

Common seafood pollutants inhibit a crucial cellular defense protein.
Abstract
The world’s oceans are a global reservoir of persistent organic pollutants to which humans and other animals are exposed. Although it is well known that these pollutants are potentially hazardous to human and environmental health, their impacts remain incompletely understood. We examined how persistent organic pollutants interact with the drug efflux transporter P-glycoprotein (P-gp), an evolutionarily conserved defense protein that is essential for protection against environmental toxicants. We identified specific congeners of organochlorine pesticides, polychlorinated biphenyls, and polybrominated diphenyl ethers that inhibit mouse and human P-gp, and determined their environmental levels in yellowfin tuna from the Gulf of Mexico. In addition, we solved the cocrystal structure of P-gp bound to one of these inhibitory pollutants, PBDE (polybrominated diphenyl ether)–100, providing the first view of pollutant binding to a drug transporter. The results demonstrate the potential for specific binding and inhibition of mammalian P-gp by ubiquitous congeners of persistent organic pollutants present in fish and other foods, and argue for further consideration of transporter inhibition in the assessment of the risk of exposure to these chemicals.
INTRODUCTION
Persistent organic pollutants (POPs) are hazardous, man-made chemicals that endure in the environment and bioaccumulate in animals. Their environmental persistence ensues from properties such as halogenation and hydrophobicity that slow degradation and promote partitioning into organisms. At the same time, these properties also favor POP bioaccumulation by slowing their elimination. Indeed, although all animals have numerous metabolic enzymes, conjugation systems, and transporter proteins that normally act to eliminate xenobiotics, these systems appear ineffective at limiting POP bioaccumulation.
A critical step toward understanding the persistence and organismal impacts of POPs is defining their interactions with xenobiotic elimination systems. Drug transporters are plasma membrane proteins that both limit the entry of foreign chemicals into the body and speed their clearance, and are already well studied for their roles in drug disposition (1). Previous studies have suggested that environmental chemicals can also interact with drug transporters, such as P-glycoprotein (P-gp), but that they are poorly transported, and that these interactions ultimately lead to inhibition of transporter function (2–10). Of concern is that this inhibition reduces the efficacy of transport, thereby sensitizing animals to toxic chemicals that would otherwise be effluxed (11, 12).
Here, we took a multilevel approach to examine transporter-pollutant interactions, from levels in the environment down to the cocrystal structure of an environmental chemical bound to the transporter. We focused on P-gp, an adenosine triphosphate (ATP)–binding cassette (ABC) transporter (13–15), which plays a major role in the disposition of xenobiotics (1, 16) and which is one of the best-studied drug transporters to date. P-gp has a large binding pocket that interacts with a wide variety of structurally divergent hydrophobic molecules (16–19), and binding within this large pocket can have different impacts on the transporter, from stimulation to inhibition of function (20, 21). P-gp is conserved and is typically expressed at environmental barrier tissues, such as the small intestine or gills (22–24).
To identify POPs that interact with P-gp, we used robust biochemical and cellular assays of mouse and human P-gp and identified specific congeners that inhibit this transporter. Using x-ray crystallography, we validated the binding of one of these chemicals, polybrominated diphenyl ether (PBDE)–100, deep within the ligand pocket of the transporter, providing the first snapshots of P-gp bound to a pollutant. To gain insight into the environmental relevance of P-gp inhibitors, we measured their levels in yellowfin tuna (Thunnus albacares) from the Gulf of Mexico (GOM) and used these data to examine the effects of a representative POP mixture on the transport function of the human P-gp.
RESULTS
Persistent pollutants inhibit P-gp
A gap in knowledge exists with regard to the identity and levels of environmental modulators of P-gp. To begin to address this question, we applied a systematic screen to test ubiquitous chemicals for effects on P-gp and to identify specific congeners most relevant to human and environmental health (Fig. 1). In this screen, we used two independent assays to test 37 pollutants for inhibitory effects, and published data on the incidence of the specific congeners in humans to identify relevant compounds (Fig. 1 and Table 1). We took advantage of mouse P-gp (fig. S1), an established model of mammalian P-gp that is amenable to high-resolution x-ray crystallography (25, 26) and has a high amino acid sequence homology with human P-gp (18).
