

Abstract
Although accumulating evidence suggests that repetitive mild TBI (rmTBI) may cause long-term cognitive dysfunction in adults, whether rmTBI causes similar deficits in the immature brain is unknown. Here we used an experimental model of rmTBI in the immature brain to answer this question. Post-natal day (PND) 18 rats were subjected to either one, two, or three mild TBIs (mTBI) or an equivalent number of sham insults 24 h apart. After one or two mTBIs or sham insults, histology was evaluated at 7 days. After three mTBIs or sham insults, motor (d1–5), cognitive (d11–92), and histological (d21–92) outcome was evaluated. At 7 days, silver degeneration staining revealed axonal argyrophilia in the external capsule and corpus callosum after a single mTBI, with a second impact increasing axonal injury. Iba-1 immunohistochemistry showed amoeboid shaped microglia within the amygdalae bilaterally after mTBI. After three mTBI, there were no differences in beam balance, Morris water maze, and elevated plus maze performance versus sham. The rmTBI rats, however, showed impairment in novel object recognition and fear conditioning. Axonal silver staining was observed only in the external capsule on d21. Iba-1 staining did not reveal activated microglia on d21 or d92. In conclusion, mTBI results in traumatic axonal injury and microglial activation in the immature brain with repeated impact exacerbating axonal injury. The rmTBI in the immature brain leads to long-term associative learning deficit in adulthood. Defining the mechanisms damage from rmTBI in the developing brain could be vital for identification of therapies for children.
Key words: : axonal injury, development, head injury, microglia
Introduction
Traumatic brain injury (TBI) in infants and children is an important public health problem. It is estimated that more than 500,000 children ranging in age from 0–14 years incur a TBI every year in the United States.1 The majority (75%) of these TBIs are mild,2 and loss of consciousness is not presented in 81–92% of these cases. Hence, most patients do not seek medical advice and subsequently present an increased risk of repeated TBI.3 Specific populations, such as victims of abusive head trauma (AHT) or athletes having sports concussions, are at risk of having repetitive mild TBI (rmTBI).
Recent reports show that rmTBI is associated with a number of neurodegenerative diseases including chronic traumatic encephalopathy, Alzheimer-like dementia, and Parkinsonism.4 The mechanisms underlying the adverse effects of multiple mTBIs in adults are largely unknown, and the knowledge gap is greater in the developing brain. Experimental models may lead to understanding the pathophysiology of rmTBI and identify potential therapeutic targets. Adult models of repeated mild closed head impact and blast injury have shown long-term neurocognitive dysfunction associated with traumatic axonal injury and microglial activation.5–7 It is not known, however, whether rmTBIs occurring early during development lead to cognitive declines that persist into adulthood.
Patients with mTBI typically do not display gross pathology, such as hemorrhage or structural abnormalities, on brain imaging by computed tomography. More advanced techniques, however, such as diffusion tensor imaging (DTI), do show abnormalities in the corpus callosum and frontal white matter corresponding with physical, emotional, and cognitive symptoms exhibited by patients with mTBI.8–11 Some of these symptoms observed in children after TBI resemble those seen in attention deficit hyperactivity disorder12 and post-traumatic stress disorder.13–16
The majority of experimental models investigating the pathophysiology of rmTBI have focused on the adult brain.5,17–19 Important differences exist in response to injury and recovery between the adult and immature brain, where the normal developmental trajectory may diverge over time after TBI. Some factors such as delayed myelination of the frontal lobes—a normal phenomenon during development20—may place the immature brain at a higher risk for worse outcome compared with adults after TBI.21 Conversely, increased plasticity observed in the immature brain may confer advantages in recovery.22 It is not known, however, whether these observations hold true with repetitive injuries.
Few studies to date have reported the effects of rmTBI on neurocognitive outcome in the developing brain. Using an infant model of closed skull controlled cortical impact (CCI) in post-natal day (PND) 11 rats, Huh and associates23 did not see differences in spatial learning between sham controls and any of the TBI groups at 14 days despite axonal swelling, white matter damage, and ventricular enlargement that worsened with repeated injuries (two or three) with 5 min intervals suggesting that the immature brain likely has enhanced repair mechanisms and plasticity. Support for this notion comes from a study that showed increasing impairment in learning and memory assessed by novel object recognition at day 2 post-injury with repeated injuries (two closed skull CCIs) with 24 h intervals in PND 35 rats.24 Whether these deficits in functional outcome persist beyond the acute period after rmTBI into adulthood is not known.
We hypothesized that rmTBI in the immature brain will result in long-term functional deficits at adulthood associated with axonal injury, microglial activation, and minimal cell death. To evaluate the effects of rmTBI on acute and long-term histological outcome and neurocognitive function in the immature brain, we developed a rmTBI model in PND 18 rats. Exposure of PND 18 rats to rmTBIs resulted in axonal injury and microglial activation in select regions of the brain and was associated with persistent learning and memory deficits lasting up to ∼3 months after the last impact.
Methods
Male Sprague-Dawley rat pups (PND 10) were purchased with lactating mothers from Harlan. The rats were housed for at least 1 week before commencement of the experiments. The rats were fed standard rat chow and provided water ad libitum. Ambient temperature was controlled at 20–22°C, and lighting was on a 12-h/12-h cycle. Experiments were performed exploring the effect of (1) mTBI on short-term histological outcome and (2) repetitive mTBI on motor, cognitive, and long-term histological outcome. The experimental design and time points for histological and neurocognitive outcome testing are summarized in Figure 1. This study was approved by the University of Pittsburgh Animal Care and Use Committee.
FIG. 1.
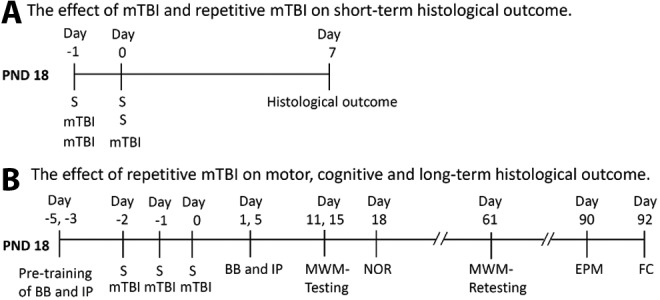
Schematic time line of the experimental design to assess: (A) The effect of mild traumatic brain injury (mTBI) and repetitive mTBI on short-term histological outcome 7 days after injury in post-natal day (PND) 18 rats, and (B) The effect of repetitive mTBI on motor, cognitive, and long-term histological outcome in the PND 18 rat. S, sham; BB, beam balance test; IP, inclined plane test; MWM, Morris water maze; NOR, novel object recognition test; EPM, elevated plus maze test; FC, fear conditioning test.
Mild traumatic brain injury (mTBI)
An adult mTBI model5 was modified for all experiments with several important changes. Briefly, rats were anesthetized via a nose cone using 2% isoflurane and a 2:1 nitrous oxide/oxygen mix. The heads were shaved, and the surgical site was disinfected with povidone-iodine. The anesthetized rats were placed onto a stereotaxic frame, secured using ear bars, an incision was made along the scalp, and a pneumatically driven piston delivered a pre-defined injury to the intact skull just above the left parietal cortex. A rubber ball (9.5 mm in diameter with a durometer of 50, Precision Associates, Inc, Minneapolis, MN) was mounted onto a bowl shaped metal tip (University of Pittsburgh Machine Shop), and the rubber tip was lowered at 23 degrees and measured to strike at 1.8 mm caudal to bregma and 3.0 mm left of the midline. The tip was retracted and lowered to a depth of 1.0 mm and fired at 4.0 ± 0.2 m/sec with duration of 50 msec.
The procedure resulted in <10% skull fractures and no immediate fatalities or apnea. After the impact, the skin was sutured and the rats were allowed to recover from anesthesia with 100% oxygen on a warming pad and then returned to their cages. Sham controls underwent similar procedures including anesthesia, except the device was fired in the air above the rat's exposed skull without making contact. After the insult, the incision was sutured.
