

Summary
Impaired Insulin and IGF-1 Signaling (iIIS) in C. elegans daf-2 mutants extends lifespan more than two-fold. Constitutively iIIS increases mitochondrial activity and reduces reactive oxygen species (ROS) levels. By contrast, acute impairment of daf-2 in adult C. elegans reduces glucose uptake and transiently increases ROS. Consistent with the concept of mitohormesis, this ROS signal causes an adaptive response by inducing ROS-defense enzymes (SOD, catalase) culminating in ultimately reduced ROS levels despite increased mitochondrial activity. Inhibition of this ROS signal by antioxidants reduces iIIS-mediated longevity by up to 60%. Induction of the ROS signal requires AAK-2 (AMPK), while PMK-1 (p38) and SKN-1 (NRF-2) are needed for the retrograde response. IIIS upregulates mitochondrial L-proline catabolism, and impairment of the latter impairs the lifespan-extending capacity of iIIS while L-proline supplementation extends C. elegans lifespan. Taken together, iIIS promotes L-proline metabolism to generate a ROS signal for the adaptive induction of endogenous stress defense to extend lifespan.
Graphical Abstract
Introduction
In many studies on the prevention of aging, extended life span is associated with increased stress resistance. Several interventions have been described to promote both longevity and stress defense mechanisms, including calorie restriction (Masoro, 1998; Weindruch and Walford, 1988; Xia et al., 1995), physical exercise (Lanza et al., 2008; Warburton et al., 2006), and impaired insulin/IGF1-signaling (IIS).
The eminent role of impaired IIS for the extension of life span has repeatedly been demonstrated across a wide evolutionary spectrum from nematodes (Friedman and Johnson, 1988; Kenyon et al., 1993; Kimura et al., 1997; Morris et al., 1996), to flies (Clancy et al., 2001; Tatar et al., 2001) and to mice (Blüher et al., 2003; Brown-Borg et al., 1996; Holzenberger et al., 2003). These mechanisms by which impaired IIS promotes life span are not well understood, but presumably involve increasing resistance against various stressors, such as thermal and oxidative stress (Brys et al., 2010; Honda and Honda, 1999; Lithgow et al., 1995; Murphy et al., 2003; Vanfleteren, 1993; Vanfleteren and De Vreese, 1995). On the other hand, impaired IIS has also been shown to increase metabolic rate and mitochondrial metabolism in both C. elegans (Houthoofd et al., 2005; Vanfleteren and De Vreese, 1995) and mice (Brown-Borg et al., 2012; Katic et al., 2007).
We have previously shown that reactive oxygen species (ROS) emanating from the mitochondria have an essential role for the life span-extending and health-promoting effects of glucose restriction in C. elegans (Schulz et al., 2007) and physical exercise in mammals (Ristow et al., 2009). In the present study, we address the hypothesis that impairment of IIS causes depletion of intracellular glucose which is sensed by AMP-activated protein kinase (AMPK) to induce oxidative non-glucose metabolism and to generate a ROS imbalance which in turn is instrumental for the life span-extending capabilities of impaired IIS in C. elegans.
Results
To study the effects of constitutively impaired IIS in a species-independent fashion we have used three different models: a C. elegans strain carrying a mutant daf-2(e1370) [insulin/IGF-1 receptor homologue] gene, mouse embryonic fibroblasts (MEFs) lacking a protein primary target of both the insulin and IGF-1 receptors, namely insulin receptor substrate 1 (IRS-1) (Brüning et al., 1997) (Figure S1A), and lastly MEFs inducibly lacking the insulin receptor (IR) in a heterozygous fashion (previously unpublished, see Experimental Procedures for details) (Figure S1B). These three models were independently analyzed regarding the following seven parameters.
Constitutively impaired IIS promotes stress resistance
Daf-2 mutant and appropriate wild-type control strains of C. elegans (N2) were exposed to the established ROS generator paraquat at a concentration of 10 mM for six days. The daf-2 mutant worms exhibited increased resistance against paraquat stress, as reflected by increased survival (Figure 1A) consistent with previously published data (Brys et al., 2010; Honda and Honda, 1999). Similarly, MEFs deficient for IRS1 (IRS1−/−) and MEFs with heterozygous inactivation of the insulin receptor (IR+/−) were more resistant to paraquat stress in vitro than control fibroblasts (Figures 1B and 1C, Figure S1C).
Figure 1. Constitutive impaired insulin-/IGF1-signaling induces mitochondrial metabolism, reduces ROS levels, and increases endogenous antioxidant defense in both C. elegans and murine embryonic fibroblasts.

(A–C) Survival on paraquat (D–F) ATP content, (G–I) oxygen consumption, (J–L) mitochondrial ROS levels, (N–O) hydrogen peroxide production, (P–R) superoxide dismutase activity, (S–U) catalase activity, each quantified in (A, D, G, J, M, P, S) daf-2(e1370) nematodes or (B, E, H, K, N, Q, T) IRS1−/− MEFs or (C, F, I, L, O, R, U) IR+/− MEFs (all depicted in black bars) relative to effects in the respective wild-type controls (white bars). All values are given as mean ± SD. *p<0.05, **p<0.01, ***p<0.001 versus respective controls.
Constitutively impaired IIS increases mitochondrial metabolism
It has been previously suggested that impaired IIS in C. elegans, due to impaired expression of DAF-2, induces an increase in metabolic rate in C. elegans (Houthoofd et al., 2005). Indeed, in the present study we find that the ATP content in daf-2 mutants is increased by 102% (Figure 1D). Likewise, despite the impairment in insulin/IGF-1 signalling, both IRS1−/− and IR+/− MEFs have increased ATP levels by 69 and 40%, respectively (Figures 1E and 1F), suggesting an increase in energy efficiency and mitochondrial activity in daf-2, IRS1−/− and IR+/−. Similarly, we observed an increase in oxygen consumption by 39% of daf-2(e1370) mutants (Figure 1G), as well as in IRS1−/− and IR+/− by 45 and 28%, respectively (Figures 1H and 1I). Taken together, these findings indicate that chronic impairment of IIS uniformly causes an induction of mitochondrial activity in daf-2, IRS1−/− and IR+/−.
Constitutively impaired daf-2 expression reduces ROS levels and induces endogenous ROS defense enzymes
Mitochondria are the main source of reactive oxygen species (ROS). Since impaired IIS promotes increased mitochondrial activity, as shown above (Figures 1D 1I), we anticipated that the daf-2 mutant worms and IRS1−/− and IR+/− MEFs would exhibit increased mitochondrial ROS levels. To test this hypothesis, we have quantified ROS levels using a redox-sensitive dye targeted that is accumulated within the mitochondrial compartment in the presence of ROS (Esposti et al., 1999). To our surprise, this revealed an unexpected reduction of ROS levels by 14 to 28% in all three models (Figures 1J–1L), despite increased mitochondrial activity (Figures 1D 1I). Furthermore when we quantified the accumulation of hydrogen peroxide in the supernatant of worms and MEFs cultures to obtain an independent estimate of ROS levels we observed a 9 to 50 % reduction in all three cases (Figures 1M 1O). Thus, using two independent approaches all three model systems of chronically impaired IIS show reduced ROS levels. Moreover, we quantified production of hydrogen peroxide in mitochondria that had been isolated from N2 and daf-2 nematodes, respectively. Consistent with the findings in alive worms (Figure 1M) we found decreased production of hydrogen peroxide in mitochondria isolated from daf-2 mutants (Figure S1D).
To resolve how increased mitochondrial activity might be associated with decreased ROS levels, we quantified activities of antioxidant enzymes in lysates of the C. elegans and MEFs. Consistent with findings of others (Vanfleteren, 1993), no significant activity of glutathione peroxidase was detected in these nematodes (data not shown). However, activities of both superoxide dismutase (SOD) (Figure 1P) and catalase (CAT) (Figure 1S) were found to be increased by 50 and 36% in daf-2 mutants, and similar findings were obtained for IRS1−/− as well as IR+/− (Figures 1Q, 1R, 1T and 1U). These findings indicate that in different states of chronically impaired IIS there is a decrease in ROS levels despite increased mitochondrial activity, and that this reduction is likely the result of increased activities of the major ROS defense enzymes, suggesting that the increase in ROS defense enzymes reflects an adaptive response to increased mitochondrial metabolism and possibly transiently increased ROS levels due to increased respiration.
Acutely impaired daf-2 expression causes reduced glucose uptake in C. elegans
To test this hypothesis we have analyzed the metabolic effects of an acute rather than constitutive daf-2 impairment in a time-resolved manner. To this end, we administered RNAi against daf-2, RNAi(daf-2) (Dillin et al., 2002) to young adult C. elegans and quantified the uptake of radioactively labelled 2-deoxy-glucose as a measure in insulin action. Employing this assay, we observe a persistent reduction of 2-deoxy-glucose uptake in RNAi(daf-2)-treated nematodes by 25% (Figure 2A), consistent with long-standing evidence for reduced glucose uptake following impaired IIS in mammals.
Figure 2. Acute impairment of daf-2 signaling transiently induces mitochondrial ROS levels to promote endogenous antioxidant defense.

(A) 2-Deoxy-glucose uptake, (B) ATP content, (C) oxygen consumption and (D) mitochondrial ROS levels each following exposure to RNAi against daf-2 (black bars) relative to effects on control RNAi-treated nematodes (white bars) at different time points. (E–G) Fluorescent microphotographs (enlargement: 10fold) of MitoTracker Red CM-H2X-treated nematodes, (E) after 24 h of control RNAi treatment, (F) after 24 h of daf-2 RNAi treatment, (G) after 1 h of paraquat treatment (positive control). (H) Hydrogen peroxide production, (I) superoxide dismutase activity, (J) catalase activity, each following exposure to RNAi against daf-2 (black bars) relative to effects on control RNAi-treated nematodes (white bars) at different time points. (K) Oxygen consumption, (L) mitochondrial ROS levels, (M) hydrogen peroxide production, (N) superoxide dismutase activity, (O) catalase activity, each following exposure to RNAi against daf-2 in the presence of the antioxidants NAC (red bars) and BHA (blue bars) relative to effects on control RNAi-treated nematodes in the presence of antioxidants (white bars) at different time points. In all panels relative values are depicted; for absolute values, please see Figure S2. All values are given as mean ± SD. * and # p<0.05, **and ## p<0.01, ***p<0.001 versus respective controls.
Acutely impaired daf-2 expression activates mitochondrial metabolism
Reduced availability of glucose has been previously shown to activate mitochondrial metabolism in both S. cerevisiae (Barros et al., 2004; Lin et al., 2002) and C. elegans (Schulz et al., 2007). To test whether impaired DAF-2 expression would also activate mitochondrial metabolism in C. elegans, we performed a time course assessing metabolism after addition of RNAi(daf-2). Twelve hours after addition of RNAi(daf-2) we observed a transient reduction in respiration and ATP levels in the nematode (Figure 2B and 2C), suggesting an initial energy deficit caused by impaired glucose uptake. Consistently following this primary reduction of ATP, we observed a secondary increase in oxygen consumption which reached a maximum at 24 to 48 hours after addition of RNAi(daf-2) (Figure 2C) and this was paralleled by an increase in ATP content (Figure 2B). Taken together, these data indicate that the nematode compensates for an energy deficit caused by decreased glucose availability by increased respiration culminating in secondarily increased ATP levels despite permanently decreased glucose uptake. The latter finding strongly suggests that energy sources other than glucose, i.e. fatty acids and/or amino acids, are likely to be metabolized.