Fig. 1. Identification of P-gp–inhibiting POPs.
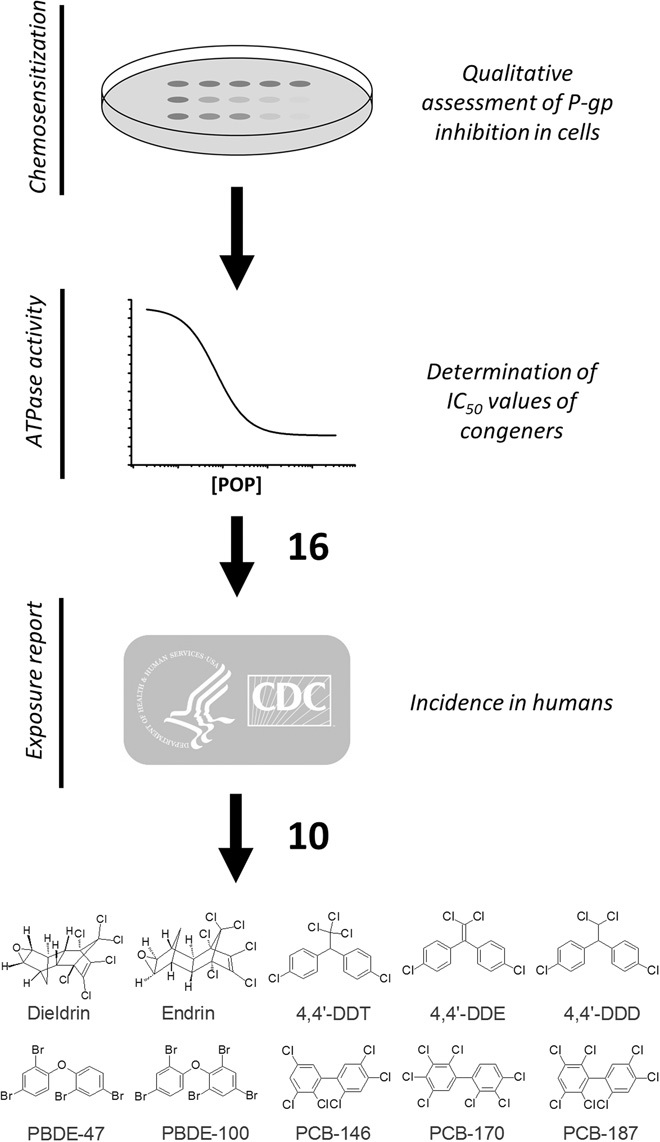
Thirty-seven pollutants were tested for interactions with mouse P-gp using two independent assays (see Table 1). Sixteen compounds were identified as inhibitors in both assays. We focused on 10 congeners reported in humans on the basis of the literature (86, 87) and the Fourth National Report on Human Exposure to Environmental Chemicals of the U.S. Centers for Disease Control and Prevention (88). DDD, dichlorodiphenyldichloroethane; DDE, dichlorodiphenyldichloroethylene; DDT, dichlorodiphenyltrichloroethane; PCB, polychlorinated biphenyl; PBDE, polybrominated diphenyl ether.
Table 1. Summary of the interaction kinetics of environmental chemicals with mouse P-gp.