Assessment of histological outcome
Rats were anesthetized with 3% isoflurane and transcardially perfused with ice-cold saline and 0.5% heparin followed by 2% paraformaldehyde (PFA). The brains were post-fixed with 2% PFA for 24 h, then transferred to 15% and 30% sucrose solutions, respectively, 24 h apart. The brains were cut with a cryostat microtome (Leica, Jung CM1800) at 40 μm, and the sections were kept in cryoprotectant. Neuropathologic evaluations were performed on four transverse sections ∼200 μm apart spanning from ∼ −3.0 mm to −4.5 mm relative to bregma25 on days 7, 21, and 92.
Silver degeneration staining
Silver staining was performed using the FD NeuroSilver Kit II (FD NeuroTechnologies, Ellicott City, MD) according to the manufacturer's instructions. Briefly, sections were placed in 4% PFA for 4 days before staining, and all steps were performed on shaker with 130 rpm.
Immunohistochemistry
For free floating peroxidase immunohistochemistry staining, 40 μm sections were placed in 2% H2O2 for 20 min and blocked with 3% normal goat serum in Tris-buffered saline (TBS) containing 0.25% Triton X (TBS-X) for 1 h. Sections were incubated overnight in TBS-X solution containing rabbit anti-Iba-1 antibody (1:1000, Wako Chemicals) and 1.5% normal goat serum. The following day, the sections were washed, incubated for 1 h with anti-rabbit HRP-conjugated secondary antibody (ABC Elite kit, PK6100, Vector Laboratories, Burlingame, CA). Sections were washed with TBS three times for 10 min each and stained with DAB for visualization (DAB, Vector Laboratories, Burlingame, CA).
Double immunofluorescence staining
For double immunofluorescence staining, 40 μm coronal sections were rinsed with TBS and incubated with TBS++ (0.1% Triton X-100 and 3% goat serum in 0.1 M TBS) for 30 min at 37°C. Sections were double stained with antibodies against Iba-1 (rabbit, 1:1000, Wako Chemicals), iNOS (monoclonal mouse,1:50, Thermo Scientific), and arginase-1 (polyclonal goat, 1:200, Santa Cruz) for 1 h at 37°C, and they were kept overnight at 4°C. The following day, sections were washed with TBS (3 × 10 min) and incubated with anti-mouse Alexa Flour 488-conjugated IgG (for INOS, 1:1000), anti-goat Alexa Flour 488-conjugated IgG (for arginase-1, 1:1000), anti-rabbit Alexa Flour 594-conjugated IgG (for Iba-1, 1:1000) for 1 h at 37°C. After rinsing, sections were mounted on slides with DAPI containing medium (Victashield, H-200). Slides were examined under fluorescence microscope (Nikon Eclipse 90i and Olympus Fluoview 1000).
Digital image analysis of axonal injury
Digital image analysis of silver staining was performed to quantify the degree of axonal injury. Two random 20× magnified pictures were taken of each region of interest (i.e., cortex, external capsule, and corpus callosum) with a light microscope (Nikon Eclipse 90i). Each brain contributed a total of four sections ∼200 μm apart spanning from ∼−3.0 mm to −4.5 mm, bregma.25 An area threshold of 0.8–20 μm2 was used to exclude artifacts and cell staining. The percentage of injured neuronal processes was defined by dividing the stained area by the area of interest.
Quantification of microglia
Iba-1 positive cells were counted as previously described.26 To estimate the number of Iba-1 labeled microglia, images were taken using a light microscope (Nikon Eclipse 90i). Each brain contributed a total of four sections ∼200 μm apart spanning from ∼−3.0 mm to −4.5 mm, bregma.25 Light intensity and area (>115 μm2) thresholds were defined to detect Iba-1 labeled cells. Images were analyzed using NIS-Elements software to calculate the number of Iba-1 labeled cells within the area of interest.
Assessment of neurocognitive outcome
Beam balance and inclined plane tests
Rats were assessed for motor function using the beam balance27 and inclined plane tasks28 for 5 days beginning the day after the last impact as described previously. Briefly, the beam balance task consists of a 1.5 cm wide and 90 cm elevated wooden beam that the rats must remain on for 60 sec. Before surgery, the rats were pre-trained on the beam. To reach criterion, each rat had to remain on the beam for 60 sec for three consecutive trials. For the inclined plane, the rats were placed on a flat board with angles increasing from 45 degrees to 85 degrees in 5 degree increments. Each degree consisted of three trials that were 10 sec each. The maximum angle at which the rats could maintain position for 10 sec was recorded. All rats were pre-trained for the inclined plane before surgery. To reach criterion, each had to remain on the incline for 10 sec, while positioned at 70 degrees for 3 consecutive trials.
Morris water maze (MWM) task
Rats underwent MWM training to assess the acquisition of spatial learning and memory retention. Briefly, rats underwent training on days 11–15 and were retested on day 61 after the last impact (sham or injury) for latency to find the hidden platform. One day of probe and visible platform trials were performed on day 16 after the last impact to assess memory retention and nonspecific visual deficits. The water maze consists of a pool that is 180 cm in diameter and 60 cm high. The pool was filled with water (26 ± 1°C in temperature) to a depth of 28 cm. A platform 10 cm in diameter and 26 cm high (i.e., 2 cm below the water's surface) was used as the hidden platform. The pool was located in a 2.5 × 2.5 meter room with numerous extramaze cues that remained constant throughout the experiment.
A video tracking system (Anymaze, San Diego Instruments, San Diego, CA) was used to record and quantify the swimming motions of the rats. The tracking system consists of a CCD camera that is mounted over the pool and is attached to a tracking detection unit interfaced to a personal computer. On each trial, the rats were placed by hand in the pool facing the wall at one of four start points. Rats were given a maximum of 120 sec to find the hidden platform. If the rat failed to find the platform after 120 sec, it was placed on the platform by the experimenter for 30 sec before being returned to a heated incubator between trials for 4 min.
For the probe trial, the hidden platform was removed, and the rat was placed in the maze from the location point most distal to the quadrant where the platform was previously situated and allowed to freely explore the pool for 30 sec. For the visible platform task, the platform was raised 2 cm above the surface of the water to assess potential nonspecific deficits in visual and motor function.
Novel object recognition (NOR)
The NOR task assesses declarative memory and consists of an opaque rectangular plastic container with approximate dimensions of 28″ L × 20″ W × 20″ H with an open top such that the experimenter can observe the rat and objects simultaneously. Three plastic objects consisting of a white PVC tube 3″ diameter and 1′ high (permanent object), red plastic jar 3″ round 3″ high (decoy object), and white plastic bottle 3″ square 4″ high (novel object) were used on every rat throughout testing. During the habituation phase (day 17), each rat was individually placed in the chamber in ambient lighting conditions to freely explore and habituate to two objects, the permanent and decoy, for a total time of 30 min. Testing began 24 h after habituation (day 18) with a reminder trial consisting of 1 min in the same container with the habituation objects. The rat was then removed and returned to the colony for 1 h. Testing occurred after a 1 h interval, and the rat was again placed in the container with the permanent object and the novel object while the experimenter measured the time spent with the novel object and the permanent object for a total time of 2 min.
Elevated plus maze
The elevated plus maze (EPM) (Stoelting Co., Wood Dale, IL) consisted of two high walls for the closed arms (wall height 40 cm) perpendicular to two open arms. Each arm had a width of 10 cm and a length of 50 cm. The open arms had 0.5 cm curbs along the edges to prevent falls. A floor lamp (40 watt bulb) illuminated the maze from above. Each arm had an average light intensity of 100 ± 10 lux. Rats were placed in the center platform of the maze facing the junction of an open and closed arm and were allowed to freely explore the maze for 5 min. A video tracking system (Anymaze, San Diego Instruments, San Diego, CA) was used to record and quantify behavior.
