

Abstract
Natural product discovery from environmental genomes (metagenomics) has largely been limited to the screening of existing environmental DNA (eDNA) libraries. Here, we have coupled a chemical-biogeographic survey of chromopyrrolic acid synthase (CPAS) gene diversity with targeted eDNA library production to more efficiently access rare tryptophan dimer (TD) biosynthetic gene clusters. A combination of traditional and synthetic biology-based heterologous expression efforts using eDNA-derived gene clusters led to the production of hydroxysporine (1) and reductasporine (2), two bioactive TDs. As suggested by our phylogenetic analysis of CPAS genes, identified in our survey of crude eDNA extracts, reductasporine (2) contains an unprecedented TD core structure: a pyrrolinium indolocarbazole core that is likely key to its unusual bioactivity profile. This work demonstrates the potential for the discovery of structurally rare and biologically interesting natural products using targeted metagenomics, where environmental samples are prescreened to identify the most phylogenetically unique gene sequences and molecules associated with these genes are accessed through targeted metagenomic library construction and heterologous expression.
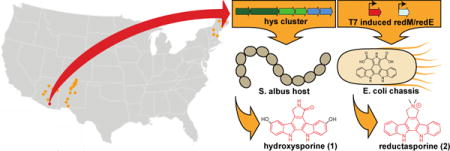
Graphical abstract
Introduction
Natural product discovery programs have long relied on screening of broth extracts from cultured bacteria to identify new bioactive small molecules.1 While extraordinarily productive, this approach has likely failed to access the majority of nature’s microbial biosynthetic potential, due to culture bias2 and gene cluster expression limitations3. Culture-independent natural product discovery methods, in which DNA extracted directly from the environment (environmental DNA, eDNA) is cloned into a cultured bacterial host, provide a means of accessing additional biosynthetic diversity from metagenomes. Most previous metagenome mining studies have focused on the analysis of gene clusters captured in pre-constructed eDNA libraries.4–5 This approach allows for the efficient discovery of novel metabolites from the biosynthetic diversity captured in existing libraries, but it likely misses the truly rare gene clusters, as they are unlikely to be represented in the small collection of existing libraries. In an effort to identify these rare gene clusters, we adopted a targeted library construction approach where sequencing is used to survey a large number of environments for the presence of rare biosynthetic gene variants microbiomes found to contain phylogenetically distinct gene sequences are specifically targeted for library production and natural product discovery. The lack of a prerequisite for library construction permits the scope of screening for novel biosynthetic genes to expand by orders of magnitude, thereby enabling the more extensive exploration of microbial biosynthesis that is needed to identify truly rare gene clusters. Here, we use this targeted metagenomics strategy to guide the discovery of tryptophan dimer (TD) gene clusters that are rare in the environment and are thus likely to encode for compounds with unprecedented structure.
TDs represent a structurally diverse class of natural products that are known to bind diverse molecular targets,6–7 leading to the notion that the oxidative coupling of two tryptophans may be a simple natural solution to generating a biologically privileged small molecule scaffold (i.e., ligand families capable of targeting a broad array of receptors).8 Despite differences in chemical structure and biological activity, biosynthetically characterized bacterial TDs share the two initial biosynthetic transformations: i) the activation (oxidation) of tryptophan by an indole-3-pyruvic acid imine synthase and ii) the dimerization of activated tryptophan by a chromopyrrolic acid (3) synthase (CPAS) (Figure 1A).9–10 Following these conserved steps, TD biosynthesis diverges to give rise to a variety of distinct core structural classes (Figure 1) that are then often highly modified by pathway-specific collections of tailoring enzymes.
Figure 1. Targeted metagenomics for tryptophan dimer pathway discovery.
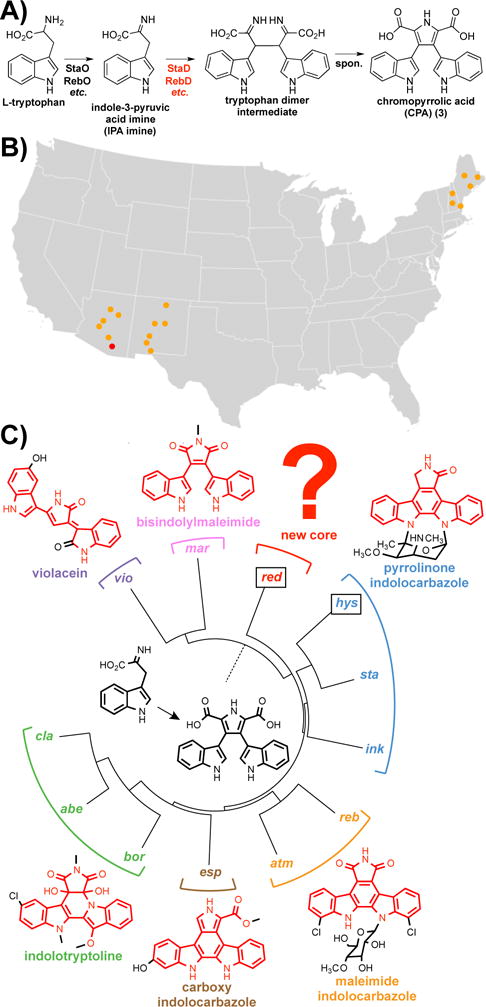
A) Conserved biosynthetic steps in pathway, with CPAS gene (red) serving as a sequence tag. B) CPAS sequencing of various soil samples (orange dots) in America, leading to AZ25 soil sample (red dot) that likely contains novel clusters. C) Phylogenetic tree of known CPAS gene sequences, alongside with the hys and red sequences (boxed) that were amplified from AZ25.
The fact that variations in core structure can result in different molecular targets led to our interest in identifying TDs with novel core structures from metagenomes.11–12 In previous studies, we have shown that CPAS gene phylogeny predicts, with very high fidelity, the core structure encoded by the gene cluster from which this gene arises (Figure 1). Based on this correlation, we developed CPAS specific degenerate PCR primers to help guide the discovery of novel TD gene clusters from archived eDNA libraries.11–12 In this study we hypothesized that by using these primers to prescreen environmental samples, followed by library construction from samples containing phylogenetically unique gene sequences, we could identify rare gene clusters that encode for compounds containing previously unknown core structures with potentially novel modes of action.
By using a chemical-biogeographic survey of geographically diverse American soils to target samples for library construction, we have discovered hydroxysporine (1) and reductasporine (2), two new bioactive TDs. As predicted from the phylogeny of the sequences that were identified in our soil prescreening efforts, hydroxysporine is based on the common pyrrolinone indolocarbazole core structure, while reductasporine is based on a structurally unprecedented pyrrolinium indolocarbazole core structure. This new core structure endows reductasporine with a bioactivity profile that differs from that seen for other core structures tested. The work presented here establishes the framework for a rare gene cluster discovery pipeline, where novel molecules with potentially rare chemical structures can be recovered from complex soil metagenomes using a combination of targeted metagenomics and synthetic biology.