Compounds that were inhibitors in both assays and present in humans are in boldface. Compounds that were inhibitors in both assays but not known to be detected in humans are italicized. Y, yes; N, no; HCH, hexachlorocyclohexane; DDD, dichlorodiphenyldichloroethane; DDE, dichlorodiphenyldichloroethylene; DDT, dichlorodiphenyltrichloroethane; PFOA, perfluorooctanoic acid; PFOS, perfluorooctane sulfonate; DEHP, di(2-ethylhexyl)phthalate; NI, noninteraction; DL, dioxin-like PCB congeners.
| Chemical | IC50 (μM)* | IMax (%)† | IYeast‡ |
| Organochlorine pesticides | |||
| Aldrin | 26.2 (±1.9) | 0.93 | Y |
| Dieldrin | 21.8 (±4.2) | 0.69 | Y |
| Endrin | 1.1 (±0.7) | 0.51 | Y |
| α-HCH | 26.8 (±18.0) | 0.09 | N |
| β-HCH | NI | 0.00 | N |
| γ-HCH (lindane) | 82.6 (±9.9) | 0.58 | N |
| Heptachlor | 10.4 (±0.9) | 0.98 | N |
| Hexachlorobenzene | NI | 0.00 | N |
| Methoxychlor | 21.7 (±2.4) | 0.57 | N |
| Mirex | 3 (±0.2) | 0.24 | N |
| 4,4′-DDD | 72.5 (±5.7) | 0.45 | Y |
| 4,4′-DDE | 31.3 (±3.7) | 0.63 | Y |
| 4,4′-DDT | 25.6 (±4.8) | 0.61 | Y |
| Polybrominated diphenyl ethers | |||
| PBDE-3 | NI | 0.00 | N |
| PBDE-47 | 22.6 (±6.2) | 0.52 | Y |
| PBDE-49 | 35.6 (±5.4) | 0.52 | Y |
| PBDE-100 | 23.2 (±2.9) | 0.68 | Y |
| PBDE-209 | 6.5 (±0.4) | 0.94 | N |
| Perfluorochemicals | |||
| PFOA | 156.5 (±6.1) | 1.20 | N |
| PFOS | NI | 0.00 | N |
| Plastic-related compounds | |||
| Bisphenol A | NI | 0.00 | N |
| DEHP | NI | 0.00 | N |
| Polychlorinated biphenyls | |||
| PCB-118DL | 15.9 (±1.0) | 0.89 | N |
| PCB-134§ | 12.5 (±0.8) | 0.82 | Y |
| PCB-142 | 6.1 (±0.7) | 0.90 | Y |
| PCB-145 | 4.4 (±0.4) | 0.92 | Y |
| PCB-146 | 12.8 (±1.9) | 0.50 | Y |
| PCB-147§ | 23.6 (±3.1) | 0.82 | Y |
| PCB-152 | 22 (±4.2) | 0.84 | N |
| PCB-153§ | 21.8 (±3.1) | 0.46 | N |
| PCB-154§ | 14.3 (±1.1) | 0.54 | N |
| PCB-161 | 43.2 (±8.3) | 0.40 | N |
| PCB-168§ | 25.8 (±3.7) | 0.66 | N |
| PCB-169DL | 9.7 (±0.5) | 0.93 | N |
| PCB-170 | 9.2 (±0.8) | 0.73 | Y |
| PCB-186 | 6.9 (±0.5) | 0.91 | N |
| PCB-187 | 11.6 (±0.6) | 0.56 | Y |
*Inhibition coefficients (IC50) (±SD) of verapamil-stimulated ATPase activity.
†Percent maximum inhibition (IMax).
‡Inhibition in the yeast cytotoxicity assay (IYeast).
§Co-elusion with other PCB congeners.
For chemosensitization assays, we expressed mouse P-gp in drug-sensitive yeast (Saccharomyces cerevisiae). In these strains, three endogenous transporters are deleted, rendering the cells sensitive to the cytotoxic effects of doxorubicin (DOX) (27). Expression of P-gp protects the cells against DOX (fig. S2), and inhibition of recombinant P-gp by exogenous inhibitors restores DOX sensitivity, causing a dose-dependent decrease in their growth (Fig. 2A). For ATPase assays, we used purified, detergent-solubilized protein and measured the liberation of inorganic phosphate in the presence or absence of chemicals (28, 29). To determine the potency of chemicals as inhibitors, P-gp was prestimulated with verapamil (30, 31) and then incubated with each pollutant individually.
Fig. 2. P-gp–inhibiting effects of POPs.