Fear conditioning (FC)
The FC apparatus consisted of a sound and light insulated box containing a smaller Plexiglas chamber (30.5 × 24.1 × 21 cm, Medical Associates, St. Albans, VT) with an electric grid floor (1.6 cm spacing, 4.8 mm diameter rods) and a camera mounted at the top. The rats were placed in the room 1 h before testing for acclimation and then placed in the box for 60 sec where they were presented with a 10 sec tone (75 dB, 32.8 kHz) coterminating with a 1 sec foot shock (1 mA) that was delivered through the grid floor. Five pairings of the tone and foot shock were given sequentially for a total training period of 410 sec, with 60 sec separating each tone. The box was cleaned with Versa Clean between rats. On day 2, 24 h after training, contextual FC was assessed by placing the rats in the box, and freezing was measured for 3 min.
At 1 h after contextual FC and 25 h after training, cue FC was assessed by placing the rats in an altered chamber. Specifically, the box was cleaned and scented with a novel vanilla scent, a paper roll lined with print was placed within the box to hide the walls and floor, and a white noise of 65 dB was given the first 2 min. After 2 min, only a cue sound (75 dB, 32.8 kHz) heard as the day before without the foot shock was delivered for 1 min followed by 1 min of silence. The rat's freezing behavior was measured for 4 min. Freezing behavior was quantified as percent of time motionless after putting a threshold of motion based on sham animals using a video-based analysis (FreezeFrame, Coulburn Instruments).
Statistical analysis
Statistical analyses were performed using Statview 5.0.1 software (Abacus Concepts, Inc., Berkeley, CA). The motor and MWM data were analyzed by repeated-measures analysis of variance (rm-ANOVA). When the overall ANOVA revealed a significant effect, the data were further analyzed with the Fisher post hoc test to determine specific group differences. All other data were analyzed either by ANOVA (quantification of silver and microglial staining) or Student t test (NOR, EPM, FC). All data are presented as the mean ± standard error of the mean. Differences were considered statistically significant when the p value was ≤0.05.
Results
The effect of mTBI and rmTBI on short-term (day 7) histological outcome
PND 18 TBI rats and age-matched sham controls were used to evaluate the effect of rmTBI on short-term histological outcome. Rats were divided into three groups and subjected to (1) two sham insults, (2) one mTBI and one sham insult, or (3) two mTBIs 24 h apart (Fig. 1A). Rats exposed to single mTBI and two mTBIs had positive axonal silver staining in the external capsule (Fig. 2A e, f) underlying the impact site and in the corpus callosum (Fig. 2A h, i). mTBI, however, did not result in visible axonal silver staining in the cortex or in any other brain region. Axonal staining was more pronounced after two impacts compared with a single impact in the external capsule (Fig. 2C).
FIG. 2.

Silver staining at 7 days after a second sham insult or mild traumatic brain injury (mTBI). Post-natal day 18 rats were divided into three groups and subjected to (1) two sham insults (S-S), (2) one mTBI, one sham insult (mTBI-S), or (3) two mTBIs (mTBI-mTBI) 24 h apart. (A) Representative views of coronal sections (scale bar = 100 μm) at approximately −3.0 to 4.5 mm bregma. Insets include higher magnification (scale bar = 10 μm) image of axonal silver staining indicated by arrows. (B) Red (cortex), blue (external capsule), and black (corpus callosum) boxes indicate areas of interest, shown in higher magnification in panels A. (C) Proportional area of positive silver staining in rat brains (n = 3/group). Mean ± standard error of the mean, *p < 0.05 vs. S-S, §p = 0.09 vs. mTBI-S. Color image is available online at www.liebertpub.com/neu
No abnormalities were seen in the brains of sham-injured controls. No significant degree of silver staining of neuronal bodies was observed, and hematoxylin and eosin staining did not show overt neuronal death in hippocampus 7 days after two insults (Supplementary Fig. 1; see online supplementary material at ftp.liebertpub.com). There was no evidence of gross pathological changes such as hemorrhage, cerebral contusion, or other visible abnormalities.
To assess whether microglial activation was present in regions with injured axons, Iba-1 immunohistochemistry was performed. Iba-1 staining showed ramified microglia that appeared to be in the resting state in sham rats (Fig. 3A a, d, g, insets). After mTBI and rmTBI, microglia acquired a more amoeboid shape with shorter projections indicating activation (Fig. 3A h, i, and insets). Microglial activation was not uniform through the brain sections examined but was most prominent within the amygdalae bilaterally (Fig. 3A h, i). Although microglial morphology differed between mTBI and sham groups, quantification of Iba-1 positive cells did not reveal differences between sham and injured rats necropsied on day 7 (Fig. 3C), suggesting that microglial proliferation was not affected by mTBI.
FIG. 3.

Immunohistochemical staining with Iba-1 antibody at 7 days after second sham insult or mild traumatic brain injury (mTBI). Post-natal day (PND) 18 rats were divided into three groups and subjected to (1) two sham insults (S-S), (2) one mTBI, one sham insult (mTBI-S), or (3) two mTBIs (mTBI-mTBI) 24 h apart. (A) Representative views of coronal sections (scale bar = 200 μm) at approximately −3.0 to 4.5 mm, bregma. Insets include higher magnification (scale bar = 25 μm) image of Iba-1 immunoreactive microglia indicated by arrows. (B) Red (cortex), blue (external capsule), and black (amygdala) boxes indicate areas of interest, shown in higher magnification in panel A. (C) Quantification of the number of Iba-1 immunoreactive cells in PND 18 brains (n = 3/group). Mean ± standard error of the mean. Color image is available online at www.liebertpub.com/neu
To assess microglial subtypes, we performed double immunostaining with antibodies against Iba-1 and iNOS for M1 and Iba-1 and arginase-1 for M2 microglia after rmTBI as described previously.29,30 iNOS positive microglia were observed in the external capsule underlying the impact whereas arginase-1 positive microglia were observed in amygdalae bilaterally at 7 days after rmTBI, suggesting a more protracted proinflammatory response to rmTBI in the external capsule (Fig. 4).
FIG. 4.

Dual immunofluorescence staining with antibodies against Iba-1 and iNOS and Iba-1 and arginase-1 at 7 days after repetitive sham or mild traumatic brain injury (rmTBI). Representative views of coronal sections (scale bar = 100 μm) at approximately −3.0 to 4.5 mm bregma. Color image is available online at www.liebertpub.com/neu
The effect of rmTBI on motor, cognitive, and long-term histological outcome in the immature brain
Previous studies in adult mouse models of mTBI and rmTBI reported abnormalities in corpus callosum and white matter areas including the external capsule associated with cognitive dysfunction5,31; thus, we tested memory acquisition and retention in PND 18 rats exposed to rmTBI. PND 18 rats were divided into two groups and underwent either sham insult or mTBI on three consecutive occasions 24 h apart (Fig. 1B). Motor function was assessed with beam-balance and inclined platform tasks; cognitive performance was evaluated using MWM and NOR, which are sensitive to motor and cognitive function/dysfunction after TBI in this age group.28
Balancing ability or inclined platform performance did not differ among groups before surgery (Fig. 5A, B), suggesting that pre-training was consistent among all groups. After surgery, there was no effect of rmTBI on beam balance performance (Fig. 5A). Although both rmTBI and sham groups were able to achieve their pre-training performance on the inclined plane after the surgery, the rmTBI group performed better than the sham group on days 2 and 3 (Fig. 5B). The performance on the inclined plane improved over time because the maximum angle that a rat climbed increased significantly over time in both the rmTBI and sham injury groups.
FIG. 5.

