
Figure 1.
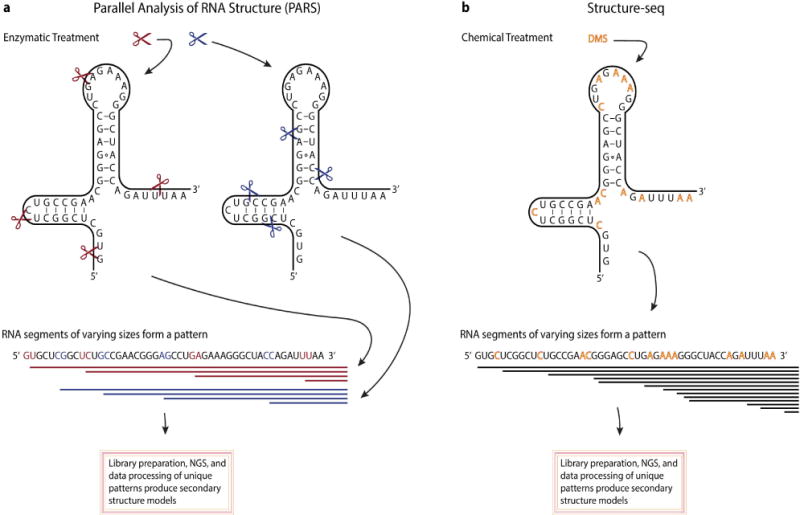
Overview of two methodologies used to determine RNA secondary structure. A. Parallel analysis of RNA structure (PARS) uses an in vitro enzymatic treatment with single strand (S1 nuclease, red scissor) and double strand (RNase V1, blue scissor) cutters to generate two pools of digested RNA. Once digested, adaptor sequences are ligated to the cleavage sites, converted into a cDNA library and subject to next-generation sequencing (NGS). Cleavages sites, identified from the sequencing data, will provide the locations of double stranded RNA regions (seen from the RNase V1 cleavage sites) or single stranded regions (seen from the S1 nuclease cleavage sites). Collectively, from these data secondary structure of RNA molecules can be determined. B. An in vivo chemical treatment, named Structure-seq, uses DMS to selectively methylate available adenines and cytosines (denoted by red letters). Reverse transcriptase activity stops one nucleotide before reaching the methylated adenine or cytosine. A cDNA library is constructed and subject to NGS. As a result, the signature of discernable stop sites can be used to infer secondary structure from NGS data.