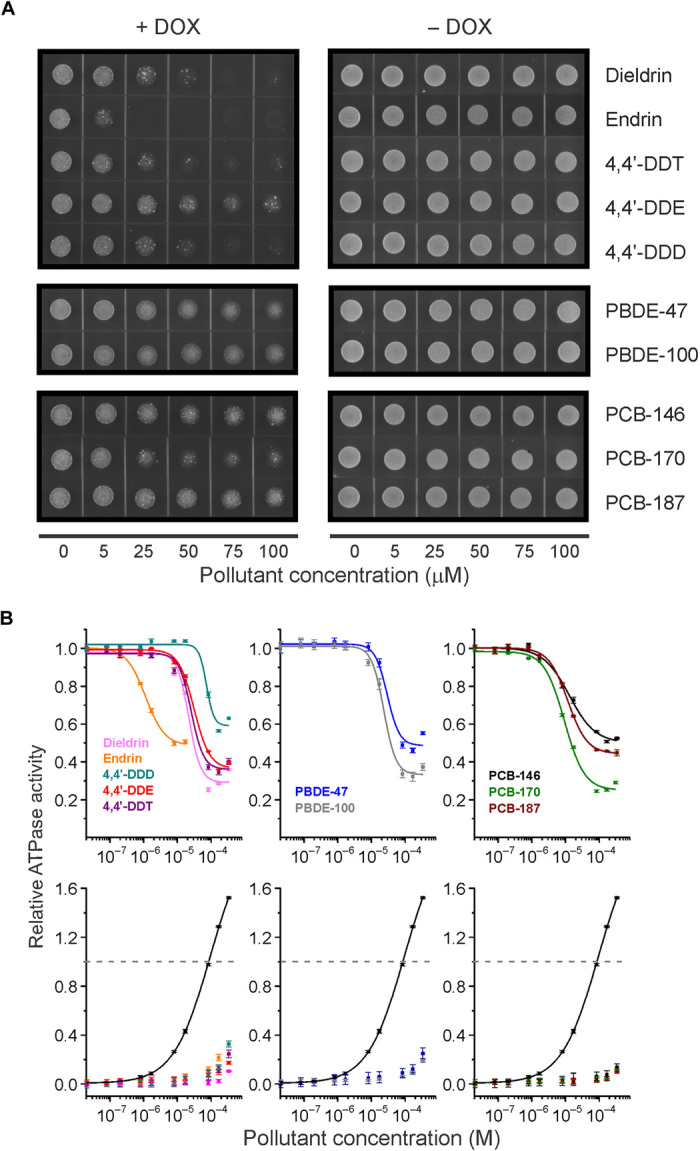
(A) Representative images showing chemosensitization of P-gp–expressing yeast by POPs. Inhibition of mouse P-gp heterologously expressed in drug (+DOX)–sensitive yeast. Inhibition is indicated by the reduction in yeast growth with increasing POP concentration. None of the POPs were toxic to yeast in the absence of DOX (−DOX). Yeast assays were replicated three times and representative micrographs are shown. (B) Upper panel: Inhibition of verapamil-stimulated P-gp adenosine triphosphatase (ATPase) activity by POPs. Graphs show P-gp ATPase inhibition kinetics with the 10 transporter-inhibiting POPs. ATPase assays were performed with purified, recombinant mouse P-gp protein. Lower panel: Lack of P-gp ATPase activation by POPs. ATPase activation of P-gp–inhibiting organochlorine pesticides (OCPs), PBDEs, and PCBs was determined with increasing concentrations of each compound and without verapamil prestimulation. The black curves show verapamil stimulation. Points were normalized to 100 μM verapamil stimulation (gray dashed line) and represent the average ATPase activity ± SD from three to six experiments. Where not visible, error bars are smaller than symbols; R2 values were all >0.99.