The effect of repetitive mild TBI (rmTBI) on gross motor function, spatial learning, and memory in post-natal day 18 rats that underwent either sham insult or mTBI on three consecutive occasions 24 h apart. Before surgery, balancing ability (A) or inclined platform performance (B) did not differ among groups. After surgery, there was no effect of rmTBI on beam balance performance (A); rmTBI group performed better than the sham group on inclined plane (B) on days 2 and 3 (n = 12–18/group, mean ± standard error of the mean (SEM), *p < 0.05 vs. S-S-S). (C) Both rmTBI and sham groups began at a similar level in the Morris water maze, and there was no difference between the two groups during acquisition on days 11–15. (D) Analysis of the time to hidden platform of the last acquisition day (day 15) and retesting day (day 61) showed that injured rats tended to take longer to find the hidden platform compared with sham rats (n = 9–12/group, mean ± SEM, p = 0.0685). (E) Rats exposed to rmTBI spent significantly less time exploring the novel object versus rats exposed to sham insults on day 18 (n = 12–18/group, mean ± SEM, *p < 0.05 vs. S-S-S).
Acquisition of spatial learning after rmTBI was conducted using a MWM task on days 11–15 after the third insult and again on day 61. Both rmTBI and sham groups began at a similar level in the water maze, and there was no difference between the two groups during acquisition (Fig. 5C). Analysis of the time to hidden platform of the last acquisition day (day 15) and retesting day (day 61) was not significantly different between rmTBI and sham groups (p = 0.0685, Fig. 5D). Similarly, the time spent searching in the target quadrant during a single probe trial on day 16 after the third insult was not statistically different between the two groups (data not shown).
To further evaluate the effect of rmTBI on declarative memory, the NOR task was performed on day 18 after the third insult. Rats exposed to rmTBI spent significantly less time exploring the novel object versus rats exposed to sham insults (Fig. 5E), suggesting that while rmTBI did not lead to deficits in the acquisition of spatial learning, it resulted in impairments in memory retention in PND 18 rats (Fig. 5E). Swim speed did not differ among groups (p = 0.12), indicating that neither water maze nor NOR performance was influenced by swimming, motor ability, or motivational deficits.
To gain further insight into the late behavioral and learning deficits caused by the rmTBI sustained at PND 18, elevated plus maze and FC tests were performed at 90 and 92 days, respectively. There was no difference between groups in the percentage of time spent on the open arms of the maze and the percentage of open arm entries between rmTBI and sham rats (Fig. 6A, B), suggesting that anxiety-like behavior and risk taking were not altered by rmTBI at this time. In fear response testing, rats exposed to rmTBI froze less than the rats exposed to repeated sham insults during both contextual FC and cued FC (Fig. 6C, D). Notably, there were no differences between the two groups in percent baseline freezing during the training phase (data not shown). Together, these data indicate that rmTBI occurring at PND 18 results in long-term associative (fear) learning deficit that is not confounded by increased anxiety or risk taking behavior.
FIG. 6.

The effect of repetitive mild TBI (rmTBI) on anxiety and associative learning in post-natal day 18 rats that underwent either sham insult or mTBI on three consecutive occasions 24 h apart. Elevated plus maze and fear conditioning tests were performed at 90 and 92 days, respectively, after the last sham insult or mTBI. There was no difference between groups in the amount of time spent on the open arms of the maze (A) and the percentage of open arm entries (B). Rats exposed to rmTBI froze less than the rats exposed to repeated sham insults during both contextual fear conditioning (C) and cued fear conditioning (D) (n = 9–12/group, mean ± standard error of the mean, *p < 0.05).
Histopathological analysis at 21 days and 92 days after the last impact revealed the presence of axonal silver degeneration staining only in the external capsule at 21 days (Fig. 7) —albeit less than the magnitude of staining observed at 7 days after double mTBI (compare with Fig. 2A, f). There was no visible axonal silver staining at 92 days after rmTBI. Iba-1 staining did not reveal visible activated microglia at 21 and 92 days (data not shown).
FIG. 7.

Silver staining in external capsule at 21 days and 92 days after third sham or mild traumatic brain injury (mTBI) in post-natal day 18 rats. Low-power (scale bar = 100 μm) views of coronal sections at approximately −3.0 to 4.5 mm, bregma. Insets include higher magnification (scale bar = 10 μm) image of positive axonal silver staining indicated by an arrow. Color image is available online at www.liebertpub.com/neu
Discussion
Our findings show that a second episode of mTBI that occurs within 24 h after the first one during development increases axonal injury in select areas in the immature brain. Previous studies in PND 11 rats23 and PND 3–5 piglets32 both thought to model the newborn period, and PND 35 rats,24 thought to model adolescence, showed increased axonal injury assessed by APP or neurofilament protein immunohistochemistry after a second mTBI that are separated by 5–20 min or up to 1 day. Despite increased axonal injury, there were no deficits in functional outcome at 12–14 days after the rmTBI in the neonatal models.23,33 PND 35 rats, however, showed deficits in memory retention 2 days after a second mTBI. Collectively, these data suggest that increased plasticity seen in the immature brain may confer advantages in recovery. Our data, however, show that deficits in memory retention and fear learning—that is not confounded by increased anxiety or risk taking behavior—occur at adulthood after experiencing rmTBI during development.
Pathophysiological mechanisms of traumatic axonal injury have been studied mainly in moderate-severe TBI and include neurofilament misalignment, mitochondrial dysfunction, and oxidative stress.34 Focal mechanoporation of the axolemma leading to plasma membrane perturbation were observed within minutes after moderate/severe fluid percussion injury and persisted up to 6 h.35 Rapid neurofilament misalignment and axonal swelling, however, occurred without plasma membrane perturbation after a single mild fluid percussion injury.36
Coordinated actions of neurofilaments and motor proteins are essential for efficient axonal transport, which supplies the distal synapse with newly synthesized proteins, lipids, and mitochondria and clears damaged proteins and mitochondria via autophagosomal transport to somal lysosomes.37 Thus, disruption of axonal transport can lead to accumulation of protein aggregates and damaged dysfunctional mitochondria at the synaptic terminals.38–40 In addition, trauma results in an increase in intra-axonal sodium and calcium concentrations, which can lead to activation of proteases and worsen cytoskeletal damage, thus impeding axonal transport.41–44
Physiologically, these processes manifest as a reduction in conduction velocity that was reported after mTBI.45–47 Collectively, disrupted axonal transport, ionic imbalance, and mitochondrial dysfunction can lead to axonal swelling,8,22–26 plasma membrane disruption, and release of intracellular contents, including mitochondria. The latter can act as damage associated molecular patterns and lead to recruitment or activation of inflammatory cells.48
The reason for specific spatial distribution of axonal silver staining in our model is likely because of primary injury where mechanical forces were the highest in the cortex and external capsule beneath the impact site. These were also the areas where activated microglia were observed. Although previous studies using in vitro mechanical stretch injury showed that microglia can be activated by direct mechanical injury,49,50 Iba-1 immunoreactive cells with activated microglial morphology most likely represent secondary response to primary traumatic axonal injury. Supporting this, iNOS positive microglia were observed in the external capsule underlying the impact. Further studies with a more detailed time course will be required to fully evaluate the inflammatory response to rmTBI in the immature brain.
Shitaka and associates5 in 2011 showed activated microglia in the same areas as the injured axons using electron microscopy and Iba-1 immunohistochemistry starting 2 days and lasting to up to 7 weeks after the last impact in an adult mouse rmTBI model.5 Mouzan and colleagues6 in 2014 also showed increased Iba-1 and APP staining 6 and 12 months after single mTBI and rmTBI.6 We did not observe persistent activation of microglia and axonal injury at a delayed time point (92 days) after rmTBI in PND 18 rats, however. It is possible that a lesser degree of microglial activation observed in the immature brain may be because of a relatively lesser degree of myelination.51
Supporting this notion, PND 29 rats exposed to the same injury parameters as these PND 18 rats show more robust axonal silver staining and microglial activation (Supplementary Figure 2 and 3; see online supplementary material at ftp.liebertpub.com). An alternative explanation could be an age-related lag in microglial development and proliferation in the immature brain. This is less likely, however, because by ∼PND 15, microglia are well distributed throughout the brain, facilitating surveillance of the majority of the parenchyma, and by PND 20, the adult population of microglia is well established.51,52 This period of microglial maturation also coincides with a time when active synaptogenesis is taking place during development53–56 with mature synaptic function being established within the rat hippocampus by PND 17 as reflected by the ability to induce long-term potentiation.57
Traumatic axonal injury and microglial activation have been described in humans after moderate and severe TBI.58–61 Further, multifocal areas of axonal injury and clusters of reactive microglia were observed on neuropathological examination of a patient who died of bronchopneumonia 13 days after a mTBI,61 suggesting that our experimental findings mirror the clinical condition.
