. Blood 2007;110(5):1587–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heath DJ, Chantry AD, Buckle CH, et al. Inhibiting Dickkopf-1 (Dkk1) removes suppression of bone formation and prevents the development of osteolytic bone disease in multiple myeloma. J Bone Miner Res 2009;24(3):425–436. [DOI] [PubMed] [Google Scholar]
- 41.Qian J, Zheng Y, Zheng C, et al. Active vaccination with Dickkopf-1 induces protective and therapeutic antitumor immunity in murine multiple myeloma. Blood 2012;119 (1):161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu Y, Sun J, Sheard MA, et al. Lenalidomide overcomes suppression of human natural killer cell anti-tumor functions by neuroblastoma microenvironment-associated IL-6 and TGFbeta1. Cancer Immunol Immunother 2013;62(10):1637–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tanner J, Tosato G. Impairment of natural killer functions by interleukin 6 increases lymphoblastoid cell tumorigenicity in athymic mice. J Clin Invest 1991;88(1):239–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aoyagi M, Furusawa S, Waga K, et al. Suppression of normal hematopoiesis in acute leukemia: effect of leukemic cells on bone marrow stromal cells and hematopoietic progenitor cells. Intern Med 1994;33(5):288–295. [DOI] [PubMed] [Google Scholar]
- 45.van den Berk LC, van der Veer A, Willemse ME, et al. Disturbed CXCR4/CXCL12 axis in paediatric precursor B-cell acute lymphoblastic leukaemia. Br J Haematol 2014;166(2):240–249. [DOI] [PubMed] [Google Scholar]
- 46.Groen RW, Noort WA, Raymakers RA, et al. Reconstructing the human hematopoietic niche in immunodeficient mice: opportunities for studying primary multiple myeloma. Blood 2012;120(3):e9–e16. [DOI] [PubMed] [Google Scholar]