From our assays, 16 chemicals that exhibited inhibitory action in both assays were identified. Of these, 10 have been reported in humans (Fig. 1 and Table 1), including DDT, DDE, DDD, dieldrin, endrin, PCB-146, PCB-170, PCB-187, PBDE-47, and PBDE-100 (Fig. 1). All 10 compounds chemosensitized P-gp–overexpressing yeast toward DOX and had IC50 (median inhibitory concentration) values of 1.1 to 72.5 μM in the ATPase assays (Fig. 2 and Table 1). We also observed that congener-specific effects were similar in the two assays, suggesting common modes of interaction with the transporter. For instance, in the cellular assay, endrin had the most pronounced chemosensitizing effect, whereas its stereoisomer, dieldrin, had a markedly lower inhibitory effect (Fig. 2A). Similarly, in ATPase assays, endrin was nearly 20 times more potent than dieldrin (Fig. 2B and Table 1).
We also examined whether the 10 compounds might stimulate the basal (that is, unstimulated) activity of P-gp (32), which is sometimes seen when P-gp is incubated with its transported substrates, and we found that none of them significantly stimulated activity (Fig. 2B). This, along with their ready bioaccumulation, suggests that POPs are not well transported; however, such a possibility is not completely ruled out because the ATPase assay does not directly measure transport. In addition, none of these 10 compounds were toxic to yeast in the absence of DOX (Fig. 2A), indicating that the observed chemosensitizing effects were not related to nonspecific effects, such as membrane perturbation.
PBDE-100 binds to a conserved region deep within the drug-binding pocket of P-gp
To further investigate these interactions, we next sought to visualize the binding of a pollutant with P-gp. Until recently, little has been known about the mechanisms by which environmental chemicals might interact with P-gp, and an unresolved question has been whether their interactions involve specific binding to the ligand-binding domain or nonspecific interactions with the transporter. Although solving structures of large transmembrane proteins bound to inhibitors is a challenge because of the resolution range of ~3 to 4 Å, this limitation can be circumvented by the use of compounds that have an anomalous x-ray scatter signal from atoms such as selenium, mercury, or bromine. For this reason, we chose to cocrystallize and solve the structure of mouse P-gp bound to the penta-brominated compound PBDE-100. The resulting crystal diffracted to 3.5 Å resolution (table S1). The PBDE-100 binding site was located on one-half of the transporter’s pseudo-symmetric dimer structure (Fig. 3, A and B). Fifteen residues from transmembrane segments 5, 6, 7, 8, and 12 mediated hydrophobic interactions with the diphenyl core of PBDE-100 (Fig. 3, C and D), burying more than 90% of its solvent-accessible surface. Eleven of these residues (Y303, Y306, A307, F310, F331, F724, I727, V731, S752, F755, and S975) were unique to this structure and not shared with those previously identified in structures of P-gp crystallized with a series of selenium-labeled cyclopeptide ligands (26). Alternative side-chain conformations were observed for all interacting residues upon PBDE-100 binding (fig. S3).
Fig. 3. POP interactions at the substrate-binding site of mouse P-gp.