Results and Discussion
Sequence tag analysis of diverse soil samples
Biosynthetically characterized bacterial tryptophan dimers can be organized into six groups according to their core structure: pyrrolinone indolocarbazole (e.g. staurosporine),13 maleimide indolocarbazole (e.g. rebeccamycin),14 violacein,15 indolotryptoline (e.g. BE-54017),16 carboxy indolocarbazole (e.g. erdasporine),11 and bisindolylmaleimide (e.g. arcyriarubin)17 (Figures 1C). The divergent evolutionary history of the gene clusters that encode these six families of structures is reflected in the fact that CPAS gene phylogeny reproduces the differences seen in TD core structure.11 This simple relationship allows sequence phylogeny to serve as a guide (i.e., sequence tag) for discovery, where eDNA-derived sequences that define deep new branches are expected to arise from gene clusters encoding novel cores and, conversely, sequences that fall in existing branches are predicted to arise from gene clusters that encode metabolites with known cores.
To identify soils that might yield new structural classes of TDs, we PCR amplified and analyzed environmental CPAS sequence from geographically diverse American soils.11 Soils containing sequences that reside outside of any known clades were identified by phylogenetically clustering the environmental sequences with known CPAS sequences. Soil sample AZ25, collected in the Sonora Desert of Southern Arizona (Figure 1B), was found in a previous survey to be rich in nonribosomal peptide and polyketide type biosynthetic diversity.18 The discovery, in the course of the present study, that this biosynthetically rich environment also contained a phylogenetically unique sequence tag that groups away from both genes from known clusters and sequences amplified in our soil survey, made this microbiome particularly attractive for targeted library construction. Ultimately, this library yielded a sequence that groups with the known pyrrolinone indolocarbazole clusters (hys tag) and a sequence (red tag) that groups away from all CPAS found in known clusters, which we predicted would be associated with the biosynthesis of a new TD core (Figure 1C; Supporting Information Figure S1).
Cloning new gene clusters from a target soil microbiome
Sequence tag screening can be used to suggest the presence of gene clusters capable of encoding novel metabolites in a microbiome; however, the utility of this analysis for natural products discovery depends on the ability to clone and heterologously express these clusters. Because of the extreme complexity of soil metagenomes, especially those predicted to be rich in secondary metabolite biosynthesis like AZ25, it was not obvious from the outset of this study that this would be possible. Previously, we have found that at least 5–10 million eDNA cosmid clones are needed to begin saturating the genetic diversity present in most soils.19 In an effort to clone the novel gene clusters predicted to be present in the AZ25 microbiome, we constructed and arrayed a ~20 million-membered cosmid library from AZ25 soil eDNA using our previously described cosmid cloning methods.20
The presence of the target genes in the AZ25 library was confirmed by using cosmid DNA isolated from the library as a template in PCR reactions with CPAS specific degenerate PCR primers. Sequencing of the resulting amplicons showed the presence of the same sequences we found in our survey of AZ25 crude eDNA. Unique cosmid clones associated with each of the two targeted tags (hys and red) were recovered from the cosmid library, sequenced, and annotated. Both cosmid P204, associated with hys tag, and cosmid P121, associated with red tag, were predicted to contain tryptophan dimer gene clusters that differ in gene content from any cluster in publicly available databases (Figure 2 and Supporting Information Figure S2–S3).
Figure 2. Novel gene clusters.
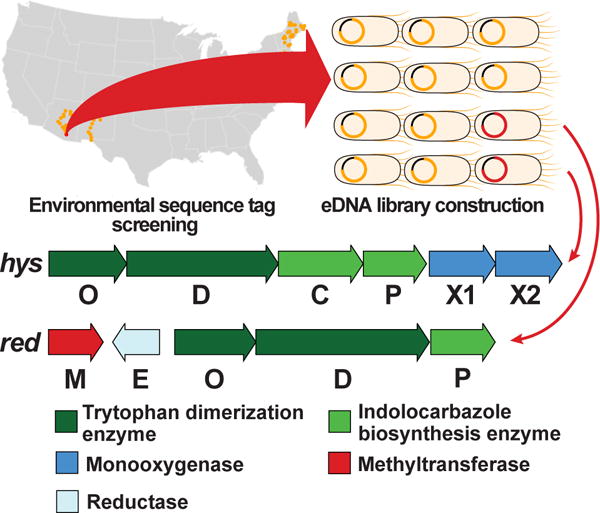
The hys and red gene clusters were discovered from a library constructed from soil (AZ25) identified from screening diverse soils for microbiomes containing phylogenetically unique CPAS gene sequences.
Bioinformatics analysis of the hys gene cluster
The predicted hys gene cluster is comprised of six biosynthetic genes organized in a single operon (Figure 2). Consistent with the observation that the hys CPAS falls in the pyrrolinone indolocarbazole clade (Figure 1C), the hys gene cluster contains all four genes required for pyrrolinone indolocarbazole biosynthesis. This includes indole-3-pyruvic acid imine (hysO) and CPAS (hysD) homologs that appear in all characterized bacterial TD gene clusters.21–22 It also includes a gene that encodes for a homolog of the cytochrome P450 (hysP) that carries out the Cβ aryl coupling needed to produce the indolocarbazole intermediate (4)23 and the flavin-dependent monooxygenase (hysC) that directs the formation of the pyrrolinone indolocarbazole core from this intermediate.24 In addition to these conserved pyrrolinone indolocarbazole biosynthesis genes, the hys cluster contains two genes predicted to encode cytochrome P450 hydroxylases (hysX1, hysX2) that are not seen in any characterized gene clusters. Based on gene content, we predicted that the hys cluster would likely encode for the biosynthesis of a di-hydroxylated pyrrolinone indolocarbazole-based metabolite.
Bioinformatics analysis of the red gene cluster
The red gene cluster is predicted to contain five biosynthetic genes (Figure 2). As seen in the hys cluster, three red genes (redO, redD, redP) are homologs of well-characterized TD biosynthetic genes that encode for the production of the indolocarbazole intermediate (4).23 The red cluster also encodes a predicted methyltransferase (redM) and an imine reductase (redE) that are not found in any known TD gene clusters. Interestingly, the red cluster does not contain a homolog of any gene known to encode the conversion of an unstable intermediate 4 to one of the known stable TD intermediates, suggesting that the red cluster likely encodes the production of an unprecedented methylated core structure.
Heterologous expression and characterization of the hys gene cluster
Heterologous expression of the hys gene cluster was achieved by first retrofitting the P204 cosmid with elements needed for conjugation and integration into Streptomyces. The retrofitted P204 construct was then introduced by conjugation into Streptomyces albus, a host that has been previously used to heterologously express a number of other TD gene clusters.25–26 LC-MS analysis of culture broth extracts from the resulting S. albus transformants identified two clone specific compounds (1 and 3). Compound 3 was easily recognized as the TD biosynthesis intermediate, chromopyrrolic acid (Figure 3A; P204, 5 day). We could not assign a structure to the second peak (1) by MS alone. Compound 1 was purified (1.1 mg/L) from large-scale culture broth extracts and the structure of the purified metabolite was elucidated using a combination of HRMS and NMR. Compound 1 is a di-hydroxylated pyrrolinone indolocarbazole that we have called hydroxysporine (1) (Figure 4 and Supporting Information Figure S4). Although hydroxysporine has been made synthetically,27–28 this is the first report of hydroxysporine as a natural product.
Figure 3. HPLC-UV chromatograms of bacterial extracts in heterologous expression studies.