The clinical presentation of single or rmTBI includes acute and chronic motor and cognitive symptoms.62 We did not observe deficits in beam balance and inclined plane within the first 5 days after rmTBI compared with shams in PND 18 rats. Interestingly, PND 18 rats exposed to rmTBI in our model showed transiently increased activity in the inclined plane within the first week after rmTBI. The reason for this is not entirely clear. Inclined platform performance did not differ among groups before surgery, suggesting that pre-training was consistent among all groups. It is possible that better performance on the inclined plane after rmTBI might be because of increased impulsivity and hyperactivity.
Previous reports showed hyperactivity early after rmTBI in adult mice.17,63 Hyperactivity is associated with attention deficit in children.64 After mTBI, behavioral symptoms of hyperactivity, anxiety, and attention deficit have been reported in children.65 Further, severity of the symptoms seem to correlate with the number of mTBI episodes; children experiencing more than two mTBIs have persistent deficits in attention and concentration.66 Corroborating this examination of cognitive function in adolescent and college students shows a decrease in verbal and visual memory, processing speed composite, and reaction time composite up to 2 weeks after concussions.67–69 Long-term cognitive problems including memory impairment, executive dysfunction, aggressive and irritable behavior, language difficulties, as well as dementia accompanied with atrophy of the frontal and medial temporal lobes of brain have also been reported after rmTBI in adults.4
Recent studies in experimental models of rmTBI sustained during adulthood show long-lasting impairments in spatial learning and memory when tested up to 6 months after injury.17,18 Our data in a developmental model slightly differ from the adult studies in that mTBI that occurs at PND 18 lead to deficits in short-term memory retention but not in spatial learning, suggesting that both cortical and hippocampus-dependent pathways were affected by rmTBI.70–72 We observed deficits in short-term memory without overt neuronal death in the hippocampus. This is in agreement with previous studies by Lyeth and coworkers73 showing spatial memory deficits in the adult rat without hippocampal neuronal death or overt axonal injury in hippocampal pathways.
It is thought that the immature brain has greater recovery potential compared with the adult brain.74,75 On the other hand, the immature brain is likely more vulnerable because the injury can alter the developmental trajectory in a way that acquisition of early skills, on which later functioning relies, may be interrupted or fail to fully develop.76,77 This might be particularly applicable to early skills such as the ability to differentiate threatening (danger) and nonthreatening (safety) behaviors through associative processes.78 Learning about danger cues can be tested with a conditioned fear paradigm that associates a contextual environment and cue stimulus to a foot shock.79 Contextual and cued learning depends on an intact hippocampus and amygdala, respectively, because lesions in these regions diminish the cognitive response. Further, only bilateral lesions but not unilateral lesions can abolish fear learning. Our findings showing amoeboid shaped microglia with shorter projections in bilateral amygdala (Fig. 3A) prompted us to test associative learning after rmTBI in PND 18 rats.
There are conflicting reports in the literature regarding deficits in FC in rodent models of TBI depending on the type and severity of the injury sustained.79,80 Elder and colleagues80 in 2012 showed enhanced contextual FC as evidenced by increased freezing time in response to the conditioned stimulus (tone) in an adult rat model after repetitive blast exposure (one exposure per day for 3 consecutive days). On the other hand Lifshitz and associates79 in 2007 showed deficits in contextual FC at 7 days after a single lateral fluid percussion brain injury that correlated with hippocampal damage. Cued FC response was not different between injured and control mice in these studies corroborating with an intact amygdala in this model.79
We found decreases in conditioned fear response to both contextual and cued testing without increased anxiety at 3 months after rmTBI. Injury to the amygdala early in life might be the reason for the associative learning deficits we observed later in life in our model. During development, there is a sensitive period in which amygdala activation to threatening stimuli is inhibited, resulting in an approach response to aversive stimuli.81 Indeed, peak neurogenesis in the amygdala begins in the first week of life, yet the functional development of the amygdala extends into adolescence.81 Thus, it is possible that sustaining rmTBI early in life may alter the developmental trajectory of amygdala from normal over time.
In our studies, we used 100% oxygen during recovery after mTBI. Hyperoxic resuscitation with 100% oxygen ventilation after acute brain injury, specifically global cerebral ischemia, has been shown to worsen neurological outcome in rodents and humans after cardiac arrest.82–84 Further, ventilation with 100% oxygen for 1 h after severe CCI was shown to increase peroxynitrite-mediated protein nitration in the hippocampus assessed at 24 h after the injury.85 Whether increased protein nitration in the hippocampus correlated with functional outcome was not evaluated in this study.
Contrary to experimental findings, however, normobaric hyperoxia is commonly used in the clinic to improve brain tissue oxygenation, to attenuate TBI-induced metabolic compromise, and reduce cytotoxic edema; it has been found to not worsen oxidative stress in patients with severe TBI.86–89 Notably, rats in the sham group also underwent similar recovery with 100% oxygen. Nevertheless, future studies evaluating recovery with 21% oxygen versus 100% oxygen could directly address the effect of 100% oxygen on injury.
Conclusion
We demonstrated that mTBI resulted in traumatic axonal injury and microglial activation in the PND 18 rat brain. A second impact increased the level of axonal injury. In addition, rmTBI sustained at PND 18 led to long-term associative learning deficit in adulthood. Defining the mechanisms underlying damage from a second impact could be vital to therapy development for AHT or sports concussion in children.
Supplementary Material
Acknowledgments
Supported, in part, by grants from the NIH (NS061817, U19AIO68021, NS076511, NS084604, NS060005, HD069620, NS084967).