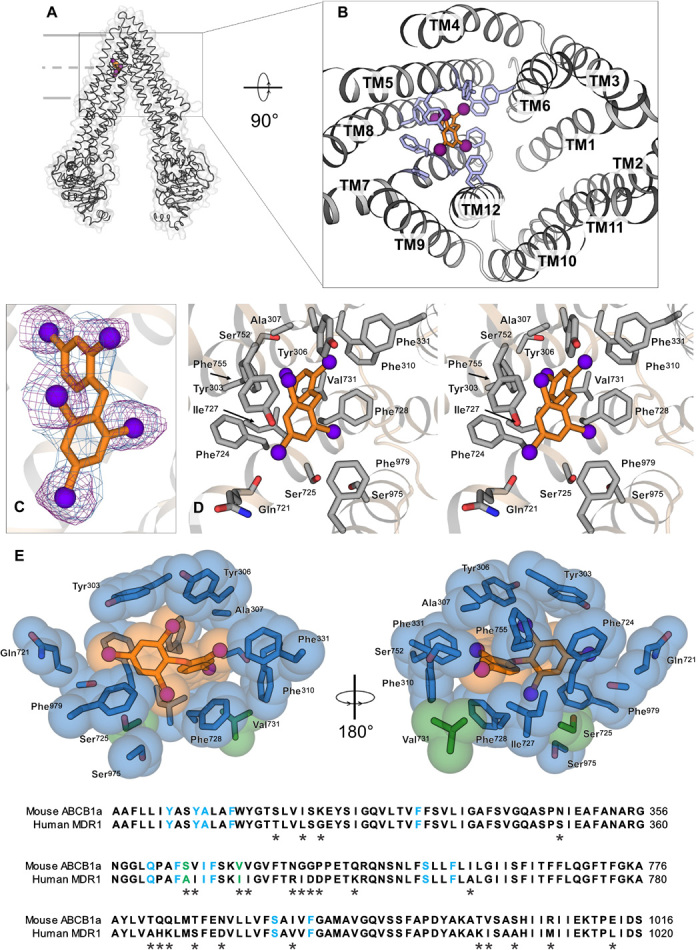
(A) Structure of mouse P-gp cocrystallized with PBDE-100. (B) Location of PBDE-100 at a distinct binding site in the internal cavity of P-gp, viewed from the intracellular side. TM, transmembrane. (C) 2mFo − DFc electron density (where m is the figure of merit and D is the Sigma-A weighting factor) for PBDE-100 (blue; contour level of 1.2σ) and anomalous difference density peaks (purple; contour level of 3.5σ). (D) Stereo view of the binding pocket, with key residues important for the interaction with the diphenyl backbone of PBDE-100 shown as sticks. (E) Conserved binding site for PBDE-100. Top: Side chains found to interact with PBDE-100 are shown in blue (conserved in human and mouse) or green (not conserved). These residues are Y303, Y306, A307, F310, F331, Q721, F724, S725, I727, F728, V731, S752, F755, S975, and F979. Bottom: Amino acid sequence alignment of mouse and human P-gp highlighting the 15 interacting residues with PBDE-100 in TM5, TM6, TM7, TM8, and TM12.
To compare PBDE-100 binding sites of human and mouse P-gp, we aligned the interacting regions identified in our cocrystal structure (Fig. 3E). In addition, we examined potential conservation or divergence of this region in human, mouse, zebrafish, and sea urchin P-gp (fig. S4). These comparisons revealed a high degree of similarity in PBDE-100 binding residues, with 11 of the 15 residues being identical in vertebrates and with 13 being identical in humans and mice (Fig. 3E). Nine of these residues were conserved in sea urchins (fig. S4), which diverge from humans at the base of the deuterostome lineage (33). Collectively, these results suggest evolutionary conservation of the newly identified PBDE-100 binding sites.
Environmentally relevant inhibitor mixtures impair the transport function of human P-gp
The chemicals that we found to inhibit P-gp are ubiquitous, legacy pollutants that have been widely reported in humans and wildlife. Humans are likely to be exposed to these compounds through consumption of contaminated food, raising questions on the environmental levels and impacts of these inhibitor mixtures. Previous studies have demonstrated that marine environments are major global repositories of POPs (34) and that fish can have high levels of these compounds as compared to other foods (35, 36). Thus, to determine the environmental levels of the 10 inhibitors, we measured them in wild-caught yellowfin tuna. We focused on tuna because they are among the most widely consumed fish in the world (37). More than 1 million metric tons of yellowfin tuna are caught annually, accounting for 27% of the global tuna catch (38).
White dorsal muscle fillets of eight fish (Fig. 4A and table S2) were collected and analyzed for levels of OCPs, PCBs, PBDEs, and perfluorochemicals (PFCs) (fig. S5). Nine of the 10 inhibitory POPs were detected in these tuna, and the mean lipid-normalized concentration of the nine inhibitors was 1.01 μM, accounting for approximately 23% of total POPs (Fig. 4, B and C, and table S2). PCBs accounted for 79% of total POPs (fig. S5 and table S2) and 44% of inhibitors (Fig. 4C and table S2).
Fig. 4. Levels of P-gp inhibitors in yellowfin tuna (T. albacares).