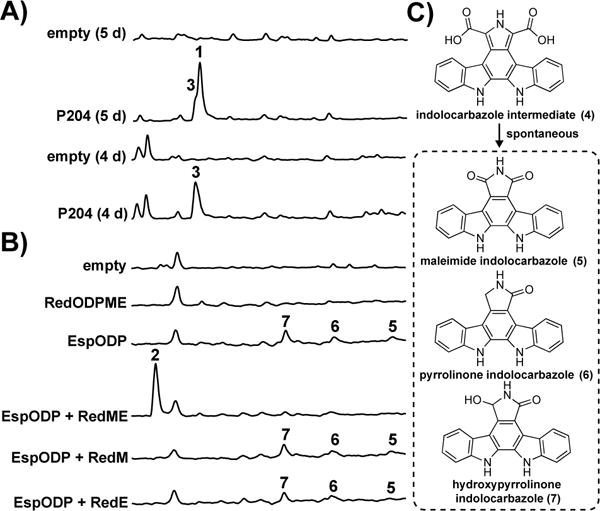
A) hys cluster in S. albus. B) red cluster in E. coli. C) Low levels of 5, 6, and 7 present in select broth extracts that are produced by spontaneous decarboxylation of 4.
Figure 4. New TD structures.
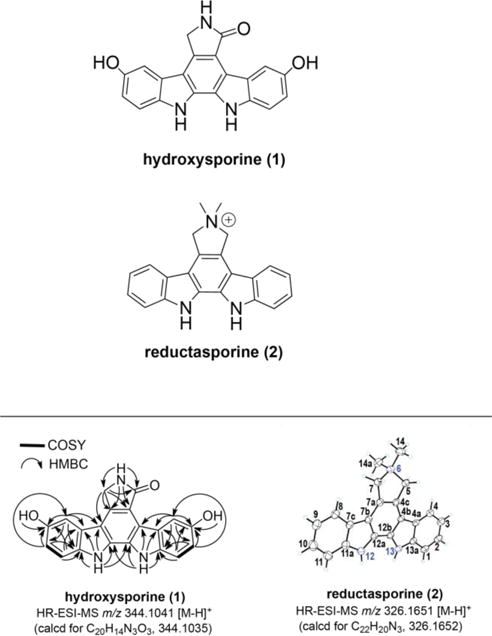
Chemical drawings of hydroxysporine (1) and reductasporine (2), along with structure elucidation data (2-D NMR for 1 and X-ray structure for 2).
In young cultures of S. albus transformed with cosmid P204, we see the initial accumulation of only chromopyrrolic acid (3) (Figure 3A; P204, 4 day). The absence of a hydroxylated tryptophan dimer intermediate suggests that HysX1/X2 are pyrrolinone indolocarbazole hydroxylases and not tryptophan hydroxylases. This is not unexpected as HysX1/X2 show higher sequence identity to hydroxylases from lankamycin29 and erythromycin30 biosynthesis, which act on complex secondary metabolite substrates, than they do to tryptophan hydroxylases. Although the number of hydroxylases in the cluster matches the number of hydroxyl groups on 1, it is not clear at this point whether only one or both predicted hydroxylases are required to produce 1.
Heterologous expression and characterization of the red gene cluster
Unlike cosmid P204, the introduction of cosmid P121 into model hosts (e.g., Escherichia coli and diverse Streptomyces spp.) did not result in the production of any detectable clone specific metabolites. Consequently, we employed a series of synthetic biology approaches focused on transcriptionally activating all of the genes in this cluster in an attempt to produce the metabolite encoded by the red cluster. Initially, each red gene was individually cloned in front of a T7 promoter and this collection of inducible genes was introduced into E. coli. As with the native clone, the refactored gene cluster also failed to confer the production of any detectable clone-specific small molecules to the host (Figure 3B; RedODPME). Surprisingly, we did not observe any of the biosynthetic intermediates (e.g., 5, 6 and 7) that are expected to accumulate with the successful expression of even just the three core TD biosynthetic genes redO, redD, and redP (Figure 3C).23 In a number of previous studies, the expression of functional redO, redD, and redP homologs in E. coli has proved challenging, with improper protein folding thought to be the key roadblock.31–32 As redO, redD, and redP homologs are present and functionally conserved in all previously characterized indolocarbazole-producing gene clusters,6 we hypothesized that we could access the metabolite encoded by the red cluster using an E. coli strain expressing a better behaved collection of “O,D,P” homologs. One such set of homologs is espO, espD, and espP from erdasporine biosynthesis.11 The expression of this gene set in E. coli leads to high-level production of the expected indolocarbazole intermediates 5, 6 and 7 (Figure 3B; EspODP). The introduction of redM and redE genes under the control of the T7 promoter into this indolocarbazole scaffold-producing strain resulted in the accumulation of a single clone-specific metabolite (2) (Figure 3B; EspODP + RedME). Compound 2 was purified (15.7 mg/L) from cultures of E. coli EspODP + RedME. Once purified, it readily crystalized from acetone and water. Its structure was initially solved by X-ray crystallography (Figure 4 and Supporting Information Figure S5) and confirmed using HRMS and NMR spectroscopic data. Compound 2 is an N,N-dimethyl-pyrrolinium indolocarbazole, which we have named reductasporine.
To the best of our knowledge, reductasporine is the first reported natural product with a pyrrolinium indolocarbazole core structure, and thus establishes a new subclass of natural tryptophan dimers. Numerous biologically significant metabolites produced by phylogenetically diverse organisms contain a quaternary ammonium cation.33–34 While a cationic pyrrole ring is seen in a number of compounds isolated from plants35–36 and to a lesser extent marine animals,37 to the best of our knowledge this functionality has not been previously seen in alkaloids isolated from bacteria.
When either RedM or RedE were individually expressed in the indolocarbazole scaffold-producing strain, only compounds 5, 6 and 7 were detected (Figure 3B; EspODP + RedM or RedE). This suggests that RedM and RedE likely act in tandem to produce 2 from the indolocarbazole intermediate 4 before it spontaneously oxidizes to 5, 6, and 7 (Figure 3C). Consistent with the potential role of RedE in the reduction of pyrole ring in 2, the closest characterized relative of the RedE reductase is the NADPH-dependent oxidoreductase Q1EQE0 that catalyzes the reduction of 2-methyl-1-pyrroline to (R)-2-methylpyrrolidine.38 Based on these observations, a biosynthetic scheme for 2 was developed (Figure 5). Homologs of RedO, RedD, and RedP are known to produce the indolocarbazole intermediate 4, which spontaneously decarboxylates to yield 5, 6, and 7 in the absence of any other biosynthetic enzymes (Figure 3C).23 In other gene clusters, downstream enzymes that “babysit” this spontaneous decarboxylation, such that specific products are formed.39 In the red pathway, we propose that RedM and RedE work in tandem to control decarboxylation of 4 and generate 2 through one reduction and two methylation events.
Figure 5. Proposed biosynthetic scheme for reductasporine (2).