Acutely impaired daf-2 expression promotes a transient increase in ROS
Increased mitochondrial activity is known to promote formation of mitochondrial ROS as an inevitable by-product of respiration and oxidative phosphorylation. Consistent with this fact, we observed increased ROS levels 24 and 48 hours after addition of RNAi(daf-2) (Figures 2D 2G) when mitochondrial activity and respiration were increased. However, after 5 days of RNAi(daf-2) treatment, both mitochondrial activity and respiration remained increased, whereas mitochondrial ROS content was found to be significantly decreased (Figure 2D). These ROS levels were confirmed for the two key time points (24 and 120 hrs) using the independent AmplexRed-based method (Figure 2H). These findings indicate that only at time points beyond 48 hours of RNAi(daf-2) addition, increased mitochondrial activity, as reflected by ATP content and respiration rates, is associated with a decrease in ROS levels (as reflected in the steady state, see Figure 1), whereas ROS levels are transiently increased at earlier time points.
Acutely impaired daf-2 expression secondarily induces endogenous ROS defense enzymes
To better understand the changing ROS levels, we quantified activities of antioxidant enzymes in lysates of whole nematodes. Both superoxide dismutase (SOD) and catalase (CAT) were found to be induced following RNAi(daf-2) treatment for 48 and 120 hours, but interestingly not at earlier time points (Figures 2I and 2J). The relatively late induction of SOD and CAT was preceded by increases in respiration, ATP and ROS (Figures 2B 2H). These findings suggest that adaptive inductions of both SOD and CAT activities efficiently counteract the primarily increased ROS levels culminating in decreased ROS exposure at 120 hours. We have previously observed this type of adaptive response in states of caloric restriction and physical exercise (Ristow and Zarse, 2010; Schulz et al., 2007), while the time-resolved analysis of ROS signals preceding increased adaptive responses have not been described before.
The daf-2-dependent transient ROS signal is required for induction of endogenous ROS defense enzymes
C. elegans carrying constitutively inactive alleles of daf-2 have repeatedly been shown to be resistant to a variety of stresses (Brys et al., 2010; Honda and Honda, 1999; Lithgow et al., 1995; Murphy et al., 2003; Vanfleteren, 1993; Vanfleteren and De Vreese, 1995). Our findings of a late, i.e. secondary, induction of SOD and CAT activities following exposure of worms to RNAi(daf-2) suggest that transiently increased ROS levels may be required to cause this increase in ROS defense. To test this hypothesis, we have treated nematodes with RNAi(daf-2) that were exposed to two mechanistically independent antioxidants, n-acetyl-cysteine (NAC, 1 mM final concentration) and butylated hydroxyanisole (BHA, 25 μM final concentration). In the presence of either NAC or BHA, we still observe an induction of respiration following RNAi(daf-2) treatment (Figure 2K). However, both the induction of ROS levels (Figures 2L and 2M) as well as the secondarily increased in activities of SOD and CAT (Figures 2N and 2O) were absent, indicating that increased ROS levels are required to induce endogenous ROS defense enzymes and antioxidant defense capacity. These findings indicate that RNAi(daf-2) is incapable of inducing SOD and CAT whenever the RNAi(daf-2)-mediated, transient increase in mitochondrial ROS is blocked by exogenous antioxidant supplements, even in the presence of increased respiration.
Exogenous antioxidants inhibit the life span-extending capabilities of DAF-2 and AGE-1 deficiency
Based on these latter findings we asked whether induction of SOD and CAT by increased ROS levels contributes to the life span-extending capabilities of impaired IIS by determining life span of NAC- or BHA-treated nematodes in the presence and absence of RNAi(daf-2). While no effect of antioxidants on life expectancy was observed in the absence of RNAi(daf-2) (Figures 3A and 3B) precluding the possibility of antioxidant toxicity on wild-type N2 nematodes, both NAC (Figure 3A) and BHA (Figure 3B) reduced life span extension caused by RNAi(daf-2) treatment. As shown in more detail in Table 1, NAC impaired the life span-extending capabilities of RNAi(daf-2) by 35.7% (maximum life span) and 36.7% (mean life span), while BHA reduced the life span-extending capabilities of RNAi(daf-2) by 37.7% and 59.9% (maximum and mean life span, respectively). Moreover, NAC (Figure 3C) and BHA (Figure 3D) produced similar reduction in the long-lived worms with constitutively inactivating daf-2(e1370) mutation (see Table 1 for quantitative effects of antioxidants on daf-2(e1370) mutations). We lastly tested constitutive age-1(hx546) mutants which affect the phosphoinositide-3-OH kinase orthologue age-1 downstream of daf-2, and very similarly found both NAC (Figure 3E) and BHA (Figure 3F) to reduce the life span-extending propensities of a constitutively inactivating age-1 mutation.
Figure 3. Exogenous antioxidants impair the life span-extending effects of daf-2 impairment.

(A) Lifespan analyses of nematodes exposed to control-RNAi (open circles) in the presence (red) or absence (black) of the antioxidant NAC; life span analyses of nematodes exposed to RNAi against daf-2 (closed triangles) in the presence (red) or absence (black) of NAC. (B) Lifespan analyses in the presence of RNAi as above replacing NAC with the antioxidant BHA (blue). (C) Lifespan analyses of daf-2(e1370) nematodes (closed triangles) in the presence (red) or absence (black) of NAC. (D) Lifespan analyses of daf-2(e1370) nematodes replacing NAC with BHA (blue). (E) Lifespan analyses of age-1(hx546) nematodes (closed triangles) in the presence (red) or absence (black) of NAC. (F) Lifespan analyses of age-1(hx546) nematodes replacing NAC with BHA (blue).
Table 1.
Life Span Assay Results and Statistical Analyses
| Strain, RNAi, Solvent | Maximum Life Span (d) (+/− SEM) | Mean Life Span (d) (+/− SEM) | P-value (vs. control, see footnotes) | Number of experiments (n) | Number of nematodes (n) |
|---|---|---|---|---|---|
| N2 (control RNAi) H2O | 28.0 +/−0.6 | 19.40 +/−0.3 | 3 | 323 | |
| N2 (control RNAi) NAC/H2O | 27.3 +/−0.9 | 18.96 +/−0.2 | n.s.a | 3 | 357 |
| N2 (daf-2 RNAi) H2O | 70.3 +/−0.9 | 46.76 +/−1.0 | <0.0001a,b | 3 | 310 |
| N2 (daf-2 RNAi) NAC/H2O | 60.3 +/−0.9 | 39.64 +/−1.1 | <0.0001a,b,c | 3 | 318 |
| N2 (control RNAi) DMSO | 31.0 +/−0.6 | 19.89 +/−0.2 | 3 | 335 | |
| N2 (control RNAi) BHA/DMSO | 28.3 +/−0.9 | 19.32 +/−0.2 | n.s.d | 3 | 331 |
| N2 (daf-2 RNAi) DMSO | 71.7 +/−1.2 | 46.00 +/−1.0 | <0.0001d,e | 3 | 342 |
| N2 (daf-2 RNAi) BHA/DMSO | 60.0 +/−0.6 | 34.16 +/−0.7 | <0.0001d,e,f | 3 | 331 |
| daf-2(e1370) H2O | 73.5 +/−1.5 | 51.09 +/−0.6 | 2 | 218 | |
| daf-2(e1370) NAC/H2O | 61.5 +/−1.5 | 44.87 +/−0.5 | <0.0001g | 2 | 248 |
| daf-2(e1370) DMSO | 73.5 +/−1.5 | 47.23 +/−1.1 | 2 | 222 | |
| daf-2(e1370) BHA/DMSO | 63.0 +/−2.0 | 31.81 +/−1.2 | <0.0001h | 2 | 253 |
| N2 (control RNAi) | 30.0 +/−0.5 | 21.0 +/− 0.1 | 2 | 386 | |
| N2 (sod-3 RNAi) | 30.0 +/−1.0 | 20,6 +/− 0.2 | n.s.a | 2 | 412 |
| N2 (daf-2 RNAi) | 72.0 +/−0.5 | 42.1 +/− 1.2 | <0.0001a | 2 | 352 |
| N2 (sod-3 RNAi/daf-2 RNAi) | 55.0 +/−1.9 | 37.1 +/− 0.7 | <0.0001a,c | 2 | 394 |
| N2 (ctl-2 RNAi) | 30.0 +/−1.0 | 20.8 +/− 0.2 | n.s.a | 2 | 452 |
| N2 (ctl-2 RNAi/daf-2 RNAi) | 61.0 +/−2.4 | 40.1 +/− 0.8 | <0.05a,c | 2 | 427 |
| age-1(hx546) H2O | 50.5 +/−0.5 | 28.90 +/−0.2 | 2 | 247 | |
| age-1(hx546) NAC/H2O | 41.0 +/−1.0 | 23.46 +/−0.2 | <0.0001i | 2 | 256 |
| age-1(hx546) DMSO | 50.5 +/−1.0 | 27,87 +/−0.2 | 2 | 259 | |
| age-1(hx546) BHA/DMSO | 44.5 +/−0.5 | 22,63 +/−1.6 | <0.0001k | 2 | 242 |
| aak-2(ok524) (control RNAi) H2O | 24.5 +/−0.5 | 16.78 +/−0.4 | 2 | 213 | |
| aak-2(ok524) (control RNAi) NAC/H2O | 24.0 +/−1.0 | 16.65 +/−0.3 | n.s.a | 2 | 234 |
| aak-2(ok524) (daf-2 RNAi) H2O | 36.0 +/−1.0 | 21.02 +/−0.1 | <0.0001a,b | 2 | 229 |
| aak-2(ok524) (daf-2 RNAi) NAC/H2O | 38.0 +/−1.0 | 21.13 +/−0.3 | <0.0001a,b,, n.s.c | 2 | 231 |
| aak-2(ok524) (control RNAi) DMSO | 24.0 +/−1.0 | 16.64 +/−0.4 | 2 | 217 | |
| aak-2(ok524) (control RNAi) BHA/DMSO | 24.0 +/−0.0 | 16.89 +/−0.7 | n.s.d | 2 | 220 |
| aak-2(ok524) (daf-2 RNAi) DMSO | 37.5 +/−1.5 | 22.53 +/−0.2 | <0.0001d,e | 2 | 202 |
| aak-2(ok524) (daf-2 RNAi) BHA/DMSO | 37.5 +/−0.5 | 23.16 +/−0.3 | <0.0001d,e,, n.s.f | 2 | 205 |
| pmk-1(km25) (control RNAi) H2O | 28.0 +/−1.0 | 19.36 +/−0.2 | 2 | 178 | |
| pmk-1(km25) (control RNAi) NAC/H2O | 29.5 +/−0.5 | 19.42 +/−0.2 | n.s.a | 2 | 160 |