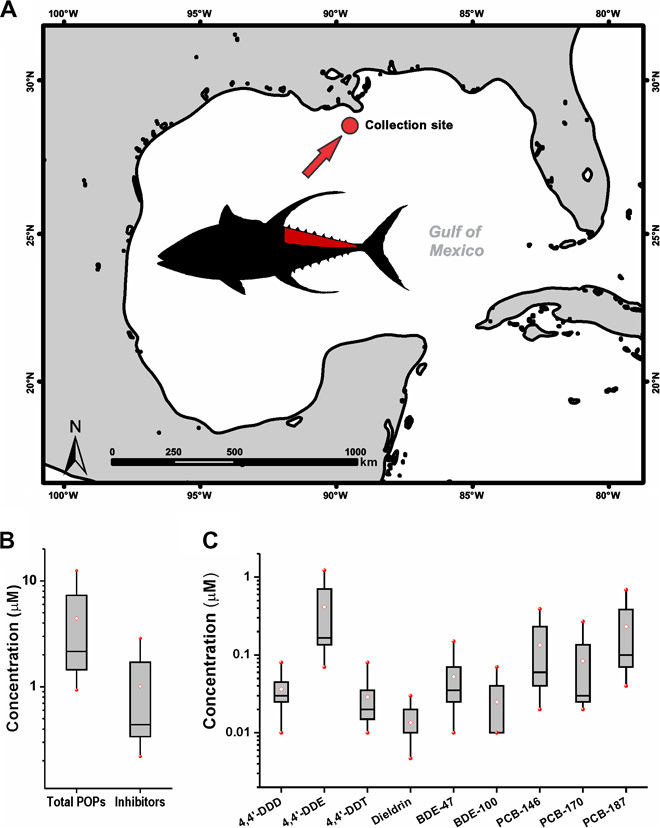
(A) Sampling site for the eight yellowfin tunas (T. albacares) caught in the GOM. The inset shows a yellowfin tuna with the sampled dorsal muscle tissue marked in red. (B) Lipid-normalized concentrations of the total POPs and the 10 P-gp inhibitors. The red-filled circles represent the minimum and maximum values. The white diamonds represent the mean value. The horizontal lines represent the 50th percentile, and the boxes represent the 25th and 75th percentiles. (C) Range of concentrations of nine inhibitory POPs measured in yellowfin tuna muscle from the GOM.
On the basis of these data, a pollutant mixture was formulated, reflecting relative levels of inhibitors found in tuna (Fig. 5A). We used this mixture to examine effects on the transport function of human P-gp. In this assay, inside-out membrane vesicles of mammalian cells overexpressing human P-gp were generated, and effects of POPs were assayed by measurement of the accumulation of the P-gp substrate N-methylquinidine (NMQ) into the vesicles (fig. S6A). In the presence of the POP mixture, NMQ transport was inhibited with an IC50 of 28.7 μM and an IC10 of 7.1 μM (Fig. 5B). We also verified the inhibition of NMQ transport by the individual compounds (fig. S6B). The effects of the POP mixture on mouse P-gp ATPase activity were similar to those seen on human P-gp, with an IC50 of 25 μM and an IC10 of 5.7 μM (Fig. 5C). Collectively, these results indicate that POPs identified in our assays are likely to exhibit similar inhibitory effects on human P-gp.
Fig. 5. An environmentally relevant POP mixture inhibits the transport function of human and mouse P-gp.

(A) Relative ratio of the mean concentrations of P-gp inhibitors found in yellowfin tuna. (B) Inhibition of human P-gp by the POP mixture. Points represent the average percentage of NMQ uptake ± SD relative to the control from nine different experiments and with increasing concentration of the POP mixture. (C) Inhibition of verapamil-stimulated ATPase activity of mouse P-gp by the POP mixture. Shown is the respective dose-response curve as ATPase activity relative to 100 μM verapamil stimulation. The ATPase activity of the purified protein was measured in the presence of increasing concentrations of the POP mixture on the basis of the relative concentration of nine inhibitory POPs identified in this study. All data were fitted using a Hill function [y = v1 + (v2 − v1) * xn/(kn + xn)]. The R2 value was >0.99.