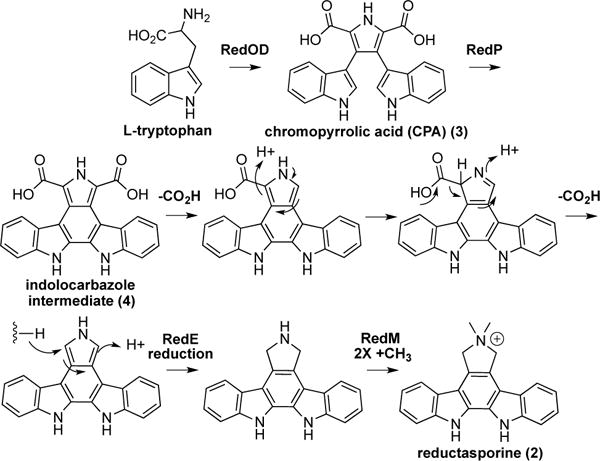
The tandem action of reductase RedE and methyltransferase RedM is proposed to be key in the formation of 2 from a common indolocarbazole intermediate.
Biological activities
Hydroxysporine (1) and reductasporine (2) were assayed for growth inhibition activity against representative Gram-positive and Gram-negative bacteria, human cells and fungi (Figure 6). A basic bioactivity profile for each core structure was developed by comparing these activities to activities observed for other simple tryptophan dimers with distinct core structures [e.g., erdasporine B (8) and arcyriarubin A (9), (Figure 7)] (Figure 6). As expected from its chemical structure, 1 was found to have a similar biological profile to that reported for staurosporine aglycone (6)40, with moderate antiproliferative and antifungal activities, but limited antibacterial activity. In contrast, 2 exhibited moderate antifungal activity, but minimal cytotoxic activity against human cells, showing no antiproliferative activity against human cell line HCT116 at the highest initial concentration tested. The lack of significant human cell cytotoxicity is in contrast to other TDs, including 8 and 9, as well as the core structures represented by staurosporine aglycone (6) and rebeccamycin aglycone (5)41 Most antifungal medications are limited by having a very narrow therapeutic index due to similarities between their molecular targets in fungal and human cells.42 While 2 is only moderately active as an antifungal agent, the disparity in cytotoxicity between C. albicans and HCT116 cells suggests the N,N-dimethyl pyrrolinium indolocarbazole core could prove useful as a lead structure for selective antifungal agents.
Figure 6. In vitro whole-cell cytotoxicity.
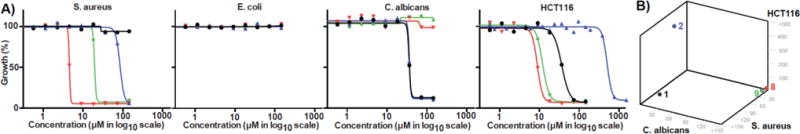
A) Dose response curves for determining the half maximal inhibitory concentration (IC50) against various organisms. Black = 1, Blue = 2, Red = 8, Green = 9. IC50 (in μM): S. aureus >150 (1), 105 (2), 4.55 (8), 19.6 (9); E. coli >150 (1), >150 (2), >150 (8), >150 (9); C. albicans 36.0 (1), 36.3 (2), >150 (8), >150 (9); Human HCT116 36.5 (1), 503 (2), 8.92 (8), 12.0 (9). B) Plot of IC50 (in μM) in S. aureus, C. albicans, and human cancer cell HCT116.
Figure 7. Overview of biosynthetically characterized tryptophan dimers.
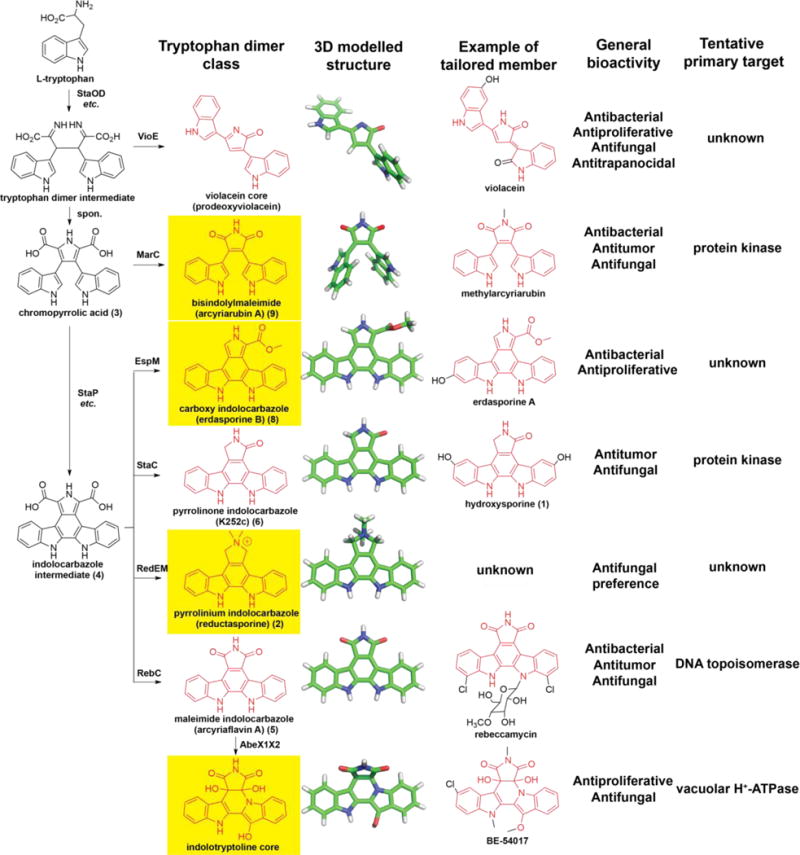
Divergent biosynthesis of bacterial TDs giving rise to seven core structural classes (first column; those whose gene clusters were characterized by metagenomics are highlighted in yellow) with diverse chemical structures (three-dimensional structure in second column; representative tailored member in third column) and bioactivities (general bioactivity profile in fourth column; tentative molecular target in fifth column).
The pyrrole ring in both indolocarbazole and bisindolylmaleimide-based structures is known to directly interact with the compound’s molecular target, rendering the structure of the pyrrole a particularly important factor in defining a TD’s mode of action.43–45 For example, while pyrrolinone indolocarbazoles primarily inhibit protein kinases,44 maleimide indolocarbazoles interact with a DNA topoisomerase46 as their primary molecular target. In the case of compound 2, the dimethylation at N6 and the complete reduction at C5 and C7 result in significant structural and hydrogen bond differences around the pyrrole ring compared to previously characterized TD cores (Figure 7). Although detailed mode of action studies will be required to confirm our hypothesis, we suspect that the interesting biological activity profile of 2 may result from its pyrrolinium structure binding a different molecular target than other, previously characterized tryptophan dimers (Figure 6 and 7).
Conclusions
The dimerization of tryptophan generates a privileged intermediate that serves as the biosynthetic precursor to a diverse collection of chemical structures with varied molecular targets (Figure 7). In this study, this is exemplified by the discovery of reductosporine (2), which contains a novel core structure with an interesting bioactivity profile. Previous cultured-based13–15 and metagenome-based11,16–17 (Figure 7) natural product discovery studies have identified and functionally characterized a number of TD gene clusters. Unfortunately, these approaches are inherently limited by the small fraction of global biosynthetic diversity currently captured in archived culture collections and metagenomic libraries.