| pmk-1(km25) (daf-2 RNAi) H2O | 38.0 +/−1.0 | 27.02 +/−1.1 | <0.0001a,b | 2 | 166 |
| pmk-1(km25) (daf-2 RNAi) NAC/H2O | 55.0 +/−2.0 | 34.13 +/−1.0 | <0.0001a,b,c | 2 | 170 |
| pmk-1(km25) (control RNAi) DMSO | 26.5 +/−1.5 | 17.49 +/−0.7 | 2 | 157 | |
| pmk-1(km25) (control RNAi) BHA/DMSO | 28.5 +/−1.5 | 17.40 +/−0.6 | n.s.d | 2 | 161 |
| pmk-1(km25) (daf-2 RNAi) DMSO | 41.5 +/−1.5 | 28.16 +/−1.2 | <0.0001d,e | 2 | 148 |
| pmk-1(km25) (daf-2 RNAi) BHA/DMSO | 54.0 +/−2.0 | 32.97 +/−1.2 | <0.0001d,e, =0.00010f | 2 | 151 |
| skn-1(zu67) (control RNAi) H2O | 21.5 +/−0.5 | 16.77 +/−0.1 | 2 | 140 | |
| skn-1(zu67) (control RNAi) NAC/H2O | 22.5 +/−0.5 | 16.57 +/−0.1 | n.s.a | 2 | 160 |
| skn-1(zu67) (daf-2 RNAi) H2O | 34.0 +/−1.0 | 21.89 +/−0.1 | <0.0001a,b | 2 | 144 |
| skn-1(zu67) (daf-2 RNAi) NAC/H2O | 45.5 +/−0.5 | 26.71 +/−1.1 | <0.0001a,b,c, | 2 | 150 |
| skn-1(zu67) (control RNAi) DMSO | 23.0 +/−0.0 | 16.78 +/−0.2 | 2 | 148 | |
| skn-1(zu67) (control RNAi) BHA/DMSO | 23.5 +/−0.5 | 16.66 +/−0.3 | n.s.d | 2 | 147 |
| skn-1(zu67) (daf-2 RNAi) DMSO | 33.5 +/−1.5 | 21.05 +/−0.3 | <0.0001d,e | 2 | 158 |
| skn-1(zu67) (daf-2 RNAi) BHA/DMSO | 45.5 +/−0.5 | 26.00 +/−1.1 | <0.0001d,e,f | 2 | 150 |
| daf-16(mu86) (control RNAi) H2O | 24.0 +/−1.0 | 16.71 +/−0.1 | 2 | 184 | |
| daf-16(mu86) (control RNAi) NAC/H2O | 22.5 +/−0.5 | 16.35 +/−0.1 | n.s.a | 2 | 195 |
| daf-16(mu86) (daf-2 RNAi) H2O | 24.5 +/−0.5 | 16.55 +/−0.1 | n.s.a.b | 2 | 166 |
| daf-16(mu86) (daf-2 RNAi) NAC/H2O | 24.0 +/−0.0 | 16.20 +/−0.1 | n.s.a.b.c | 2 | 186 |
| daf-16(mu86) (control RNAi) DMSO | 24.5 +/−0.5 | 14.68 +/−0.1 | n.s. | 2 | 162 |
| daf-16(mu86) (control RNAi) BHA/DMSO | 23.5 +/−0.5 | 14.62 +/−0.1 | n.s.d | 2 | 176 |
| daf-16(mu86) (daf-2 RNAi) DMSO | 24.5 +/−0.5 | 14.87 +/−0.1 | n.s.d.e | 2 | 180 |
| daf-16(mu86) (daf-2 RNAi) BHA/DMSO | 24.0 +/−0.0 | 14.64 +/−0.1 | n.s.d.e.f | 2 | 171 |
| N2 (control RNAi) | 33.0 +/−0.5 | 22.6 +/− 0.3 | 3 | 350 | |
| N2 (B0513.5 RNAi) | 32.0 +/−1.0 | 22.6 +/− 0.1 | n.s.a | 2 | 240 |
| N2 (daf-2 RNAi) | 69.0 +/−1.0 | 36.4 +/− 1.9 | <0.0001a | 3 | 423 |
| N2 (B0513.5 RNAi/daf-2 RNAi) | 55.0 +/−1.0 | 31.1 +/− 1.1 | <0.0001a,c | 3 | 345 |
| N2 (H2O) | 29.5 +/−0.5 | 20.46 +/−0.3 | 2 | 312 | |
| L-Proline (5 μM) | 33.5 +/−0.5 | 21.65 +/−0.1 | <0.0001i | 2 | 256 |
Controls:
(control RNAi) H2O,
(control RNAi) NAC/H2O,
(daf-2 RNAi) H2O,
(control RNAi) DMSO,
(control RNAi) BHA/DMSO,
(daf-2 RNAi) DMSO,
daf-2 (e1370) H2O,
daf-2 (e1370) DMSO,
(H2O)
These findings indicate that antioxidants reduce the life span-extending capacity produced by impaired insulin/IGF signaling in the daf-2 and age-1 mutants, and that a transient induction of ROS levels following impairment of daf-2 and age-1 signaling is required to induce an adaptive response cumulating in increased stress resistance and maximum longevity. It should be noted, however, that both RNAi(daf-2) exposure and the daf-2(e1370) as well as age-1(hx546) mutations still cause a significant extension of life span in the presence of antioxidants in comparison to antioxidant treated wild-type N2 nematodes. Altogether this shows that exogenous antioxidants significantly lower the life span-extending capabilities of impaired IIS by up to 59.9%, whereas some other daf-2 and age-1 effects are independent of increased ROS levels.
The AMP-activated protein kinase AAK-2 is essential for induction of the transient ROS signal
AAK-2, the AMPK homologue, is the energy sensor in C. elegans (Apfeld et al., 2004; Greer et al., 2007; Schulz et al., 2007). As shown in Figure 2B, there is an initial reduction of nematodal ATP levels after addition of RNAi(daf-2) suggesting a reciprocal increase in nematodal AMP content. Direct assessment of AMP in C. elegans lysates by HPLC demonstrated an increased AMP to ATP ratio in wild-type N2 nematodes following RNAi(daf-2) exposure for 12 hours (AMP/ATP = 14.1% +6,5% SD, p=0.038). This strongly suggests that AMP-activated AAK-2 is induced by RNAi(daf-2), culminating in increased mitochondrial respiration, as previously shown for states of dietary restriction (Schulz et al., 2007), and possibly here for increased ROS production.
Accordingly, we tested whether RNAi(daf-2) could still induce respiration and/or ROS levels in aak-2(ok524) mutants. However, this was not the case (Figures 4A and 4B), indicating that aak-2 is required to generate a ROS signal in states of impaired IIS in nematodes. Nevertheless, RNAi(daf-2) was capable of extending life span to some extent in aak-2(ok524) nematodes (Figures 4C and 4D). However, it should be noted that the relative capacity of RNAi(daf-2) to extend life span in aak-2(ok524) is severely reduced compared to relative effects of RNAi(daf-2) on wild-type N2 (see Table 1 for quantification), indicating that a significant proportion of the effects of RNAi(daf-2) on life span is mediated by AAK-2 and the generation of the ROS signal as shown above (Figures 4B vs. 2D).
Figure 4. Molecular regulation of ROS-dependent extension of life span following daf-2 impairment.

(A) Oxygen consumption, (B) mitochondrial ROS levels in aak2(ok524) nematodes following exposure to RNAi against daf-2 (black bars) relative to effects on control RNAi-treated aak2(ok524) nematodes (white bars) at different time points. In both panels relative values are depicted; for absolute values, see Figure S3. (C) Lifespan analyses of aak-2(ok524) nematodes exposed to control-RNAi (open circles) in the presence (red) or absence (black) of the antioxidant NAC; life span analyses of aak2(ok524) nematodes exposed to RNAi against daf-2 (closed triangles) in the presence (red) or absence (black) of NAC. (D) Lifespan analyses in the presence of mutation and RNAi as in C, replacing NAC with the antioxidant BHA (blue). (E) Lifespan analyses of pmk-1(km25) nematodes exposed to control-RNAi (open circles) in the presence (red) or absence (black) of NAC; life span analyses of pmk-1(km25) nematodes exposed to RNAi against daf-2 (closed triangles) in the presence (red) or absence (black) of NAC; (F) Lifespan analyses in the presence of mutation and RNAi as in E, replacing NAC with BHA (blue). (G) Lifespan analyses of skn-1(zu67) nematodes exposed to control-RNAi (open circles) in the presence (red) or absence (black) of NAC; life span analyses of skn-1(zu67) nematodes exposed to RNAi against daf-2 (closed triangles) in the presence (red) or absence (black) of NAC. (H) Lifespan analyses in the presence of mutation and RNAi as in G, replacing NAC with BHA (blue). Activities of superoxide dismutase (I) and catalase (J) in wild-type nematodes and mutants for pmk-1 and skn-1 without (white bars) and with (black bars) daf-2 RNAi treatment for 5 days. In panels I and J relative values are depicted; for absolute values, see Figure S3. (K) Lifespan analyses of N2 nematodes exposed to control RNAi (empty circles) and daf-2 RNAi (black triangles) in comparison to exposure against sod-3 RNAi (purple circles) alone and co-incubation with daf-2 RNAi (purple triangles). (L) Lifespan analyses of N2 nematodes exposed to control RNAi (empty circles) and daf-2 RNAi (black triangles) in comparison to exposure against ctl-2 RNAi (purple circles) alone and co-incubation with daf-2 RNAi (purple triangles). (M) Lifespan analyses of daf-16(mu86) nematodes exposed to control-RNAi (open circles) in the presence (red) or absence (black) of NAC; lifespan analyses of daf-16(mu86) nematodes exposed to RNAi against daf-2 (closed triangles) in the presence (red) or absence (black) of NAC. (N) Lifespan analyses in the presence of mutation and RNAi as in M, replacing NAC with BHA (blue). In panels A, B, I and J values are given as mean ± SD. *p<0.05, **p<0.01, ***p<0.001 versus respective controls.
AAK-2 has been previously shown to be required for increased respiration in states of impaired glycolysis (Schulz et al., 2007). We here show that AAK-2 is also required for generating the transient ROS signal (Figure 4B). Therefore, we hypothesized that antioxidants would exert no effects on RNAi(daf-2)-mediated life span extension in aak-2(ok524) mutants, if AAK-2 is initiating the only ROS signal in the RNAi(daf-2) treated worm. Indeed, both NAC (Figure 4C) and BHA (Figure 4D) had no effect on the RNAi(daf-2)-mediated limited extension of life span, again indicating that AAK-2 is required to generate a transient increase in ROS following RNAi(daf-2) treatment (see Table 1 for quantification). Taken together, these findings indicate that AAK-2 (AMPK) is an indispensable mechanistic link between impaired IIS and transiently increased ROS levels in nematodes.