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Faul M., Xu L., Wald M.M., and Coronado V.G. (2010). Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations and Deaths 2002–2006. Centers for Disease Control and Prevention, National Center for Injury Prevention and Control: Atlanta, GA [Google Scholar]
- 2.National Center for Injury Prevention and Control (2003). Report to Congress on Mild Traumatic Brain Injury in the United States: Steps to Prevent a Serious Public Health Problem. Centers for Disease Control and Prevention: Atlanta, GA [Google Scholar]
- 3.Daneshvar D.H., Riley D.O., Nowinski C.J., McKee A.C., Stern R.A., and Cantu R.C. (2011). Long-term consequences: effects on normal development profile after concussion. Phys. Med. Rehabil. Clin. N. Am. 22, 683–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stern R.A., Riley D.O., Daneshvar D.H., Nowinski C.J., Cantu R.C., and McKee A.C. (2011). Long-term consequences of repetitive brain trauma: chronic traumatic encephalopathy. PM R 3, Suppl 2, S460–S467 [DOI] [PubMed] [Google Scholar]
- 5.Shitaka Y., Tran H.T., Bennett R.E., Sanchez L., Levy M.A., Dikranian K., and Brody D.L. (2011). Repetitive closed-skull traumatic brain injury in mice causes persistent multifocal axonal injury and microglial reactivity. J. Neuropathol. Exp. Neurol. 70, 551–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mouzon B.C., Bachmeier C., Ferro A., Ojo J.O., Crynen G., Acker C.M., Davies P., Mullan M., Stewart W., and Crawford F. (2014). Chronic neuropathological and neurobehavioral changes in a repetitive mild traumatic brain injury model. Ann. Neurol. 75, 241–254 [DOI] [PubMed] [Google Scholar]
- 7.Donovan V., Kim C., Anugerah A.K., Coats J.S., Oyoyo U., Pardo A.C., and Obenaus A. (2014). Repeated mild traumatic brain injury results in long-term white-matter disruption. J. Cereb. Blood Flow. Metab. 34, 715–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kraus M.F., Susmaras T., Caughlin B.P., Walker C.J., Sweeney J.A., and Little D.M. (2007). White matter integrity and cognition in chronic traumatic brain injury: a diffusion tensor imaging study. Brain 130, 2508–2519 [DOI] [PubMed] [Google Scholar]
- 9.Wilde E.A., McCauley S.R., Hunter J.V., Bigler E.D., Chu Z., Wang Z.J., Hanten G.R., Troyanskaya M., Yallampalli R., Li X., Chia J., and Levin H.S. (2008). Diffusion tensor imaging of acute mild traumatic brain injury in adolescents. Neurology 70, 948–955 [DOI] [PubMed] [Google Scholar]
- 10.Wozniak J.R., Krach L., Ward E., Mueller B.A., Muetzel R., Schnoebelen S., Kiragu A., and Lim K.O. (2007). Neurocognitive and neuroimaging correlates of pediatric traumatic brain injury: a diffusion tensor imaging (DTI) study. Arch. Clin. Neuropsychol. 22, 555–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shenton M.E., Hamoda H.M., Schneiderman J.S., Bouix S., Pasternak O., Rathi Y., Vu M.A., Purohit M.P., Helmer K., Koerte I., Lin A.P., Westin C.F., Kikinis R., Kubicki M., Stern R.A., and Zafonte R. (2012). A review of magnetic resonance imaging and diffusion tensor imaging findings in mild traumatic brain injury. Brain Imaging Behav. 6, 137–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anderson V., Eren S., Dob R., Le Brocque R., Iselin G., Davern T.J., McKinlay L., and Kenardy J. (2012). Early attention impairment and recovery profiles after childhood traumatic brain injury. J. Head Trauma Rehabil. 27, 199–209 [DOI] [PubMed] [Google Scholar]
- 13.Max J.E., Castillo C.S., Robin D.A., Lindgren S.D., Smith W.L., Jr., Sato Y., and Arndt S. (1998). Posttraumatic stress symptomatology after childhood traumatic brain injury. J. Nerv. Ment. Dis. 186, 589–596 [DOI] [PubMed] [Google Scholar]
- 14.Levi R.B., Drotar D., Yeates K.O., and Taylor H.G. (1999). Posttraumatic stress symptoms in children following orthopedic or traumatic brain injury. J. Clin. Child Psychol. 28, 232–243 [DOI] [PubMed] [Google Scholar]
- 15.Gerring J.P., Slomine B., Vasa R.A., Grados M., Chen A., Rising W., Christensen J.R., Denckla M.B., and Ernst M. (2002). Clinical predictors of posttraumatic stress disorder after closed head injury in children. J. Am. Acad. Child. Adolesc. Psychiatry 41, 157–165 [DOI] [PubMed] [Google Scholar]
- 16.Daviss W.B., Mooney D., Racusin R., Ford J.D., Fleischer A., and McHugo G.J. (2000). Predicting posttraumatic stress after hospitalization for pediatric injury. J. Am. Acad. Child. Adolesc. Psychiatry 39, 576–583 [DOI] [PubMed] [Google Scholar]
- 17.Luo J., Nguyen A., Villeda S., Zhang H., Ding Z., Lindsey D., Bieri G., Castellano J.M., Beaupre G.S., and Wyss-Coray T. (2014). Long-term cognitive impairments and pathological alterations in a mouse model of repetitive mild traumatic brain injury. Front. Neurol. 5, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mannix R., Meehan W.P., Mandeville J., Grant P.E., Gray T., Berglass J., Zhang J., Bryant J., Rezaie S., Chung J.Y., Peters N.V., Lee C., Tien L.W., Kaplan D.L., Feany M., and Whalen M. (2013). Clinical correlates in an experimental model of repetitive mild brain injury. Ann. Neurol. 74, 65–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fujita M., Wei E.P., and Povlishock J.T. (2012). Intensity- and interval-specific repetitive traumatic brain injury can evoke both axonal and microvascular damage. J. Neurotrauma 29, 2172–2180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sowell E.R., Thompson P.M., Tessner K.D., and Toga A.W. (2001). Mapping continued brain growth and gray matter density reduction in dorsal frontal cortex: inverse relationships during postadolescent brain maturation. J. Neurosci. 21, 8819–8829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prins M.L., and Giza C.C. (2012). Repeat traumatic brain injury in the developing brain. Int. J. Dev. Neurosci. 30, 185–190 [DOI] [PubMed] [Google Scholar]
- 22.Giza C.C., Kolb B., Harris N.G., Asarnow R.F., and Prins M.L. (2009). Hitting a moving target: Basic mechanisms of recovery from acquired developmental brain injury. Dev. Neurorehabil. 12, 255–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huh J.W., Widing A.G., and Raghupathi R. (2007). Basic science; repetitive mild non-contusive brain trauma in immature rats exacerbates traumatic axonal injury and axonal calpain activation: a preliminary report. J. Neurotrauma 24, 15–27 [DOI] [PubMed] [Google Scholar]
- 24.Prins M.L., Hales A., Reger M., Giza C.C., and Hovda D.A. (2010). Repeat traumatic brain injury in the juvenile rat is associated with increased axonal injury and cognitive impairments. Dev. Neurosci. 32, 510–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Paxinos G., Kus L., Ashwell K.W., and Watson C. (1998). Chemoarchitectonic Atlas of the Rat Forebrain. 1st ed. Academic Press: Waltham, MA [Google Scholar]