Here we show the power of a targeted metagenomic discovery approach to systematically survey and access nature’s rare biosynthetic diversity. Using a targeted discovery approach, we were able to identify novel TDs based on both known (1) and novel (2) core structures. The discovery of the red pathway adds a novel branch to the global biosynthetic scheme of bacterial TDs, where the controlled decarboxylation, methylation and reduction of an indolocarbazole intermediate give rise to the novel pyrrolinium indolocarbazole core with a bioactivity profile that differs from that seen for other core structures.
The pyrrolinium indolocarbazole core structure of 2 is not seen in any previously characterized TDs. The detection of this, apparently rare, tryptophan dimer class was made possible by the incorporation of a large-scale sequence tag based prescreening of environmental samples into the metagenomic discovery pipeline. While conventional natural product genome mining approaches largely rely on whole genome sequencing data to identify novel gene clusters, acquiring equivalent information from the global microbiome remains economically and technically impractical. Based on the strong predictive power of sequence tag phylogeny and the cost effective scaling of this approach, environmental sequence tag screening is likely to be a rewarding long-term approach for identifying rare biosynthetic gene clusters in the global microbiome, which can then be heterologously expressed using either traditional or synthetic biology techniques.
Methods
Environmental sequence tag screening
Our chemical-biogeographic survey of tryptophan dimer (TD) gene cluster in American soil environments was conducted using a modified approach from a previous study.11 Briefly, top soils from diverse locations in Arizona (AZ), New Mexico (NM), and New England (NE) (6 sites each; 18 total) were collected. Crude eDNA was prepared from each soil sample through resuspension in lysis buffer [100 mM Tris-HCl, 100 mM EDTA, 1.5 M NaCl, 1% (w/v) CTAB, 2% (w/v) SDS, pH 8.0], heating for 2 hr at 70 °C, centrifugation to remove soil particulates (30 min, 4000 × g, 4°C), eDNA precipitation by adding 0.7 vol of isopropanol to the resulting supernatant, pelleting (30 min, 4000 × g, 4°C) and washing with 70% ethanol.
Crude environmental DNA (eDNA) extracts were resuspended in Tris-EDTA and the TD biosynthetic diversity contained in each metagenome was analyzed by PCR amplification and sequencing of representative TD sequence tags. The degenerate primer set, StaDVF and StaDVR (Supporting Information Table S1), was designed based on a conserved region of five known chromopyrrolic acid synthase (CPAS) genes (NCBI accession no.: vioB AF172851.1, staD AB088119.1, rebD AJ414559.1, inkD DQ399653.1, atmD DQ297453.1). Sequence tags were amplified by PCR with each reaction consisting of 8.3 μl of water, 10 μl of Fail-Safe PCR Buffer D (Epicentre), 0.5 μl each of StaDVF/StaDVR primers (final concentration of 2.5 μM each), 0.5 μl of template crude eDNA (100 ng), and 0.2 μl Taq DNA polymerase (New England Biolabs). PCR cycling condition – 1 cycle of 95°C for 5 min; 30 cycles of 95°C for 30 sec, 59°C for 30 sec, 72°C for 40 sec; 1 cycle of 72°C for 7 min; 4°C hold. For AZ soil samples, the amplicons were sequenced after first TOPO cloning the resulting amplicons. Amplicons of the correct size (~560 base pairs) were gel purified, cloned into TOPO-TA vector, and individual clones (~5) were sequenced from each sample following the manufacturer’s directions (Life Technologies) such that two unique sequence tags homologous to CPAS gene from each sample were identified. For NM and NE soil samples, amplicons were sequenced using multiplexed 454 sequencing (Roche). PCR reactions were conducted using the same reagents and conditions, except the StaDVF primer was modified at the 5’ end with an addition of a 454 sequencing adapter (CGT ATC GCC TCC CTC GCG CCA TCA G), followed by a unique 8 base pair barcode for each soil sample. The correctly sized amplicons were gel purified and processed for single-end read sequencing on the 454 GS-GLX Titanium platform. After processing the raw reads using the Qiime software suite (version 1.6), the two most abundant sequences (clustered at 95% identity) showing homology to CPAS genes based on BLASTX (NCBI) were chosen as representative sequence tags for each NM and NE soil sample. AZ, NM, and NE sequence tags are summarized in Supporting Information Table S2.
For phylogenetic analysis, a ClustalW alignment (MacVector version 12.0.3, Open Gap Penalty: 10.0; Extend Gap Penalty: 5.0; Pairwise Alignment Mode: Slow) was conducted using the AZ, NM, and NE sequence tags and known representative CPAS genes (vioB, staD, rebD, inkD, atmD, marB KF551863.1, espD KF551865.1, borD JX827455.1, abeD JF439215.1, claD JN165773.1). Using the hypothetical gene THITH_03330 (NCBI accession no.: CP007029.1) as the outgroup for rooting, a phylogenetic tree was constructed (Best Tree Mode; Tree Building Method: Neighbor Joining; Distance: Tajima-Nei) and reformatted to a circular display using iTOL (Supporting Information Figure S1).
Soil eDNA library construction
A cosmid-based library was constructed from AZ25 soil crude eDNA using standard methods described previously.20 Briefly, high molecular weight (HMW; >25 kb) eDNA was purified from crude eDNA by gel electrophoresis (1% agarose gel, 16 hr, 20 V). Upon electroelution (2 hr, 100V), HMW eDNA was concentrated (100 KDa molecular weight cut off; Amicon), blunt-ended (End-It), ligated into vector pWEB-TNC, packed into λ phage (MaxPlax), and transfected into E. coli (EC100, Epicentre). To facilitate pathway recovery, the AZ25 eDNA library (~20,000,000 members total) was archived into 384 unique ~50,000 membered sublibraries with matching glycerol stocks and samples of miniprepped DNA.
Recovery and analysis of TD encoding eDNA clones
Specific PCR primers were designed to recognize the tag AZ25a (hys) and AZ25b (red) and used to recover eDNA clones harboring TD pathways associated with these tags (Supporting Information Table S1). Cosmid clones were recovered from PCR positive library wells using a dilution PCR strategy. PCR reagents and cycling conditions were equivalent to those used for environmental sequence tag screening. The sublibrary glycerol stocks were arrayed as 60 μl LB aliquots (~25 cells) in 384 well plates. Plates were grown overnight (37°C) and then each well was screened by whole cell PCR. The PCR positive aliquot was spread onto LB plates and screened by colony PCR to identify the specific clone harboring the sequence tag of interest.
Cosmid clones P204 and P121, recovered using AZ25a and AZ25b primer sets, respectively, were de novo sequenced on the Ion Torrent PGM platform (Life Technologies). Sequences assembled using Newbler (version 2.6; Roche) were annotated using FGENESB (Softberry) for gene prediction and BLASTP (NCBI) for protein homology relationships. The predicted hys cluster from P204 and red cluster from P121 are shown in Supporting Information Figures S2 and S3, respectively. Complete sequences for clone P204 and P121 were deposited in the GenBank database under accession no. KP274855 and KP274854, respectively. The upstream and downstream boundaries of each gene cluster were defined as the points at which genes predicted to encode primary metabolic enzymes first appear outside of operons containing biosynthetic genes associated with previously characterized TD gene clusters.