The p38 MAP kinase PMK-1 and the transcription factor SKN-1 (NRF-2) are required for sensing of the transient ROS signal
Next, we questioned which pathways may be involved in ROS-sensing, i.e., are required to fully exert the life span extending effects of RNAi(daf-2) due to increased ROS levels. Consistent with previously published findings (Inoue et al., 2005; Schmeisser et al., 2011) we found that pmk-1, an orthologue of the stress-inducible mammalian p38 MAP kinase gene, was involved in sensing of the ROS signal generated by RNAi(daf-2). Thus, while RNAi(daf-2) caused a limited extension of life span in pmk-1(km25) nematodes in the absence of antioxidants (Table 1), both NAC (Figure 4E) and BHA (Figure 4F) were capable of further promoting the life span-extending capacity of RNAi(daf-2) (Table 1). Similar results were obtained for skn-1(zu67) mutants, lacking a functional orthologue of the mammalian transcription factor NRF-2, which was previously shown to act downstream of PMK-1 in response to oxidative stress (Inoue et al., 2005; Tullet et al., 2008): While RNAi(daf-2) caused an equally limited extension of life span in skn-1-mutated C. elegans in the absence of antioxidants (Table 1), both NAC (Figure 4G) and BHA (Figure 4H) were able to further induce the life span-extending capacity of RNAi(daf-2) (Table 1). Consistently, the daf-2-mediated induction of both SOD- and catalase activities (Figure 2I and 2J) was found to be reduced in nematodes constitutively deficient for PMK-1 or SKN-1 (Figure 4I and J), respectively. Moreover, we have quantified differentially expressed genes in N2 wild-type nematodes in comparison to daf-2(e1370) mutants by applying RNA sequencing technology.
Out of the genes involved into antioxidant defense, we found a number of mRNAs upregulated in daf-2 mutants, including sod-3 (22.6 fold, p=5.9*10−88), sod-5 (62.2 fold, p=5.7*10−75), ctl-2 (2.12 fold, p=1.5*10−13), and ctl-3 (1.74 fold, p=0.00012). To test whether induction of antioxidant enzymes, and in particular sod-3 or ctl-2 is necessary for the lifespan extension, we treated N2 nematodes with RNAi against the superoxide dismutase isoform sod-3 (Figure 4K) and the catalase isoform ctl-2 (Figure 4L), and co-treated them with daf-2 RNAi. We observed a reduction of lifespan-extending capabilities of daf-2 RNAi in both sod-3 as well as ctl-2 RNAi-treated worms (Figures 4K and 4L) indicating that induction of antioxidant defense enzymes are, at least in part, required for the lifespan extension due to reduced daf-2 signaling.
Lastly, we questioned whether daf-16, an orthologue of a mammalian forkhead transcription factor (FoxO), may be involved into the lifespan-promoting role of transiently increased ROS formation. Consistent with previously published findings, RNAi(daf-2) had no lifespan-extending effect in daf-16(mu86) nematodes (Figures 4M and N). Accordingly, neither NAC (Figure 4M) nor BHA (Figure 4N) had any detectable influence on this phenotype.
Taken together, these findings indicate that PMK-1 and SKN-1 act as transducers of the initial ROS signal following RNAi(daf-2) treatment to extend life span, and that this effect is mediated by transiently increased ROS levels due to the daf-2 inactivation.
Trans-species transcriptome analysis of models of impaired IIS
The findings depicted on Figure 1 suggest that impaired IIS causes a reduction of intracellular glucose availability which then induces mitochondrial metabolism of alternate energy substrates, i.e. fatty acids and/or amino acids, culminating in transiently increased ROS levels, as analyzed in the findings depicted in Figs. 2 to 4. To identify a potential common metabolic denominator for the ROS-dependent fraction of IIS-related life span extension, we subjected RNA samples from daf-2 nematodes and MEFs (IRS1−/− and IR+/−) to transcriptome profiling using deep sequencing (Figures 5A–5C). When analyzing those genes that were consistently either up- or down-regulated in all three models, we identified three functional groups of genes (Figure 5D and Table S1). Notably, one of the up-regulated groups was the MAP kinase signalling pathway (Figure 5D), strikingly consistent with the fact that disruption of the MAP kinase pmk-1 severely impaired the effects of impaired IIS, as well as transiently increased ROS levels, as shown in Figures 4E and 4F.
Figure 5. Trans-species transcriptome analysis identifies increased L-proline catabolism as a life span extending response to reduced glucose metabolism in states of impaired insulin-/IGF1-signaling.

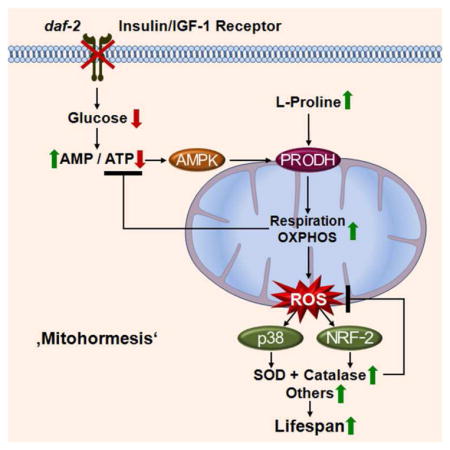
Differentially expressed genes as quantified by deep sequencing analysis for (A) daf-2 nematodes, (B) IRS1−/− MEFs and (C) IR+/− MEFs, all in comparison to respective controls; black dots indicate no differential regulation, red and green dots indicate regulation according to one statistical method only. Blue dots indicate regulation according to both statistical methods (used for further analysis). (D) functional groups of down-regulated and up-regulated genes (cut-off for at least two out of three sets: p=0.05). For details, please see also Table S1. (E) Venn analyses of differentially expressed genes derived from daf-2 nematodes, IRS1−/− MEFs and IR+/− MEFs (cut-off for all three sets: p=0.05). For details, please see also Table 2. (F) Lifespan analyses of N2 nematodes exposed to control RNAi (empty circles) and daf-2 RNAi (filled triangles) in comparison to exposure against B0513.5 RNAi (closed circles) alone and co-incubation with daf-2 RNAi (empty triangles, respectively). (G) Lifespan analysis of N2 nematodes exposed to L-proline (filled triangles) or solvent (empty circles). (H) Expression of B0513.5/prodh mRNA in the absence (white bars) and presence (black bars bars) of daf-2 RNAi for 48 h in wild-type (N2) and aak-2 mutant nematodes. (I) Oxygen consumption in N2 nematodes treated with control RNAi (white bars) or daf-2 RNAi (black bars), and the additional presence of RNAi against B0513.5/prodh (grey/striped bars), all after 48 h of treatment. (J) ROS levels in nematodes treated with RNAis as in Panel (I) for 48 h; mev-1(kn1) mutants and paraquat treatment for 1 h serve as positive controls. In panels I and J relative values are depicted; for absolute values, see Figure S4. In panels H J values are given as mean ± SD. *p<0.05, **p<0.01, ***p<0.001 versus respective controls. (K) Impaired insulin-/IGF1-signaling promotes L-proline catabolism to employ ROS as a mitochondrial second messenger culminating in extended life span: Impaired IIS causes reduction of glucose uptake in C. elegans, which leads to an intermittent energy deficit that activates mitochondrial respiration by increasing L-proline catabolism in an aak-2 dependent manner. This induction of mitochondrial respiration generates a transient ROS signal, which is sensed by PMK-1 and SKN-1 to secondarily cause an adaptive response to increase the respective activities of superoxide dismutase and catalase which ultimately terminate the initial ROS signal, in parallel leading to increased stress resistance and extended C. elegans life span.
Mitochondrial L-proline catabolism extends life span in states of IIS and decreased glucose availability
By employing RNAseq, we also found that metabolism of short-chain organic acids is upregulated in states of impaired IIS. Notably, ech-6 (encoding enoyl Coenzyme A hydratase 6)/echs1 (encoding enoyl Coenzyme A hydratase, short chain, 1, mitochondrial) reflecting a checkpoint for catabolism of both fatty acids and amino acids was found to be up-regulated in all three models (Figure 5D and Table 2). To even better define the genes responsible for the effects of impaired IIS in a trans-species fashion, we performed a Venn-analysis for all three models (Figure 5E and Table 2). Among the genes involved in the effects of impaired IIS in both mouse and worm (Tables 2 and S1) revealed the orthologous pair of genes coding for a L-proline dehydrogenase, (M. musculus: prodh, C. elegans: B0513.5). This enzyme is essential for catabolism of one single amino acid, L-proline, to make it available for mitochondrial oxidation and oxidative phosphorylation-based, non-glucose-based energy generation.
Table 2.