- 26.Liaury K., Miyaoka T., Tsumori T., Furuya M., Wake R., Ieda M., Tsuchie K., Taki M., Ishihara K., Tanra A.J., and Horiguchi J. (2012). Morphological features of microglial cells in the hippocampal dentate gyrus of Gunn rat: a possible schizophrenia animal model. J. Neuroinflammation 9, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kline A.E., McAloon R.L., Henderson K.A., Bansal U.K., Ganti B.M., Ahmed R.H., Gibbs R.B., and Sozda C.N. (2010). Evaluation of a combined therapeutic regimen of 8-OH-DPAT and environmental enrichment after experimental traumatic brain injury. J. Neurotrauma 27, 2021–2032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ji J., Kline A.E., Amoscato A., Samhan-Arias A.K., Sparvero L.J., Tyurin V.A., Tyurina Y.Y., Fink B., Manole M.D., Puccio A.M., Okonkwo D.O., Cheng J.P., Alexander H., Clark R.S., Kochanek P.M., Wipf P., Kagan V.E., and Bayir H. (2012). Lipidomics identifies cardiolipin oxidation as a mitochondrial target for redox therapy of brain injury. Nat. Neurosci. 15, 1407–1413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miron V.E., Boyd A., Zhao J.W., Yuen T.J., Ruckh J.M., Shadrach J.L., van Wijngaarden P., Wagers A.J., Williams A., Franklin R.J., and ffrench-Constant C. (2013). M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat. Neurosci. 16, 1211–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Loane D.J., Stoica B.A., Tchantchou F., Kumar A., Barrett J.P., Akintola T., Xue F., Conn P.J., and Faden A.I. (2014). Novel mGluR5 positive allosteric modulator improves functional recovery, attenuates neurodegeneration, and alters microglial polarization after experimental traumatic brain injury. Neurotherapeutics 11, 857–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spain A., Daumas S., Lifshitz J., Rhodes J., Andrews P.J., Horsburgh K., and Fowler J.H. (2010). Mild fluid percussion injury in mice produces evolving selective axonal pathology and cognitive deficits relevant to human brain injury. J. Neurotrauma 27, 1429–1438 [DOI] [PubMed] [Google Scholar]
- 32.Raghupathi R., Mehr M.F., Helfaer M.A., and Margulies S.S. (2004). Traumatic axonal injury is exacerbated following repetitive closed head injury in the neonatal pig. J. Neurotrauma 21, 307–316 [DOI] [PubMed] [Google Scholar]
- 33.Friess S.H., Ichord R.N., Owens K., Ralston J., Rizol R., Overall K.L., Smith C., Helfaer M.A., and Margulies S.S. (2007). Neurobehavioral functional deficits following closed head injury in the neonatal pig. Exp. Neurol. 204, 234–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buki A., and Povlishock J.T. (2006). All roads lead to disconnection?—Traumatic axonal injury revisited. Acta Neurochir. (Wien) 148, 181–194 [DOI] [PubMed] [Google Scholar]
- 35.Pettus E.H., and Povlishock J.T. (1996). Characterization of a distinct set of intra-axonal ultrastructural changes associated with traumatically induced alteration in axolemmal permeability. Brain Res. 722, 1–11 [DOI] [PubMed] [Google Scholar]
- 36.Pettus E.H., Christman C.W., Giebel M.L., and Povlishock J.T. (1994). Traumatically induced altered membrane permeability: its relationship to traumatically induced reactive axonal change. J. Neurotrauma 11, 507–522 [DOI] [PubMed] [Google Scholar]
- 37.Perlson E., Maday S., Fu M.M., Moughamian A.J., and Holzbaur E.L. (2010). Retrograde axonal transport: pathways to cell death? Trends Neurosci. 33, 335–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smith D.H., Wolf J.A., Lusardi T.A., Lee V.M., and Meaney D.F. (1999). High tolerance and delayed elastic response of cultured axons to dynamic stretch injury. J. Neurosci. 19, 4263–4269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tang-Schomer M.D., Johnson V.E., Baas P.W., Stewart W., and Smith D.H. (2012). Partial interruption of axonal transport due to microtubule breakage accounts for the formation of periodic varicosities after traumatic axonal injury. Exp. Neurol. 233, 364–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tang-Schomer M.D., Patel A.R., Baas P.W., and Smith D.H. (2010). Mechanical breaking of microtubules in axons during dynamic stretch injury underlies delayed elasticity, microtubule disassembly, and axon degeneration. FASEB J. 24, 1401–1410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Iwata A., Stys P.K., Wolf J.A., Chen X.H., Taylor A.G., Meaney D.F., and Smith D.H. (2004). Traumatic axonal injury induces proteolytic cleavage of the voltage-gated sodium channels modulated by tetrodotoxin and protease inhibitors. J. Neurosci. 24, 4605–4613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Staal J.A., Dickson T.C., Gasperini R., Liu Y., Foa L., and Vickers J.C. (2010). Initial calcium release from intracellular stores followed by calcium dysregulation is linked to secondary axotomy following transient axonal stretch injury. J. Neurochem. 112, 1147–1155 [DOI] [PubMed] [Google Scholar]
- 43.Wolf J.A., Stys P.K., Lusardi T., Meaney D., and Smith D.H. (2001). Traumatic axonal injury induces calcium influx modulated by tetrodotoxin-sensitive sodium channels. J. Neurosci. 21, 1923–1930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yuen T.J., Browne K.D., Iwata A., and Smith D.H. (2009). Sodium channelopathy induced by mild axonal trauma worsens outcome after a repeat injury. J. Neurosci. Res. 87, 3620–3625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nuwer M.R., Hovda D.A., Schrader L.M., and Vespa P.M. (2005). Routine and quantitative EEG in mild traumatic brain injury. Clin. Neurophysiol. 116, 2001–2025 [DOI] [PubMed] [Google Scholar]
- 46.Baker A.J., Phan N., Moulton R.J., Fehlings M.G., Yucel Y., Zhao M., Liu E., and Tian G.F. (2002). Attenuation of the electrophysiological function of the corpus callosum after fluid percussion injury in the rat. J. Neurotrauma 19, 587–599 [DOI] [PubMed] [Google Scholar]
- 47.Kumar R., Husain M., Gupta R.K., Hasan K.M., Haris M., Agarwal A.K., Pandey C.M., and Narayana P.A. (2009). Serial changes in the white matter diffusion tensor imaging metrics in moderate traumatic brain injury and correlation with neuro-cognitive function. J. Neurotrauma 26, 481–495 [DOI] [PubMed] [Google Scholar]
- 48.Galluzzi L., Kepp O., and Kroemer G. (2012). Mitochondria: master regulators of danger signalling. Nat. Rev. Mol. Cell Biol. 13, 780–788 [DOI] [PubMed] [Google Scholar]
- 49.Eder C., Klee R., and Heinemann U. (1998). Involvement of stretch-activated Cl- channels in ramification of murine microglia. J. Neurosci. 18, 7127–7137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ellis E.F., Willoughby K.A., Sparks S.A., and Chen T. (2007). S100B protein is released from rat neonatal neurons, astrocytes, and microglia by in vitro trauma and anti-S100 increases trauma-induced delayed neuronal injury and negates the protective effect of exogenous S100B on neurons. J. Neurochem 101, 1463–1470 [DOI] [PubMed] [Google Scholar]
- 51.Harry G.J. (2013). Microglia during development and aging. Pharmacol. Ther. 139, 313–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Perry V.H., Hume D.A., and Gordon S. (1985). Immunohistochemical localization of macrophages and microglia in the adult and developing mouse brain. Neuroscience 15, 313–326 [DOI] [PubMed] [Google Scholar]
- 53.Rice D., and Barone S., Jr. (2000). Critical periods of vulnerability for the developing nervous system: evidence from humans and animal models. Environ. Health Perspect. 108, Suppl 3, 511–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Izumi Y., and Zorumski C.F. (1995). Developmental changes in long-term potentiation in CA1 of rat hippocampal slices. Synapse 20, 19–23 [DOI] [PubMed] [Google Scholar]
- 55.Jackson P.S., Suppes T., and Harris K.M. (1993). Stereotypical changes in the pattern and duration of long-term potentiation expressed at postnatal days 11 and 15 in the rat hippocampus. J. Neurophysiol. 70, 1412–1419 [DOI] [PubMed] [Google Scholar]
- 56.Harris K.M., Jensen F.E., and Tsao B. (1992). Three-dimensional structure of dendritic spines and synapses in rat hippocampus (CA1) at postnatal day 15 and adult ages: implications for the maturation of synaptic physiology and long-term potentiation. J. Neurosci. 12, 2685–2705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nakazawa K., Quirk M.C., Chitwood R.A., Watanabe M., Yeckel M.F., Sun L.D., Kato A., Carr C.A., Johnston D., Wilson M.A., and Tonegawa S. (2002). Requirement for hippocampal CA3 NMDA receptors in associative memory recall. Science 297, 211–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nagamoto-Combs K., McNeal D.W., Morecraft R.J., and Combs C.K. (2007). Prolonged microgliosis in the rhesus monkey central nervous system after traumatic brain injury. J. Neurotrauma 24, 1719–1742 [DOI] [PubMed] [Google Scholar]