Production and purification of compounds from hys cluster
For retrofitting, clone P204 was linearized by AanI digest and ligated with the 6.8 kb DraI fragment of E. coli/Streptomyces shuttle vector pOJ436. This construct (P204/pOJ436) and the empty pOJ436 as a negative control were separately transformed into E. coli S17.1 and then conjugated into S. albus using published protocols.47 Exconjugants selected on mannitol soy flour medium (MS) using an apramycin (25 μg/ml) and naladixic acid (25 μg/ml) overlay were re-struck on MS and grown for 5 days before harvesting spores with 10% glycerol.
For small-scale analysis of metabolite production, S. albus spores were innoculated in 50 ml of R5A media14 in 200 ml baffled flasks and shaken at 30°C (200 rpm). 4 to 5 day old cultures were extracted with equal volumes of acidified (pH 3–4) ethyl acetate, dried in vacuo, resuspended in methanol and subjected to reversed phase LC/MS analysis (150×4.6 mm, 5 μm XBridge C18, linear gradient of 80:20 water/methanol to 100% methanol with 0.1% formic acid). Analytical LC/MS data was recorded on a Micromass ZQ mass spectrometer (Waters).
For hys specific compound isolation, 5 liters of S. albus:P204/pOJ436 was grown as described above (50 ml aliquots in 200 ml baffled flasks, 30°C, 200 rpm). Five-day-old cultures were pooled and extracted with equal volumes of ethyl acetate. After concentrating in vacuo, the crude extract was subjected to preparative reversed phase HPLC (150×10 mm, 5 μm XBridge C18, isocratic 85:15 water/acetonitrile) to yield 1 (1.1 mg/L).
Engineering of TD-scaffold producing E. coli chassis
The espO, espD, espP genes responsible for the biosynthesis of indolocarbazole core were amplified from the previously characterized esp gene cluster (NCBI accession no. KF551865.1)11 using Phusion Hot Start Flex DNA polymerase (New England Biolabs) and primers as listed in Supporting Information Table S1. PCR cycling condition – 1 cycle of 95°C for 5 min; 30 cycles of 95°C for 10 sec, 62°C for 30 sec, 72°C for 30 sec/kb sec; 1 cycle of 72°C for 7 min; 4°C hold. The resulting amplicons were digested and cloned into the NcoI/NotI sites of pCOLADuet-1 for espO, NdeI/MfeI sites of espO/pCOLADuet-1 for espD, and NcoI/HindIII sites of pETDuet-1 for espP. The espOD/pCOLADuet-1 and espP/pETDuet-1 constructs were introduced into E. coli BL21(DE3) cells by electroporation.
The redO, redD, redP, redM, and redE genes from the red gene cluster were amplified using primers listed in Supporting Information Table S1 and PCR conditions outlined above. The resulting amplicons were digested and cloned into NdeI/MfeI site of pCOLADuet-1 for redO, NcoI/HindIII site of pCOLADuet-1 for redD, NcoI/HindIII site of pETDuet-1 for redP, NcoI/HindIII site of pCDFDuet-1 for redM, NdeI/MfeI site of pCDFDuet-1 for redE. Constructs with various combinations of red genes were made and electroporated into native E. coli BL21(DE3) cells or the BL21(DE3) strain producing the TD-scaffold (espODP harboring strain).
Production and purification of compounds from red cluster
For small-scale analysis of metabolite production, construct-harboring E. coli cultures grown in 50 ml of LB media in 200 ml baffled flasks to log phase (OD600 ~0.5) were induced with the addition of IPTG (0.1 mM final concentration). After growth for 36 h (200 rpm, 25°C), cultures were extracted with equal volumes of ethyl acetate, dried in vacuo, resuspended in methanol, and analyzed by reversed phase LC/MS. All instrumentation was identical to that described for the analysis of hys cluster extracts.
For red clone specific compound isolation, 5 liters of espODP harboring E. coli BL21 (“E. coli chassis”) transformed with redME/pCDFDuet-1 was grown under the same conditions as above, except the cultures were grown as 1 liter aliquots of LB media in 2.5 liter baffled flasks. 36 h after IPTG induction (0.1 mM final concentration), cultures were pooled, extracted with an equal volume of ethyl acetate, concentrated in vacuo, and subjected to preparative reversed phase HPLC for purification (150×10 mm, 5 μm XBridge C18, linear gradient of 80:20 to 50:50 water/methanol with 0.1% trifluoroacetic acid). Compound 2 eluted with 58:42 water/methanol (15.7 mg/L). An LTQ-Orbitrap mass spectrometer (Thermo Scientific) and a 600 MHz spectrometer (Bruker) were used to acquire HRMS and NMR data, respectively.
Structure elucidation
A 2996 photodiode array detector (Waters), an LTQ-Orbitrap mass spectrometer (Thermo Scientific) and a 600 MHz spectrometer (Bruker) were used to acquire UV, HRMS and NMR data, respectively.
Hydroxysporine (1) – UV (methanol) λmax 227 (sh), 296, 349, 375 (sh); 1H NMR (600 MHz, DMSO-d6) δ4.87 (2H, s, H2-7), 6.91 (1H, dd, J = 8.5, 2.4 Hz, H-2), 6.96 (1H, dd, J = 8.5, 2.4 Hz, H-10), 7.32 (1H, d, J = 2.4 Hz, H-8), 7.47 (1H, d, J = 8.5 Hz, H-1), 7.55 (1H, d, J = 8.5 Hz, H-11), 8.34 (1H, brs, NH-6), 8.61 (1H, d, J = 2.3 Hz, H-4), 8.90 (1H, brs, OH-3), 9.10 (1H, brs, OH-9), 11.13 (1H, brs, NH-13), 11.33 (1H, brs, NH-12); 13C NMR (150 MHz, DMSO-d6) δ45.0 (C-7), 105.6 (C-8), 109.7 (C-4), 111.4 (C-1), 112.2 (C-11), 113.5 (C-7b), 114.2 (C-10), 114.7 (C-2), 115.1 (C-4b), 118.1 (C-4c), 123.4 (C-7c), 123.6 (C-4a), 126.2 (C-12b), 128.7 (C-12a), 132.5 (C-7a), 133.2 (C-11a), 133.3 (C-13a), 150.3 (C-3), 151.3 (C-9), 172.6 (C-5); HR-ESI-MS m/z 344.1041 [M-H]+ (calcd for C20H14N3O3, 344.1035).
Redutasporine (2) – UV (methanol) λmax 227 (sh), 256 (sh), 274, 314 (sh), 325, 340 (sh); 1H NMR (600 MHz, DMSO-d6) δ3.53 (6H, s, H3-14, H3-14a), 5.53 (4H, s, H2-5, H2-7), 7.28 (2H, t, J = 7.2 Hz, H-3, H-9), 7.47 (2H, t, J = 7.4 Hz, H-2, H-10), 7.74 (2H, d, J = 8.1 Hz, H-1, H-11), 7.96 (2H, d, J = 7.8 Hz, H-4, H-8), 11.85 (2H, brs, NH-12, NH-13); 13C NMR (150 MHz, DMSO-d6) δ53.2 (C-14, C-14a), 69.8 (C-5, C-7), 111.9 (C-1, C-11), 114.4 (C-4b, C-7b), 117.2 (C-4c, C-7a), 119.4 (C-3, C-9), 120.7 (C-4, C-8), 122.0 (C-4a, C-7c), 125.2 (C-2, C-10), 126.0 (C-12a, C-12b), 139.2 (C-11a, C-13a); HR-ESI-MS m/z 326.1651 [M-H]+ (calcd for C22H20N3, 326.1652).