Differentially expressed RNAs derived from three models of impaired insulin/IGF1-signaling (cutoff for all three sets p=0.05)
| Differentially expressed genes, upregulated | ||||||||
|---|---|---|---|---|---|---|---|---|
| C. elegans Gene | daf-2 log2Fold | daf-2 p-value | M. Musculus gene | gene name | IRS1−/− log2Fold | IRS1 −/− p-value | IR +/− log2Fold | IR +/− p-value |
| F25B4.8 | 1.629 | 3.012E-06 | Cenpv | centromere protein V | 2.277 | 8.758E-16 | 3.681 | 6.070E-15 |
| Y71G12B.11 | 1.396 | 2.906E-11 | Tln2 | talin 2 | 0.438 | 6.274E-06 | 1.738 | 1.656E-18 |
| bicd-1 | 0.710 | 0.0007230 | Bicd1 | bicaudal D homolog 1 (Drosophila) | 1.883 | 3.091E-23 | 0.972 | 3.731E-05 |
| ser-1 | 1.216 | 0.0001637 | Htr2c | 5-hydroxytryptamine (serotonin) receptor 2C | 1.326 | 0.0008178 | 3.517 | 3.375E-19 |
| lec-3 | 2.469 | 3.536E-26 | Lgals9 | lectin, galactose binding, soluble 9 | 1.593 | 2.012E-43 | 0.698 | 0.0009901 |
| lec-5 | 2.622 | 1.051E-28 | ||||||
| lec-4 | 1.466 | 1.166E-11 | ||||||
| lec-1 | 0.776 | 5.248E-05 | ||||||
| B0513.5 | 0.731 | 0.0020163 | Prodh | proline dehydrogenase | 3.228 | 5.120E-32 | 1.758 | 1.245E-06 |
| ech-6 | 0.650 | 0.0006435 | Echs1 | enoyl Coenzyme A hydratase, short chain, 1, mitochondrial | 0.563 | 1.288E-07 | 0.606 | 0.0044002 |
| exp-2 | 2.264 | 2.368E-12 | Kcnf1 | potassium voltage-gated channel, subfamily F, member 1 | 1.280 | 6.495E-13 | 0.966 | 0.0050530 |
| Y55F3C.3 | 0.995 | 0.0006997 | ||||||
| C32C4.1 | 1.121 | 0.0016399 | ||||||
| F44A2.2 | 1.492 | 0.0002509 | ||||||
| Y48A6B.6 | 1.078 | 0.0052666 | ||||||
| kvs-1 | 0.931 | 0.0065899 | ||||||
| prk-2 | 1.956 | 1.115E-10 | Pim1 | proviral integration site 1 | 0.535 | 0.0035235 | 0.818 | 0.0015448 |
| ras-1 | 1.498 | 2.272E-06 | Rras2 | related RAS viral (r-ras) oncogene homolog 2 | 0.398 | 4.561E-05 | 0.575 | 0.0052454 |
| cah-2 | 1.365 | 6.083E-05 | Car10 | carbonic anhydrase 10 | 1.886 | 6.360E-17 | 1.159 | 0.0058154 |
| cah-1 | 0.947 | 0.0041779 | ||||||
| Y73B6BL.19 | 2.105 | 8.671E-10 | Kcnd2 | potassium voltage-gated channel, Shal-related family, member 2 | 1.659 | 5.739E-08 | 1.186 | 0.0218384 |
| C27F2.1 | 1.572 | 1.122E-05 | Wdr60 | WD repeat domain 60 | 1.057 | 2.887E-22 | 0.527 | 0.0249645 |
| egl-3 | 1.162 | 1.113E-08 | Pcsk2 | proprotein convertase subtilisin/kexin type 2 | 1.478 | 3.103E-05 | 1.194 | 0.0296169 |
| epac-1 | 2.081 | 6.325E-08 | Rapgef4 | Rap guanine nucleotide exchange factor (GEF) 4 | 0.648 | 0.0040905 | 0.877 | 0.0298008 |
| exc-5 | 1.032 | 9.899E-06 | Fgd3 | FYVE, RhoGEF and PH domain containing 3 | 1.764 | 1.718E-56 | 0.443 | 0.0400597 |
| T03G6.3 | 2.356 | 2.263E-21 | Enpp6 | ectonucleotide pyrophosphatase/phosphodiesterase 6 | 1.473 | 0.0001826 | 1.316 | 0.0482077 |
| F47F2.1 | 0.727 | 0.0091527 | Prkx | protein kinase, X-linked | 0.372 | 0.0038382 | 0.454 | 0.0373704 |
| ceh-31 | 2.777 | 1.650E-09 | Barhl2 | BarH-like 2 (Drosophila) | Inf | 0.0201619 | Inf | 0.0469553 |
| ceh-30 | 1.530 | 8.221E-05 | ||||||
| tub-2 | 0.428 | 0.0269801 | Tulp4 | similar to mKIAA1397 protein; tubby like protein 4 | 0.2171 | 0.0137626 | 0.410 | 0.0445156 |
| Differentially expressed genes, downregulated | ||||||||
| C. elegans gene | daf-2 log2Fold | daf-2 p-value | M. Musculus gene | gene name | IRS1−/− log2Fold | IRS1 −/− p-value | IR +/− log2Fold | IR +/− p-value |
| nuc-1 | −1.880 | 2.085E-11 | Dnase2a | Dnase2a deoxyribonuclease II alpha | −0.581 | 0.0006621 | −0.643 | 0.0181585 |
| F35G2.1 | −0.758 | 0.0243548 | Qsox1 | quiescin Q6 sulfhydryl oxidase 1 | −0.323 | 0.0091651 | −0.851 | 0.0002569 |
| M01F1.9 | −0.911 | 0.0050494 | Rgp1 | RGP1 retrograde golgi transport homolog (S. cerevisiae) | −0.366 | 0.0035610 | −0.567 | 0.0262451 |
| C01B10.9 | −0.696 | 0.0435967 | Enpp4 | ectonucleotide pyrophosphatase/phosphodiesterase 4 | −1.539 | 8.893E-17 | −2.300 | 8.870E-20 |
| T13C2.4 | −1.075 | 0.0006532 | Ssu72 | Ssu72 RNA polymerase II CTD phosphatase homolog (yeast) | −0.515 | 0.0000335 | −0.521 | 0.0430715 |
| F53F10.3 | −0.716 | 0.0367228 | Brp44 | brain protein 44 | −0.633 | 0.0000076 | −0.604 | 0.0313828 |
To test the possibility that breakdown of L-proline may contribute to the effects of impaired IIS in a systemic manner, we treated both wild-type and daf-2 nematodes with RNAi against B0513.5, the C. elegans orthologue of prodh. While knock-down of this gene has no effect on life span of N2 wild-type worms (Figure 5F), the life span-extending capability of RNAi(daf-2) was reduced by 14.6 and 20.3% (mean and maximum life span) following addition of RNAi(B0513.5) (Figure 5F).
Additionally, we have treated wild-type N2 nematodes with the amino acid L-proline in the growth media to test whether increased L-proline availability was capable of extending life span. As shown in Figure 5G, this was indeed the case with an increase in life span by 5.8 and 13.6% (mean and maximum life span). Thus while B0513.5 expression is dispensable for life span in wild type worms (Figure 5F), supplementation with L-proline can extend life span to a limited but significant extent (Figure 5G).
To additionally test whether AAK-2 (Figures 4A 4D) is responsible for the induction of L-proline metabolism in states of IIS, we quantified expression of B0513.5/prodh mRNA expression in N2 as well as aak-2 mutants in the absence and presence of daf-2 RNAi. Induction of B0513.5 expression was abolished in aak-2 mutants (Figure 5H) indicating that AAK-2 activation is located upstream of PRODH. Moreover, induction of respiration by daf-2 RNAi (Figure 2C) was abolished by co-treatment with RNAi against B0513.5 (Figure 5I). Likewise, RNAi against B0513.5 abolished the ROS signal (Figure 5J) that was initially shown to be induced by daf-2 RNAi (Figure 2D).
Together, these findings indicate that impaired IIS reduces glucose metabolism and induces L-proline catabolism in a compensatory manner to culminate in a transient ROS signal and extended life span, as summarized in Figure 5K.
Discussion
We here hypothesized that reduced glucose metabolism due to impaired IIS causes a transient energy deficit and a compensatory induction of mitochondrial non-glucose metabolism to promote stress resistance and to increase longevity, both following the generation of one or several mitochondrial signaling molecules.
The findings in this study indicate that a major component, i.e. up to 59.9% of the life span extension created by reduced IIS requires a transient increase in ROS levels. ROS then act as signalling molecules to promote life span extension. We furthermore show that this ROS signal is generated by AAK-2, an orthologue of mammalian AMPK, and is sensed by the C. elegans orthologues of p38 MAP kinase and the transcription factor NRF-2, PMK-1 and SKN-1. These sensors ultimately cause an increase in endogenous ROS defense, indicating that the ROS signal is capable of self terminating by inducing an adaptive response with increased activities of SOD and CAT, culminating in life span-extending stress resistance (Figure 5K). It is, however, likely that additional genes are activated following translocation of SKN-1, which have not been studied in the present study.
It has been proposed that ROS may act as messenger molecules in a variety of biological systems (Finkel, 2011; Rhee et al., 2003; Veal et al., 2007; Woo and Shadel, 2011). Consistently, evidence has recently emerged that ROS may also act as a life span-extending (Brys et al., 2010; Pan et al., 2011; Schulz et al., 2007) and health-promoting (Loh et al., 2009; Owusu-Ansah et al., 2008; Ristow et al., 2009) signalling molecule. This is in agreement with previous observations that calorie restriction acts by inducing low-level stress which culminates in increased stress resistance and ultimately longevity (Barros et al., 2004; Masoro, 1998; Xia et al., 1995). In analogy to the findings presented here, this reflects a dose-dependent adaptive response commonly defined as hormesis (Calabrese et al., 2007; Southam and Ehrlich, 1943), which was later extended to the concept of mitohormesis (Ristow and Zarse, 2010; Tapia, 2006) when describing low-dose stressors emanating from the mitochondrial compartment.
The current study additionally shows for the first time that acute daf-2 impairment reduces glucose availability in C. elegans, implying the fact that both glucose restriction (Schulz et al., 2007) and impaired IIS (current study) similarly cause an initial energy deficit. While some authors propose that impaired insulin/IGF-1 signaling extend life span independently of pathways activated by calorie restriction (Bartke et al., 2001; Houthoofd et al., 2003; Lakowski and Hekimi, 1998; Min et al., 2008), others have suggested that impaired IIS may share mechanistic features of caloric restriction and hence decreased energy availability, at least to some extent (Al-Regaiey et al., 2005; Bonawitz et al., 2007; Clancy et al., 2002; Greer et al., 2007; Narasimhan et al., 2009; Yen et al., 2009). Our current findings strongly support and extend this latter notion, and possibly provide a common metabolic denominator for impaired IIS, calorie restriction and physical exercise.
In this regard it is interesting to note that nematodes switch to mitochondrial non-glucose metabolism, as predicted in states of impaired glycolysis (Schulz et al., 2007) as well as impaired IIS (this study), by activation of AAK-2 (AMPK). Unlike for glucose, ATP generation from fatty acids and/or amino acids can only take place in the mitochondrial compartment, and requires oxidative phosphorylation. Hence and unlike glycolysis, mitochondrial catabolism of organic acids and specifically amino acids will inevitably generate ROS which may act as signalling molecules.
One of the interesting aspects of our findings came from the combined gene expression analysis using data from three different models systems with impaired IIS, including one model in C. elegans and two in different murine lines. This revealed that metabolism of short-chain organic acids is upregulated in states of impaired IIS. Notably, ech-6 (enoyl Coenzyme A hydratase 6)/echs1 (enoyl Coenzyme A hydratase, short chain, 1, mitochondrial) reflecting a metabolic checkpoint for catabolism of both fatty acids and amino acids was found to be upregulated in all three models (Tables 2 and S1). Moreover, and unexpectedly, a major portion of the IIS-mediated effects on life span appears to depend on a dehydrogenase, B0513.5, which is specifically responsible for catabolism of a single amino acid, L-proline. This is supported by the RNAi-mediated knockdown of this protein in C. elegans which impairs the life span-extending capabilities of impaired IIS, but does not affect life span in wild-type worms, indicating that L-proline catabolism specifically contributes to induction of mitochondrial metabolism following impairment of IIS, as shown in the current study (Figure 5K). Moreover and - in a very general sense - somewhat consistent with our findings, increased L-proline oxidation has been linked to stress resistance in tomatoes (Chen et al., 2006), as well as to increased ROS formation (Donald et al., 2001) and nutrient deprivation (Pandhare et al., 2009), both in colon cancer cell lines.
Our findings on increased mitochondrial L-proline metabolism are consistent with the fact that constitutive inactivation of IIS causes increased stress resistance in C. elegans (Brys et al., 2010; Honda and Honda, 1999; Lithgow et al., 1995; Murphy et al., 2003; Vanfleteren, 1993; Vanfleteren and De Vreese, 1995), possibly by increasing metabolic activity (Houthoofd et al., 2005; Vanfleteren and De Vreese, 1995). This is also consistent with the finding that exogenous antioxidants, such as NAC and BHA, significantly impair the life span-extending capabilities of the constitutive daf-2 or age-1 mutants. This indicates that the transient increase in mitochondrial ROS observed in the current study is essential for inducing endogenous ROS defense in long-lived mutants. Similarly, in D. melanogaster, calorie restriction did not affect ROS production, and genetically decreased ROS production did not extend life span in flies (Miwa et al., 2004). Consistently, altering ROS production in various model organisms has widely failed to modulate life span (Doonan et al., 2008; Jang and van Remmen, 2009; Lapointe et al., 2009; Van Raamsdonk and Hekimi, 2009). Moreover, long-lived mutants of C. elegans show increased stress resistance which is paralleled by increased metabolic activity (Houthoofd et al., 2005; Vanfleteren and De Vreese, 1995).
Take together with our previous data, impaired IIS, calorie restriction and physical exercise share, at least in part, a common metabolic denominator, i.e. activation of mitochondrial L-proline metabolism and transiently increased ROS levels inducing an adaptive response that culminates in increased stress resistance, antioxidant defense and extends life span.