- 59.Maxwell W.L., MacKinnon M.A., Smith D.H., McIntosh T.K., and Graham D.I. (2006). Thalamic nuclei after human blunt head injury. J. Neuropathol. Exp. Neurol. 65, 478–488 [DOI] [PubMed] [Google Scholar]
- 60.Johnson V.E., Stewart J.E., Begbie F.D., Trojanowski J.Q., Smith D.H., and Stewart W. (2013). Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain 136, 28–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Oppenheimer D.R. (1968). Microscopic lesions in the brain following head injury. J. Neurol. Neurosurg. Psychiatry 31, 299–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jordan B.D. (2013). The clinical spectrum of sport-related traumatic brain injury. Nat. Rev. Neurol. 9, 222–230 [DOI] [PubMed] [Google Scholar]
- 63.Kane M.J., Angoa-Perez M., Briggs D.I., Viano D.C., Kreipke C.W., and Kuhn D.M. (2012). A mouse model of human repetitive mild traumatic brain injury. J. Neurosci. Methods 203, 41–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Konrad K., Gauggel S., Manz A., and Scholl M. (2000). Inhibitory control in children with traumatic brain injury (TBI) and children with attention deficit/hyperactivity disorder (ADHD). Brain Inj. 14, 859–875 [DOI] [PubMed] [Google Scholar]
- 65.Mittenberg W., Wittner M.S., and Miller L.J. (1997). Postconcussion syndrome occurs in children. Neuropsychology 11, 447–452 [DOI] [PubMed] [Google Scholar]
- 66.Moser R.S., Schatz P., and Jordan B.D. (2005). Prolonged effects of concussion in high school athletes. Neurosurgery 57, 300–306 [DOI] [PubMed] [Google Scholar]
- 67.McClincy M.P., Lovell M.R., Pardini J., Collins M.W., and Spore M.K. (2006). Recovery from sports concussion in high school and collegiate athletes. Brain Inj. 20, 33–39 [DOI] [PubMed] [Google Scholar]
- 68.Field M., Collins M.W., Lovell M.R., and Maroon J. (2003). Does age play a role in recovery from sports-related concussion? A comparison of high school and collegiate athletes. J. Pediatr. 142, 546–553 [DOI] [PubMed] [Google Scholar]
- 69.Zuckerman S.L., Lee Y.M., Odom M.J., Solomon G.S., Forbes J.A., and Sills A.K. (2012). Recovery from sports-related concussion: days to return to neurocognitive baseline in adolescents versus young adults. Surg. Neurol. Int. 3, 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.O'Brien N., Lehmann H., Lecluse V., and Mumby D.G. (2006). Enhanced context-dependency of object recognition in rats with hippocampal lesions. Behav. Brain. Res. 170, 156–162 [DOI] [PubMed] [Google Scholar]
- 71.D'Hooge R., and De Deyn P.P. (2001). Applications of the Morris water maze in the study of learning and memory. Brain Res. Brain Res. Rev. 36, 60–90 [DOI] [PubMed] [Google Scholar]
- 72.Antunes M., and Biala G. (2012). The novel object recognition memory: neurobiology, test procedure, and its modifications. Cogn. Process 13, 93–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lyeth B.G., Jenkins L.W., Hamm R.J., Dixon C.E., Phillips L.L., Clifton G.L., Young H.F., and Hayes R.L. (1990). Prolonged memory impairment in the absence of hippocampal cell death following traumatic brain injury in the rat. Brain Res. 526, 249–258 [DOI] [PubMed] [Google Scholar]
- 74.Dunkley C., Kung J., Scott R.C., Nicolaides P., Neville B., Aylett S.E., Harkness W., and Cross J.H. (2011). Epilepsy surgery in children under 3 years. Epilepsy Res. 93, 96–106 [DOI] [PubMed] [Google Scholar]
- 75.Duhaime A.C., Hunter J.V., Grate L.L., Kim A., Golden J., Demidenko E., and Harris C. (2003). Magnetic resonance imaging studies of age-dependent responses to scaled focal brain injury in the piglet. J. Neurosurg. 99, 542–548 [DOI] [PubMed] [Google Scholar]
- 76.Anderson V., Spencer-Smith M., Coleman L., Anderson P., Williams J., Greenham M., Leventer R.J., and Jacobs R. (2010). Children's executive functions: are they poorer after very early brain insult. Neuropsychologia 48, 2041–2050 [DOI] [PubMed] [Google Scholar]
- 77.Stamm J.M., Bourlas A.P., Baugh C.M., Fritts N.G., Daneshvar D.H., Martin B.M., McClean M.D., Tripodis Y., and Stern R.A. (2015). Age of first exposure to football and later-life cognitive impairment in former NFL players. Neurology 84, 1114–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shechner T., Hong M., Britton J.C., Pine D.S., and Fox N.A. (2014). Fear conditioning and extinction across development: Evidence from human studies and animal models. Biol. Psychol. 100, 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lifshitz J., Witgen B.M., and Grady M.S. (2007). Acute cognitive impairment after lateral fluid percussion brain injury recovers by 1 month: evaluation by conditioned fear response. Behav. Brain Res. 177, 347–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Elder G.A., Dorr N.P., De Gasperi R., Gama Sosa M.A., Shaughness M.C., Maudlin-Jeronimo E., Hall A.A., McCarron R.M., and Ahlers S.T. (2012). Blast exposure induces post-traumatic stress disorder-related traits in a rat model of mild traumatic brain injury. J. Neurotrauma 29, 2564–2575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Landers M.S., and Sullivan R.M. (2012). The development and neurobiology of infant attachment and fear. Dev. Neurosci. 34, 101–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kilgannon J.H., Jones A.E., Shapiro N.I., Angelos M.G., Milcarek B., Hunter K., Parrillo J.E., and Trzeciak S. (2010). Association between arterial hyperoxia following resuscitation from cardiac arrest and in-hospital mortality. JAMA 303, 2165–2171 [DOI] [PubMed] [Google Scholar]
- 83.Kochanek P.M., and Bayir H. (2010). Titrating oxygen during and after cardiopulmonary resuscitation. JAMA 303, 2190–2191 [DOI] [PubMed] [Google Scholar]
- 84.Walson K.H., Tang M., Glumac A., Alexander H., Manole M.D., Ma L., Hsia C.J., Clark R.S., Kochanek P.M., Kagan V.E., and Bayr H. (2011). Normoxic versus hyperoxic resuscitation in pediatric asphyxial cardiac arrest: effects on oxidative stress. Crit. Care Med. 39, 335–343 [DOI] [PubMed] [Google Scholar]
- 85.Ahn E.S., Robertson C.L., Vereczki V., Hoffman G.E., and Fiskum G. (2008). Normoxic ventilatory resuscitation following controlled cortical impact reduces peroxynitrite-mediated protein nitration in the hippocampus. J. Neurosurg 108, 124–131 [DOI] [PubMed] [Google Scholar]
- 86.Nortje J., Coles J.P., Timofeev I., Fryer T.D., Aigbirhio F.I., Smielewski P., Outtrim J.G., Chatfield D.A., Pickard J.D., Hutchinson P.J., Gupta A.K., and Menon D.K. (2008). Effect of hyperoxia on regional oxygenation and metabolism after severe traumatic brain injury: preliminary findings. Crit. Care Med. 36, 273–281 [DOI] [PubMed] [Google Scholar]
- 87.Beynon C., Kiening K.L., Orakcioglu B., Unterberg A.W., and Sakowitz O.W. (2012). Brain tissue oxygen monitoring and hyperoxic treatment in patients with traumatic brain injury. J. Neurotrauma 29, 2109–2123 [DOI] [PubMed] [Google Scholar]
- 88.Veenith T.V., Carter E.L., Grossac J., Newcombe V.F., Outtrim J.G., Nallapareddy S., Lupson V., Correia M.M., Mada M.M., Williams G.B., Menon D.K., and Coles J.P. (2014). Use of diffusion tensor imaging to assess the impact of normobaric hyperoxia within at-risk pericontusional tissue after traumatic brain injury. J. Cereb. Blood Flow Metab. 34, 1622–1627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Puccio A.M., Hoffman L.A., Bayir H., Zullo T.G., Fischer M., Darby J., Alexander S., Dixon C.E., Okonkwo D.O., and Kochanek P.M. (2009). Effect of short periods of normobaric hyperoxia on local brain tissue oxygenation and cerebrospinal fluid oxidative stress markers in severe traumatic brain injury. J. Neurotrauma 26, 1241–1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.