The UV spectrum collected for 1 closely resembles that of K252c,48 suggesting that 1 contained pyrrolinone indolocarbazole core structure. The chemical formula (C20H14N3O3) determined by HRMS differs from K252c by two oxygen atoms. The 1H and 13C spectra of 1 (Supporting Information Figure S4) closely resembles that of K252c,48 but differs by the appearance of two hydroxyl protons (δ8.90, 9.10) in place of two aromatic protons, as well as the downfield shift of two aromatic carbons (δ150.3, 151.3) that suggest the attachment of hydroxyl substituents on the indolocarbazole core. 2-D NMR correlations, in particular the HMBC correlations from H-1/H-11, H-2/H-10, H-4/H-8, and the hydroxyl protons to C-3/C-9, define the positions of the hydroxyl groups and confirm the chemical structure of 1 to be a dihydroxylated derivative of K252c, hydroxysporine.
Compound 2 readily crystallized by slow evaporation from a mixture of acetone and water and thus its structure was elucidated by X-ray diffraction (Supporting Information Figure S5). We thank Emil Lobkovsky at Cornell University for collecting and processing X-ray data for 2. Briefly, a small plate-like (0.30 × 0.15 × 0.05 mm3) single crystal was chosen, mounted on a Bruker X8 APEX II diffractometer (MoKα radiation) and cooled to −100°C. Data collection and reduction were done using Bruker APEX2 and SAINT+ software packages. An empirical absorption correction was applied with SADABS program (University of Göttingen). Structure was solved by direct methods and refined on F2 by full matrix least-squares techniques using Bruker SHELXTL software package. All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were found in a difference Fourier map and refined isotropically. CF3 group of the triflouroacetic acid was disordered in a usual way (staggered F atoms). Solvent molecule was also disordered and was SQEEZEd by using PLATON software package. Overall 23583 reflections were collected, 5544 of which were symmetry independent (Rint = 0.0355); with 3644 ‘strong’ reflections (with Fo > 4sFo). Final R1 = 4.86%. The X-ray structure data was deposited in the Cambridge Crystallographic Data Centre (CCDC) (Deposition number CCDC 1043291). The Oak Ridge Thermal Ellipsoil Plot (ORTEP) diagram of 2 and X-ray diffraction parameters are presented in Supporting Figure S5. The X-ray structure is consistent with the 2-D NMR data (Supporting Information Figure S6).
Biological activity
The biological activity profile was analyzed by measuring activity against gram-negative bacteria Escherichia coli EC100, gram-positive bacteria Bacillus subtilis Sr168, fungi Candida albicans CAI4, and human colon cancer cell line HCT116 (ATCC: CCL-247). For E. coli, B. subtilis, and C. albicans, an overnight culture grown in LB media for bacteria or YEPD media for yeast was diluted 106 fold and distributed in 100 μl aliquots across a 96-well plate. Compounds resuspended in DMSO (and DMSO alone as negative control) were added to the first well at an initial concentration of 50 μg/ml and were serially diluted 2-fold across the plate such that the final concentrations of the wells were 50, 25, 13, 6.3, 3.1, 1.6, 0.78, 0.39, 0.20, 0.098, 0.049, 0.024 μg/ml. After outgrowth for 18 hr for bacteria or 36 hr for fungi (300 rpm, 30°C), the absorbance (OD595) of each well was measured using a microplate reader (Epoch Microplate Spectrophotometer; BioTek). For human cells, HCT116 grown in McCoy’s 5A Media (modified, Invitrogen) supplemented with 10% fetal bovine serum and 1% (w/v) Penicillin/Streptomycin were seeded as 100 μl aliquots with approximate 1,000 cells per well across a 96-well plate and incubated for 24 hr (37°C, 5% CO2). Compounds in DMSO (and DMSO alone as negative control) were dissolved in fresh media and added to the cells such that the final concentrations of the wells were 50, 25, 13, 6.3, 3.1, 1.6, 0.78, 0.39, 0.20, 0.098, 0.049, 0.024 μg/ml. For compound 2, a second round of assays was also conducted with final concentrations of 500, 250, 130, 63, 31, 16, 7.8, 3.9, 2, 0.98, 0.49, 0.24 μg/ml. After growth for an additional 72 hr, the cells were fixed and stained with crystal violet as described in published protocols.49 The absorbance (OD590) of the crystal violet dye extract in each well was measuring using the same instrument. The normalized absorbance values in all assays were plotted and curve fitted (Graphpad Prism) to determine the half maximal inhibitory concentrations (IC50). The three-dimensional plot of IC50 was generated using ’Excel 3D Scatter Plot’ macros (Doka Life Cycle Assessments).
Supplementary Material
Acknowledgments
We thank Emil Lobkovsky (Cornell University) for his assistance with X-ray crystallography. This work was supported by NIH grants GM077516.
Footnotes
Supporting Information.
This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interest.
References
- 1.Sterner O. Methods Mol Biol. 2012;864:393. doi: 10.1007/978-1-61779-624-1_15. [DOI] [PubMed] [Google Scholar]
- 2.Whitman WB, Coleman DC, Wiebe WJ. Proc Natl Acad Sci U S A. 1998;95:6578. doi: 10.1073/pnas.95.12.6578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scherlach K, Hertweck C. Org Biomol Chem. 2009;7:1753. doi: 10.1039/b821578b. [DOI] [PubMed] [Google Scholar]
- 4.Wilson MC, Piel J. Chem Biol. 2013;20:636. doi: 10.1016/j.chembiol.2013.04.011. [DOI] [PubMed] [Google Scholar]
- 5.Milshteyn A, Schneider JS, Brady SF. Chem Biol. 2014;21:1211. doi: 10.1016/j.chembiol.2014.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sanchez C, Mendez C, Salas JA. Nat Prod Rep. 2006;23:1007. doi: 10.1039/b601930g. [DOI] [PubMed] [Google Scholar]
- 7.Nakano H, Omura S. J Antibiot (Tokyo) 2009;62:17. doi: 10.1038/ja.2008.4. [DOI] [PubMed] [Google Scholar]
- 8.Evans BE, Rittle KE, Bock MG, DiPardo RM, Freidinger RM, Whitter WL, Lundell GF, Veber DF, Anderson PS, Chang RS, Lotti VJ, Cerino DJ, Chen TB, Kling PJ, Kunkel KA, Springer JP, Hirshfield J. J Med Chem. 1988;31:2235. doi: 10.1021/jm00120a002. [DOI] [PubMed] [Google Scholar]