Experimental Procedures
C. elegans strains, maintenance and life span assays
C. elegans strains used in this work were provided by the Caenorhabditis Genetics Center (Univ. of Minnesota). Maintenance and synchronization (Lithgow et al., 1995) as well as RNAi treatments and life span assays (Dillin et al., 2002; Schulz et al., 2007) have been previously described, and were performed in the absence of 5′-fluorouridine. The B0513.5 RNAi clone was from the Ahringer library (Source BioScience, Nottingham, UK), sod-3 and ctl-2 RNAis were from the ORF-RNAi library (OpenBiosystems Inc., Lafayette, CO, USA) and the daf-2 RNAi clone was a kind gift by Cynthia Kenyon (Dillin et al., 2002). NAC and BHA (both Sigma-Aldrich, St. Louis, MO, USA) were dissolved in water (NAC, 500-fold stock solution, 500 mM) and DMSO (BHA, 1000-fold stock solution, 25 mM), respectively. Nematodes (wild-type Bristol N2 and respective mutants) were propagated on agar plates containing antioxidants or respective solvent for four generations before initiation of experiments. For L-proline (Sigma-Aldrich, St. Louis, MO, USA) supplementation experiments, the amino acid was added to autoclaved agar at 50°C as an aqueous 5 mM stock solution to obtain a final concentration of 5 μM.
Culture conditions of IRS1−/− MEFs
IRS1−/− MEFs and control fibroblasts have been derived from two different, genetically distinct fetuses derived from the same mother as previously described (Brüning et al., 1997) and were maintained in Dulbeccos’s modified Eagle medium (DMEM) (Sigma-Aldrich, St. Louis, MO, USA) containing 10 mM D-glucose and 10% (v/v) fetal bovine serum (Biochrom AG, Berlin, Germany).
Generation and culture conditions of IR+/− MEFs
Mice homozygous for the IRloxP mutation (Brüning et al., 1998) were bred with mice heterozygous for the tamoxifen-inducible Cre recombinase (CreERT2, Taconic Farms Inc., Hudson, NY, USA) (Seibler et al., 2003)), and MEFs were obtained from a single fetus that was heterozygous for both IRloxP and CreERT2. Cells then were continuously passaged and underwent crisis, all following previously described protocols (Brüning et al., 1997). Cells were then aliquoted and frozen, further serving as control fibroblasts. One aliquot of these fibroblasts was exposed to tamoxifen (Sigma-Aldrich, St. Louis, MO, USA) at a concentration of 1 μM for 7 days to obtain a heterozygous disruption of the insulin receptor. All cells were maintained and analyzed in Dulbeccos’s modified Eagle medium (DMEM) (Sigma-Aldrich, St. Louis, MO, USA) containing 10 mM D-glucose and 10% (v/v) fetal bovine serum (Biochrom AG, Berlin, Germany). MEFs were cultured at 37 °C in a humidified atmosphere of 5% CO2.
Paraquat stress resistance assay (C. elegans)
Resistance to lethal oxidative stress derived from paraquat was determined with minor modifications as previously described (Schulz et al., 2007). Six days (after L4) old N2 and daf-2(e1370) nematodes were transferred manually to fresh NGM plates containing 10 mM paraquat (Acros Organics, Geel, Belgium) spotted with a lawn of heat-inactivated OP50 (30 min at 65°C in a water bath) following by daily determining the survival rate until all nematodes were death. As described for life span analysis, worms were count as censored in case of internal hatching, crawling off and bursting.
Paraquat stress resistance assay (fibroblasts)
100,000 cells were seeded into each well of 12-well plates (TPP AG, Trasadingen, Switzerland). After 24 h medium was changed to DMEM (Sigma-Aldrich, St. Louis, MO, USA) containing 10 mM D-glucose and 10% (v/v) fetal bovine serum (Biochrom AG, Berlin, Germany) supplemented with 1 mM Paraquat (Acros Organics, Geel, Belgium). After 48 h cell death was determined by propidium iodide (PI) (10 μg/ml) and Hoechst 33258 (10 μg/ml) (both Sigma-Aldrich, St. Louis, MO, USA) double fluorescent staining. Cells were examined under a fluorescence microscope (Axiovert 100, Zeiss, Oberkochen, Germany) and photographed with a digital camera (Fuji FinePix S602, Tokyo, Japan).
2-Deoxyglucose uptake assays
Nematodes were incubated in S-medium containing 500 μM unlabeled 2-deoxy-glucose (Sigma-Aldrich, St. Louis, MO, USA) and 2.5 μCi per ml uniformly 14C-labeled 2-deoxy-glucose (GE Healthcare Little Chalfont, UK), washed five times, then sonicated and centrifuged. Radioactivity in supernatant was quantified using a Beckman LS6000 liquid scintillation counter (Beckman Coulter, Brea, CA, USA). An aliquot was used for protein determination for normalization.
Determinations of ATP and AMP were for C. elegans-derived samples performed by HPLC as previously described (Schulz et al., 2007) with minor modifications. For quantification of ATP content in fibroblasts, the CellTiter Glo kit (Promega, Fitchburg, WI, USA) was used according to the manufacturer’s instructions, and ATP values were normalized to protein content.
Respiration assays were performed using a Clark-type electrode for C. elegans (Schulz et al., 2007) and mammalian cells (Ristow et al., 2000).
Mitochondrial ROS levels
Prior to ROS measurement MitoTracker Red CM-H2X ROS (Invitrogen, Carlsbad, CA, USA) incubation plates were prepared as followed. For each treatment 500 μl heat inactivated OP50 (65°C, 30 min) were mixed with 100 ul MitoTracker Red CM-H2X stock solution (100 μM) and spotted on a large NGM agar plate which was allowed to dry for ~1 h. Nematodes were incubated with appropriate RNAis for time periods as indicated, then washed off the plates with S-Basal and allowed to settle by gravitation to remove offspring. Worms were washed two additional times with S-Basal and centrifuged (300g, 30 sec). The worm pellet was transferred to freshly prepared MitoTracker Red CM-H2X and incubated for 2 h at 20°C. To remove excessive dye from the gut, worms were transferred to NGM agar plates with appropriate RNAis or as a positive control, to plates containing 10 mM paraquat for additional 1 h at 20°C. Aliquots of 100 μl worm suspension were distributed into 96-well FLUOTRACTM plate (Greiner Bio-One, Frickenhausen, Germany). Fluorescence intensity was measured in a microplate reader (FLUOstar Optima, BMG Labtech, Offenburg, Germany) using well-scanning mode (ex: 570 nm, em: 610 nm). To normalize fluorescence signal, remaining worm suspension was used for protein determination. For measuring mitochondrial ROS levels in fibroblasts, 10,000 cells were seeded into each well of 96-well plate. After 24 h cells were incubated with medium containing 1 μM MitoTracker Red CM-H2X ROS for 30 min. Cells were then washed two times with PBS and fluorescence intensity was measured at same conditions described above.
Amplex Red-based quantification of supernatant hydrogen peroxide (C. elegans)
Worms were removed from plates with 0.05M sodium-phosphate buffer pH 7.4, washed twice and transferred into an upright plexiglas cylinder (1.5 ml volume) with continuous stirring at low speed (100 rpm) at 20°C. Firstly determination of fluorescence was done without horse radish peroxidase (HRP) (Sigma-Aldrich, St. Louis, MO, USA) only in the presence of 1 μM Amplex Red (Invitrogen, Carlsbad, CA, USA) to detect possible unspecific increase in fluorescence (which was not observed). Next, 0.01 U/ml HRP was added and changes of fluorescence were recorded with a fluorescence detector (LF402 ProLine, IOM, Berlin, Germany) for at least 15 minutes at excitation and emission wavelengths of 571 nm and 585 nm, respectively. Immediately afterwards, worms were removed and collected for protein determination to normalize fluorescence values. For determination of hydrogen peroxide production in fibroblasts, 10,000 cells were seeded into each well of 96-well plate. After 24 h Amplex Red Assay was performed according to the manufacturer’s instructions. Fluorescence intensity was measured in a microplate reader (FLUOstar Optima, BMG Labtech, Offenburg, Germany) using well-scanning mode (ex: 570 nm, em: 590 nm).
Isolation of mitochondria
Isolation of mitochondria was performed as previously described (Kayser et al., 2001) except for the initial rupture of nematodes was done by a Potter/Elvehjem tissue grinder at 400 rpm with 3 slow up-and-down strokes. 50 μg of isolated mitochondria were transferred into an upright plexiglas cylinder (1.5 ml volume) with continuous stirring at low speed (100 rpm) at 30°C. Measurement of fluorescence increase due to Amplex Red oxidation was carried out exactly as described above (for whole worms), while only 0.1 U/ml horseradish peroxidase were used. 2.5 mM pyruvate and 1.25 mM malate (final concentrations) were added simultaneously as substrates for the respiratory chain.
Antioxidant enzyme activities (SOD, CAT) in both nematodes and MEFs were determined by standard photometric assays as previously described (Schulz et al., 2007) with minor modifications.
Fluorescent microscopy
Worms were treated with MitoTracker Red CM-H2X ROS exactly as described above. Individual worms were placed on agarose pads and paralyzed with 1 mg/ml tetramisole (Sigma-Aldrich, St. Louis, MO, USA). Worms were examined under a fluorescence microscope (Axiovert 100, Zeiss, Oberkochen, Germany) using the filter set (BP546/12, FT580, LP590) and pictures were taken with a digital camera (Moticam 2300, Motic, Xiamen, P.R. of China).
Protein content in both nematodes and MEFs was determined by standard methods as previously described (Schulz et al., 2007) with minor modifications.
Extraction of total RNA from C. elegans and fibroblasts
RNA isolation was performed using a commercially available kit (Qiagen, Hilden, Germany, Rneasy Mini Kit) based on the phenol-chloroform extraction method according to the manufacturer’s instructions.
Real time-PCR
Reverse transcription and quantitative real-time PCR was carried out using the GoTaq 2-Step RT-qPCR System (Promega, Madison, WI, USA) according to the manufacturer’s instructions on LightCycler 480 system (Roche, Mannheim, Germany). Data were normalized to cdc-42 (Hoogewijs et al., 2008) and analyzed using the ΔΔCT method. Primer sequences used for B0513.3 and cdc-42 are: fwd 5′AAGCCAGCGGCGATGACACC and rev 5′AACACCCTGCCGCCGATCTC as well as fwd 5′CTGCTGGACAGGAAGATTACG and rev 5′CTCGGACATTCTCGAATGAAG.
Transcriptome profiling using deep sequencing
For library preparation an amount of 5 μg of total RNA per sample was processed using Illumina’s mRNA-Seq sample prep kit (Illumina; San Diego; CA, USA) following the manufacturer’s instruction. The libraries were sequenced using an Illumina GAIIx, in a single 76 nt read approach. Each library was sequenced on a single lane ends up with around 30–40 mio reads per sample. Sequence data were extracted in FastQ format and used for mapping. Reads which passed the quality filtering were mapped against the C. elegans genome and an exon junction splice database using Bowtie (Langmead et al., 2009). Only uniquely mapped reads were used for counting. The RefSeq annotation was used to assign mapping positions to exons, transcripts and genes. The data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE36041 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE36041).
Bioinformatics of RNA expression data
Raw counts for the transcripts were analyzed using the R Statistical Computing Environment (R Development Core Team, 2008) and the Bioconductor packages DESeq (Anders and Huber, 2010) and edgeR (Robinson et al., 2010). Both packages provide statistical routines for determining differential expression in digital gene expression data using a model based on the negative binomial distribution. The resulting p-values were adjusted using the Benjamini and Hochberg’s approach for controlling the false discovery rate (FDR) (Benjamini and Hochberg, 1995). Transcripts with an adjusted p-value smaller than 0.01 found by both packages (intersection), were assigned as differentially expressed.
Statistical analyses
Data are expressed as means ± SD unless otherwise indicated. Statistical analyses for all data except life-span and stress resistance assays in C. elegans were performed by Student’s t-test (unpaired, two-tailed) after testing for equal distribution of the data and equal variances within the data set. For comparing significant distributions between different groups in the life-span assays and stress resistance assays, statistical calculations were carried out using the log-rank test. All calculations were performed using Excel 2007 (Microsoft, Albuquerque, NM, USA) and SPSS version 13.0 (IBM, Armonk, NY, USA). A P-value below 0.05 was considered as statistically significant.
Supplementary Material
Research highlights.
Acute impairment of daf-2 (insulin/IGF1) signaling causes a transient ROS signal.
Different antioxidants reduce daf-2-mediated lifespan extension by up to 60%.
Reduced daf-2 signaling impairs glucose uptake and promotes proline metabolism.
Nutritional supplementation with the amino acid L-proline extends lifespan.
Acknowledgments
C. elegans strains used in this work were provided by the Caenorhabditis Genetics Center (Univ. of Minnesota), which is funded by the NIH National Center for Research Resources (NCRR). The authors thank Cynthia Kenyon for the RNAi construct against daf-2 (Dillin et al., 2002). The excellent technical assistance of Ivonne Heinze, Beate Laube, Annett Müller and Waltraud Scheiding, as well as the excellent secretarial assistance of Mandy Schalowski are gratefully acknowledged. This work is part of the research program of the Jena Centre for Systems Biology of Ageing (JenAge) funded by the German Ministry for Education and Research (Bundesministerium für Bildung und Forschung; support code: BMBF 0315581) and at the Joslin Diabetes Center by NIH grants DK31036 and DK33201 to CRK. Funding for this project was denied by the German Research Association (Deutsche Forschungsgemeinschaft, DFG), grant application number RI 1976/3-1.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Al-Regaiey KA, Masternak MM, Bonkowski M, Sun L, Bartke A. Long-lived growth hormone receptor knockout mice: interaction of reduced insulin-like growth factor i/insulin signaling and caloric restriction. Endocrinology. 2005;146:851–860. doi: 10.1210/en.2004-1120. [DOI] [PubMed] [Google Scholar]
- Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apfeld J, O’Connor G, McDonagh T, DiStefano PS, Curtis R. The AMP-activated protein kinase aak-2 links energy levels and insulin-like signals to lifespan in C. elegans. Genes Dev. 2004;18:3004–3009. doi: 10.1101/gad.1255404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barros MH, Bandy B, Tahara EB, Kowaltowski AJ. Higher respiratory activity decreases mitochondrial reactive oxygen release and increases life span in Saccharomyces cerevisiae. J Biol Chem. 2004;279:49883–49888. doi: 10.1074/jbc.M408918200. [DOI] [PubMed] [Google Scholar]
- Bartke A, Wright JC, Mattison JA, Ingram DK, Miller RA, Roth GS. Extending the lifespan of long-lived mice. Nature. 2001;414:412. doi: 10.1038/35106646. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Statist Soc B. 1995;57:289–300. [Google Scholar]
- Blüher M, Kahn BB, Kahn CR. Extended longevity in mice lacking the insulin receptor in adipose tissue. Science. 2003;299:572–574. doi: 10.1126/science.1078223. [DOI] [PubMed] [Google Scholar]
- Bonawitz ND, Chatenay-Lapointe M, Pan Y, Shadel GS. Reduced TOR signaling extends chronological life span via increased respiration and upregulation of mitochondrial gene expression. Cell Metab. 2007;5:265–277. doi: 10.1016/j.cmet.2007.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown-Borg HM, Borg KE, Meliska CJ, Bartke A. Dwarf mice and the ageing process. Nature. 1996;384:33. doi: 10.1038/384033a0. [DOI] [PubMed] [Google Scholar]
- Brown-Borg HM, Johnson WT, Rakoczy SG. Expression of oxidative phosphorylation components in mitochondria of long-living Ames dwarf mice. Age (Dordr) 2012;34:43–57. doi: 10.1007/s11357-011-9212-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brüning JC, Michael MD, Winnay JN, Hayashi T, Hörsch D, Accili D, Goodyear LJ, Kahn CR. A muscle-specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol Cell. 1998;2:559–569. doi: 10.1016/s1097-2765(00)80155-0. [DOI] [PubMed] [Google Scholar]
- Brüning JC, Winnay J, Cheatham B, Kahn CR. Differential signaling by insulin receptor substrate 1 (IRS-1) and IRS-2 in IRS-1-deficient cells. Mol Cell Biol. 1997;17:1513–1521. doi: 10.1128/mcb.17.3.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brys K, Castelein N, Matthijssens F, Vanfleteren JR, Braeckman BP. Disruption of insulin signalling preserves bioenergetic competence of mitochondria in ageing Caenorhabditis elegans. BMC Biol. 2010;8:91. doi: 10.1186/1741-7007-8-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese EJ, Bachmann KA, Bailer AJ, Bolger PM, Borak J, Cai L, Cedergreen N, Cherian MG, Chiueh CC, Clarkson TW, Cook RR, Diamond DM, Doolittle DJ, Dorato MA, Duke SO, Feinendegen L, Gardner DE, Hart RW, Hastings KL, Hayes AW, Hoffmann GR, Ives JA, Jaworowski Z, Johnson TE, Jonas WB, Kaminski NE, Keller JG, Klaunig JE, Knudsen TB, Kozumbo WJ, Lettieri T, Liu SZ, Maisseu A, Maynard KI, Masoro EJ, McClellan RO, Mehendale HM, Mothersill C, Newlin DB, Nigg HN, Oehme FW, Phalen RF, Philbert MA, Rattan SI, Riviere JE, Rodricks J, Sapolsky RM, Scott BR, Seymour C, Sinclair DA, Smith-Sonneborn J, Snow ET, Spear L, Stevenson DE, Thomas Y, Tubiana M, Williams GM, Mattson MP. Biological stress response terminology: Integrating the concepts of adaptive response and preconditioning stress within a hormetic dose-response framework. Toxicol Appl Pharmacol. 2007;222:122–128. doi: 10.1016/j.taap.2007.02.015. [DOI] [PubMed] [Google Scholar]
- Chen C, Wanduragala S, Becker DF, Dickman MB. Tomato QM-like protein protects Saccharomyces cerevisiae cells against oxidative stress by regulating intracellular proline levels. Appl Environ Microbiol. 2006;72:4001–4006. doi: 10.1128/AEM.02428-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy DJ, Gems D, Hafen E, Leevers SJ, Partridge L. Dietary restriction in long-lived dwarf flies. Science. 2002;296:319. doi: 10.1126/science.1069366. [DOI] [PubMed] [Google Scholar]
- Clancy DJ, Gems D, Harshman LG, Oldham S, Stocker H, Hafen E, Leevers SJ, Partridge L. Extension of life-span by loss of CHICO, a Drosophila insulin receptor substrate protein. Science. 2001;292:104–106. doi: 10.1126/science.1057991. [DOI] [PubMed] [Google Scholar]
- Dillin A, Crawford DK, Kenyon C. Timing requirements for insulin/IGF-1 signaling in C. elegans. Science. 2002;298:830–834. doi: 10.1126/science.1074240. [DOI] [PubMed] [Google Scholar]
- Donald SP, Sun XY, Hu CA, Yu J, Mei JM, Valle D, Phang JM. Proline oxidase, encoded by p53-induced gene-6, catalyzes the generation of proline-dependent reactive oxygen species. Cancer Res. 2001;61:1810–1815. [PubMed] [Google Scholar]
- Doonan R, McElwee JJ, Matthijssens F, Walker GA, Houthoofd K, Back P, Matscheski A, Vanfleteren JR, Gems D. Against the oxidative damage theory of aging: superoxide dismutases protect against oxidative stress but have little or no effect on life span in Caenorhabditis elegans. Genes Dev. 2008;22:3236–3241. doi: 10.1101/gad.504808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposti MD, Hatzinisiriou I, McLennan H, Ralph S. Bcl-2 and mitochondrial oxygen radicals. New approaches with reactive oxygen species-sensitive probes. J Biol Chem. 1999;274:29831–29837. doi: 10.1074/jbc.274.42.29831. [DOI] [PubMed] [Google Scholar]
- Finkel T. Signal transduction by reactive oxygen species. J Cell Biol. 2011;194:7–15. doi: 10.1083/jcb.201102095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman DB, Johnson TE. A mutation in the age-1 gene in Caenorhabditis elegans lengthens life and reduces hermaphrodite fertility. Genetics. 1988;118:75–86. doi: 10.1093/genetics/118.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer EL, Dowlatshahi D, Banko MR, Villen J, Hoang K, Blanchard D, Gygi SP, Brunet A. An AMPK-FOXO pathway mediates longevity induced by a novel method of dietary restriction in C. elegans. Curr Biol. 2007;17:1646–1656. doi: 10.1016/j.cub.2007.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzenberger M, Dupont J, Ducos B, Leneuve P, Geloen A, Even PC, Cervera P, Le Bouc Y. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature. 2003;421:182–187. doi: 10.1038/nature01298. [DOI] [PubMed] [Google Scholar]
- Honda Y, Honda S. The daf-2 gene network for longevity regulates oxidative stress resistance and Mn-superoxide dismutase gene expression in Caenorhabditis elegans. FASEB J. 1999;13:1385–1393. [PubMed] [Google Scholar]
- Hoogewijs D, Houthoofd K, Matthijssens F, Vandesompele J, Vanfleteren JR. Selection and validation of a set of reliable reference genes for quantitative sod gene expression analysis in C. elegans. BMC Mol Biol. 2008;9:9. doi: 10.1186/1471-2199-9-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houthoofd K, Braeckman BP, Johnson TE, Vanfleteren JR. Life extension via dietary restriction is independent of the Ins/IGF-1 signalling pathway in Caenorhabditis elegans. Exp Gerontol. 2003;38:947–954. doi: 10.1016/s0531-5565(03)00161-x. [DOI] [PubMed] [Google Scholar]
- Houthoofd K, Fidalgo MA, Hoogewijs D, Braeckman BP, Lenaerts I, Brys K, Matthijssens F, De Vreese A, Van Eygen S, Munoz MJ, Vanfleteren JR. Metabolism, physiology and stress defense in three aging Ins/IGF-1 mutants of the nematode Caenorhabditis elegans. Aging Cell. 2005;4:87–95. doi: 10.1111/j.1474-9726.2005.00150.x. [DOI] [PubMed] [Google Scholar]