- 9.Ryan KS, Drennan CL. Chem Biol. 2009;16:351. doi: 10.1016/j.chembiol.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alkhalaf LM, Ryan KS. Chem Biol. 2015;22:317. doi: 10.1016/j.chembiol.2015.02.005. [DOI] [PubMed] [Google Scholar]
- 11.Chang FY, Ternei MA, Calle PY, Brady SF. J Am Chem Soc. 2013;135:17906. doi: 10.1021/ja408683p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang FY, Brady SF. Proc Natl Acad Sci U S A. 2013;110:2478. doi: 10.1073/pnas.1218073110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Onaka H, Taniguchi S, Igarashi Y, Furumai T. J Antibiot (Tokyo) 2002;55:1063. doi: 10.7164/antibiotics.55.1063. [DOI] [PubMed] [Google Scholar]
- 14.Sanchez C, Butovich IA, Brana AF, Rohr J, Mendez C, Salas JA. Chem Biol. 2002;9:519. doi: 10.1016/s1074-5521(02)00126-6. [DOI] [PubMed] [Google Scholar]
- 15.Pemberton JM, Vincent KM, Penfold RJ. Current Microbiology. 1991;22:355. [Google Scholar]
- 16.Chang FY, Brady SF. J Am Chem Soc. 2011;133:9996. doi: 10.1021/ja2022653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chang FY, Brady SF. Chembiochem. 2014;15:815. doi: 10.1002/cbic.201300756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Charlop-Powers Z, Owen JG, Reddy BV, Ternei MA, Brady SF. Proc Natl Acad Sci U S A. 2014;111:3757. doi: 10.1073/pnas.1318021111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Banik JJ, Brady SF. Proc Natl Acad Sci U S A. 2008;105:17273. doi: 10.1073/pnas.0807564105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brady SF. Nat Protoc. 2007;2:1297. doi: 10.1038/nprot.2007.195. [DOI] [PubMed] [Google Scholar]
- 21.Howard-Jones AR, Walsh CT. Biochemistry. 2005;44:15652. doi: 10.1021/bi051706e. [DOI] [PubMed] [Google Scholar]
- 22.Nishizawa T, Gruschow S, Jayamaha DH, Nishizawa-Harada C, Sherman DH. J Am Chem Soc. 2006;128:724. doi: 10.1021/ja056749x. [DOI] [PubMed] [Google Scholar]
- 23.Howard-Jones AR, Walsh CT. J Am Chem Soc. 2007;129:11016. doi: 10.1021/ja0743801. [DOI] [PubMed] [Google Scholar]
- 24.Groom K, Bhattacharya A, Zechel DL. Chembiochem. 2011;12:396. doi: 10.1002/cbic.201000580. [DOI] [PubMed] [Google Scholar]
- 25.Sanchez C, Zhu L, Brana AF, Salas AP, Rohr J, Mendez C, Salas JA. Proc Natl Acad Sci U S A. 2005;102:461. doi: 10.1073/pnas.0407809102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Salas AP, Zhu L, Sanchez C, Brana AF, Rohr J, Mendez C, Salas JA. Mol Microbiol. 2005;58:17. doi: 10.1111/j.1365-2958.2005.04777.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kleinschroth, J.; Hartenstein, J.; C., S.; Rudolph, C.; Marme, D.; Paetzold, S.; Offen., G., Ed. 1993.
- 28.Yamada, R.; Hayashi, Y. 1993.
- 29.Arakawa K, Kodama K, Tatsuno S, Ide S, Kinashi H. Antimicrob Agents Chemother. 2006;50:1946. doi: 10.1128/AAC.00016-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cupp-Vickery J, Anderson R, Hatziris Z. Proc Natl Acad Sci U S A. 2000;97:3050. doi: 10.1073/pnas.050406897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chiu HT, Chen YL, Chen CY, Jin C, Lee MN, Lin YC. Mol Biosyst. 2009;5:1180. doi: 10.1039/b905293c. [DOI] [PubMed] [Google Scholar]
- 32.Chiu HT, Lin YC, Lee MN, Chen YL, Wang MS, Lai CC. Mol Biosyst. 2009;5:1192. doi: 10.1039/b912395b. [DOI] [PubMed] [Google Scholar]
- 33.Sohlenkamp C, Lopez-Lara IM, Geiger O. Prog Lipid Res. 2003;42:115. doi: 10.1016/s0163-7827(02)00050-4. [DOI] [PubMed] [Google Scholar]
- 34.Fougere F, Lerudulier D. Journal of General Microbiology. 1990;136:2503. doi: 10.1099/00221287-136-1-157. [DOI] [PubMed] [Google Scholar]
- 35.Yin J, Zhang ZW, Yu WJ, Liao JY, Luo XG, Shen YJ. Am J Chin Med. 2010;38:157. doi: 10.1142/S0192415X10007737. [DOI] [PubMed] [Google Scholar]
- 36.Ueda JY, Takagi M, Shin-ya K. J Nat Prod. 2009;72:2181. doi: 10.1021/np900580f. [DOI] [PubMed] [Google Scholar]
- 37.Roll DM, Ireland CM, Lu HSM, Clardy J. J Org Chem. 1988;53:3276. [Google Scholar]
- 38.Rodriguez-Mata M, Frank A, Wells E, Leipold F, Turner NJ, Hart S, Turkenburg JP, Grogan G. Chembiochem. 2013;14:1372. doi: 10.1002/cbic.201300321. [DOI] [PubMed] [Google Scholar]
- 39.Goldman PJ, Ryan KS, Hamill MJ, Howard-Jones AR, Walsh CT, Elliott SJ, Drennan CL. Chem Biol. 2012;19:855. doi: 10.1016/j.chembiol.2012.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sancelme M, Fabre S, Prudhomme M. J Antibiot (Tokyo) 1994;47:792. doi: 10.7164/antibiotics.47.792. [DOI] [PubMed] [Google Scholar]
- 41.Sanchez-Martinez C, Shih C, Zhu G, Li T, Brooks HB, Patel BK, Schultz RM, DeHahn TB, Spencer CD, Watkins SA, Ogg CA, Considine E, Dempsey JA, Zhang F. Bioorg Med Chem Lett. 2003;13:3841. doi: 10.1016/s0960-894x(03)00792-3. [DOI] [PubMed] [Google Scholar]
- 42.Ghannoum MA, Rice LB. Clin Microbiol Rev. 1999;12:501. doi: 10.1128/cmr.12.4.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bailly C, Qu X, Anizon F, Prudhomme M, Riou JF, Chaires JB. Mol Pharmacol. 1999;55:377. doi: 10.1124/mol.55.2.377. [DOI] [PubMed] [Google Scholar]
- 44.Prade L, Engh RA, Girod A, Kinzel V, Huber R, Bossemeyer D. Structure. 1997;5:1627. doi: 10.1016/s0969-2126(97)00310-9. [DOI] [PubMed] [Google Scholar]
- 45.Komander D, Kular GS, Schuttelkopf AW, Deak M, Prakash KRC, Bain J, Elliott M, Garrido-Franco M, Kozikowski AP, Alessi DR, Van Aalten DMF. Structure. 2004;12:215. doi: 10.1016/j.str.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 46.Staker BL, Feese MD, Cushman M, Pommier Y, Zembower D, Stewart L, Burgin AB. J Med Chem. 2005;48:2336. doi: 10.1021/jm049146p. [DOI] [PubMed] [Google Scholar]
- 47.Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA. Practical streptomyces genetics. John Innes Foundation; 2000. [Google Scholar]
- 48.Yasuzawa T, Iida T, Yoshida M, Hirayama N, Takahashi M, Shirahata K, Sano H. J Antibiot (Tokyo) 1986;39:1072. doi: 10.7164/antibiotics.39.1072. [DOI] [PubMed] [Google Scholar]
- 49.Zivadinovic D, Gametchu B, Watson CS. Breast Cancer Research. 2005;7:R101. doi: 10.1186/bcr958. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.