- Inoue H, Hisamoto N, An JH, Oliveira RP, Nishida E, Blackwell TK, Matsumoto K. The C. elegans p38 MAPK pathway regulates nuclear localization of the transcription factor SKN-1 in oxidative stress response. Genes Dev. 2005;19:2278–2283. doi: 10.1101/gad.1324805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang YC, van Remmen H. The mitochondrial theory of aging: Insight from transgenic and knockout mouse models. Exp Gerontol. 2009;44:256–260. doi: 10.1016/j.exger.2008.12.006. [DOI] [PubMed] [Google Scholar]
- Katic M, Kennedy AR, Leykin I, Norris A, McGettrick A, Gesta S, Russell SJ, Bluher M, Maratos-Flier E, Kahn CR. Mitochondrial gene expression and increased oxidative metabolism: role in increased lifespan of fat-specific insulin receptor knock-out mice. Aging Cell. 2007;6:827–839. doi: 10.1111/j.1474-9726.2007.00346.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayser EB, Morgan PG, Hoppel CL, Sedensky MM. Mitochondrial expression and function of GAS-1 in Caenorhabditis elegans. J Biol Chem. 2001;276:20551–20558. doi: 10.1074/jbc.M011066200. [DOI] [PubMed] [Google Scholar]
- Kenyon C, Chang J, Gensch E, Rudner A, Tabtiang R. A C. elegans mutant that lives twice as long as wild type. Nature. 1993;366:461–464. doi: 10.1038/366461a0. [DOI] [PubMed] [Google Scholar]
- Kimura KD, Tissenbaum HA, Liu Y, Ruvkun G. daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science. 1997;277:942–946. doi: 10.1126/science.277.5328.942. [DOI] [PubMed] [Google Scholar]
- Lakowski B, Hekimi S. The genetics of caloric restriction in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 1998;95:13091–13096. doi: 10.1073/pnas.95.22.13091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanza IR, Short DK, Short KR, Raghavakaimal S, Basu R, Joyner MJ, McConnell JP, Nair KS. Endurance exercise as a countermeasure for aging. Diabetes. 2008;57:2933–2942. doi: 10.2337/db08-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapointe J, Stepanyan Z, Bigras E, Hekimi S. Reversal of the mitochondrial phenotype and slow development of oxidative biomarkers of aging in long-lived Mclk1+/− mice. J Biol Chem. 2009;284:20364–20374. doi: 10.1074/jbc.M109.006569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SJ, Kaeberlein M, Andalis AA, Sturtz LA, Defossez PA, Culotta VC, Fink GR, Guarente L. Calorie restriction extends Saccharomyces cerevisiae lifespan by increasing respiration. Nature. 2002;418:344–348. doi: 10.1038/nature00829. [DOI] [PubMed] [Google Scholar]
- Lithgow GJ, White TM, Melov S, Johnson TE. Thermotolerance and extended life-span conferred by single-gene mutations and induced by thermal stress. Proc Natl Acad Sci U S A. 1995;92:7540–7544. doi: 10.1073/pnas.92.16.7540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh K, Deng H, Fukushima A, Cai X, Boivin B, Galic S, Bruce C, Shields BJ, Skiba B, Ooms LM, Stepto N, Wu B, Mitchell CA, Tonks NK, Watt MJ, Febbraio MA, Crack PJ, Andrikopoulos S, Tiganis T. Reactive oxygen species enhance insulin sensitivity. Cell Metab. 2009;10:260–272. doi: 10.1016/j.cmet.2009.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masoro EJ. Hormesis and the antiaging action of dietary restriction. Exp Gerontol. 1998;33:61–66. doi: 10.1016/s0531-5565(97)00071-5. [DOI] [PubMed] [Google Scholar]
- Min KJ, Yamamoto R, Buch S, Pankratz M, Tatar M. Drosophila lifespan control by dietary restriction independent of insulin-like signaling. Aging Cell. 2008;7:199–206. doi: 10.1111/j.1474-9726.2008.00373.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miwa S, Riyahi K, Partridge L, Brand MD. Lack of correlation between mitochondrial reactive oxygen species production and life span in Drosophila. Ann N Y Acad Sci. 2004;1019:388–391. doi: 10.1196/annals.1297.069. [DOI] [PubMed] [Google Scholar]
- Morris JZ, Tissenbaum HA, Ruvkun G. A phosphatidylinositol-3-OH kinase family member regulating longevity and diapause in Caenorhabditis elegans. Nature. 1996;382:536–539. doi: 10.1038/382536a0. [DOI] [PubMed] [Google Scholar]
- Murphy CT, McCarroll SA, Bargmann CI, Fraser A, Kamath RS, Ahringer J, Li H, Kenyon C. Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature. 2003;424:277–283. doi: 10.1038/nature01789. [DOI] [PubMed] [Google Scholar]
- Narasimhan SD, Yen K, Tissenbaum HA. Converging pathways in lifespan regulation. Curr Biol. 2009;19:R657–666. doi: 10.1016/j.cub.2009.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owusu-Ansah E, Yavari A, Mandal S, Banerjee U. Distinct mitochondrial retrograde signals control the G1-S cell cycle checkpoint. Nat Genet. 2008;40:356–361. doi: 10.1038/ng.2007.50. [DOI] [PubMed] [Google Scholar]
- Pan Y, Schroeder EA, Ocampo A, Barrientos A, Shadel GS. Regulation of yeast chronological life span by TORC1 via adaptive mitochondrial ROS signaling. Cell Metab. 2011;13:668–678. doi: 10.1016/j.cmet.2011.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandhare J, Donald SP, Cooper SK, Phang JM. Regulation and function of proline oxidase under nutrient stress. J Cell Biochem. 2009;107:759–768. doi: 10.1002/jcb.22174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee SG, Chang TS, Bae YS, Lee SR, Kang SW. Cellular regulation by hydrogen peroxide. J Am Soc Nephrol. 2003;14:S211–215. doi: 10.1097/01.asn.0000077404.45564.7e. [DOI] [PubMed] [Google Scholar]
- Ristow M, Pfister MF, Yee AJ, Schubert M, Michael L, Zhang CY, Ueki K, Michael MD, 2nd, Lowell BB, Kahn CR. Frataxin activates mitochondrial energy conversion and oxidative phosphorylation. Proc Natl Acad Sci U S A. 2000;97:12239–12243. doi: 10.1073/pnas.220403797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ristow M, Zarse K. How increased oxidative stress promotes longevity and metabolic health: The concept of mitochondrial hormesis (mitohormesis) Exp Gerontol. 2010;45:410–418. doi: 10.1016/j.exger.2010.03.014. [DOI] [PubMed] [Google Scholar]
- Ristow M, Zarse K, Oberbach A, Klöting N, Birringer M, Kiehntopf M, Stumvoll M, Kahn CR, Blüher M. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc Nat Acad Sci. 2009;106:8665–8670. doi: 10.1073/pnas.0903485106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmeisser S, Zarse K, Ristow M. Lonidamine extends lifespan of adult Caenorhabditis elegans by increasing the formation of mitochondrial reactive oxygen species. Horm Metab Res. 2011;43:687–692. doi: 10.1055/s-0031-1286308. [DOI] [PubMed] [Google Scholar]
- Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M, Ristow M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007;6:280–293. doi: 10.1016/j.cmet.2007.08.011. [DOI] [PubMed] [Google Scholar]
- Seibler J, Zevnik B, Kuter-Luks B, Andreas S, Kern H, Hennek T, Rode A, Heimann C, Faust N, Kauselmann G, Schoor M, Jaenisch R, Rajewsky K, Kuhn R, Schwenk F. Rapid generation of inducible mouse mutants. Nucleic Acids Res. 2003;31:e12. doi: 10.1093/nar/gng012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southam CM, Ehrlich J. Effects of extract of western red-cedar heartwood on certain wood-decaying fungi in culture. Phytopathology. 1943;33:517–524. [Google Scholar]
- Tapia PC. Sublethal mitochondrial stress with an attendant stoichiometric augmentation of reactive oxygen species may precipitate many of the beneficial alterations in cellular physiology produced by caloric restriction, intermittent fasting, exercise and dietary phytonutrients: ‘Mitohormesis’ for health and vitality. Med Hypotheses. 2006;66:832–843. doi: 10.1016/j.mehy.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Tatar M, Kopelman A, Epstein D, Tu MP, Yin CM, Garofalo RS. A mutant Drosophila insulin receptor homolog that extends life-span and impairs neuroendocrine function. Science. 2001;292:107–110. doi: 10.1126/science.1057987. [DOI] [PubMed] [Google Scholar]
- R Development Core Team. R: A language and environment for statistical computing. Vienna: 2008. [Google Scholar]
- Tullet JM, Hertweck M, An JH, Baker J, Hwang JY, Liu S, Oliveira RP, Baumeister R, Blackwell TK. Direct inhibition of the longevity-promoting factor SKN-1 by insulin-like signaling in C. elegans. Cell. 2008;132:1025–1038. doi: 10.1016/j.cell.2008.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Raamsdonk JM, Hekimi S. Deletion of the mitochondrial superoxide dismutase sod-2 extends lifespan in Caenorhabditis elegans. PLoS Genet. 2009;5:e1000361. doi: 10.1371/journal.pgen.1000361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanfleteren JR. Oxidative stress and ageing in Caenorhabditis elegans. Biochem J. 1993;292(Pt 2):605–608. doi: 10.1042/bj2920605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanfleteren JR, De Vreese A. The gerontogenes age-1 and daf-2 determine metabolic rate potential in aging Caenorhabditis elegans. FASEB J. 1995;9:1355–1361. doi: 10.1096/fasebj.9.13.7557026. [DOI] [PubMed] [Google Scholar]
- Veal EA, Day AM, Morgan BA. Hydrogen peroxide sensing and signaling. Mol Cell. 2007;26:1–14. doi: 10.1016/j.molcel.2007.03.016. [DOI] [PubMed] [Google Scholar]
- Warburton DE, Nicol CW, Bredin SS. Health benefits of physical activity: the evidence. Can Med Ass J (CMAJ) 2006;174:801–809. doi: 10.1503/cmaj.051351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weindruch R, Walford RL. The retardation of aging and disease by dietary restriction. Springfield, Illinois: Charles C Thomas Pub Ltd; 1988. [Google Scholar]
- Woo DK, Shadel GS. Mitochondrial stress signals revise an old aging theory. Cell. 2011;144:11–12. doi: 10.1016/j.cell.2010.12.023. [DOI] [PubMed] [Google Scholar]
- Xia E, Rao G, Van Remmen H, Heydari AR, Richardson A. Activities of antioxidant enzymes in various tissues of male Fischer 344 rats are altered by food restriction. J Nutr. 1995;125:195–201. doi: 10.1093/jn/125.2.195. [DOI] [PubMed] [Google Scholar]
- Yen K, Patel HB, Lublin AL, Mobbs CV. SOD isoforms play no role in lifespan in ad lib or dietary restricted conditions, but mutational inactivation of SOD-1 reduces life extension by cold. Mech Ageing Dev. 2009;130:173–178. doi: 10.1016/j.mad.2008.11.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.