DISCUSSION
Here, we show the direct binding of a pollutant to the ligand-binding pocket of P-gp and the potential for POPs to inhibit this transporter. POPs are ubiquitous contaminants and P-gp is a key protein for xenobiotic elimination in all animals. P-gp, along with cytochrome P450, is typically expressed on apical plasma membranes at sites of xenobiotic uptake (39, 40), such as the intestine, and plays a key role in first-pass elimination of dietary toxins. Thus, the inhibition of P-gp by POPs could represent a mechanism by which this critical cellular defense becomes compromised.
A notable finding of this study was that the levels of transporter inhibitors were sometimes high, raising concerns about transporter inhibition after consumption of highly contaminated foods and about exposure to inhibitors in vulnerable populations of humans and wildlife. High exposure could result from consumption of individual fish with elevated levels of inhibitors. For instance, in this study, although the mean tuna inhibitor concentration was 1.1 μM, two of our eight fish had 2.8 and 2.9 μM POP inhibitors and 11.9 and 12.6 μM total POPs (table S2). This is notable considering that we measured levels in lean cuts of tuna and that more fatty cuts or other types of fish (41) could have even higher levels.
Vulnerable populations could include human neonates, who can have limited intestinal xenobiotic metabolism activity in the first several months of life (42, 43) and, in certain places, can be exposed to high levels of POPs through breast milk. Indeed, in areas where DDT was used for malaria control, high concentrations (>28 μM) were reported in milk fat (44, 45). In wildlife, P-gp inhibitors may pose similar dangers to vulnerable life history stages and geographic populations. For instance, in the GOM, there are high levels of oil hydrocarbons (46, 47) and P-gp could be important for protection against their toxic effects (48–51). Inhibition of P-gp by POPs could be especially problematic for early life history stages because the developing fish heart is sensitive to polycyclic aromatic hydrocarbons (52, 53), and high levels of POPs could be maternally loaded through the yolk (54, 55).
Although our study sheds light on the potential effects of specific POPs on P-gp, the mechanisms of their inhibitory interactions remain incompletely understood. The compounds that we identified are hydrophobic halogenated compounds (table S3), with relatively high potential for passive movement from the environment into tissues (56). Although we did not directly determine the binding constants of these POPs to P-gp, the cocrystal structure demonstrates that a representative compound from our study can form intimate interactions with the ligand-binding domain of the transporter. The high passive flux of POPs into cells and their specific binding properties could be factors that reduce the effectiveness of P-gp in eliminating POPs (57–59). In addition, the conformational changes of the protein that occur upon ligand binding (fig. S3) might also play some as yet unknown role in the observed inhibition by POPs (60, 61).
Finally, it will be important for future studies to consider how metabolism could alter the inhibitory potential and/or transport of pollutants. For instance, at sites of excretion, metabolism pathways, including those involved in conjugation and oxidation, might mitigate some of the inhibitory effects of pollutants and alter their potential for transport. Indeed, in mice, PBDE-47 is conjugated to carrier proteins that improve its efflux by P-gp into urine (62). In addition, other ABC transporters, such as ABCC1, ABCC2, and ABCG2, show a broad tissue distribution and can serve as an additional line of defense by expelling modified and/or unmodified pollutants (16, 24, 40, 63, 64).
CONCLUSIONS
The idea that environmental pollutants could perturb P-gp was first postulated nearly 20 years ago (65); however, the precise mechanisms and environmental relevance have largely remained a mystery. Here, we show the inhibition of the transporter by ubiquitous pollutants found in fish, and likely in other foods. Transporter interactions are already among the criteria evaluated in assessing drug availability (1, 66), and our results argue for consideration of these interactions in risk assessment of environmental chemicals.
An important step taken by this study was solving the cocrystal structure of P-gp bound to a pollutant. The structure revealed a high degree of conservation in the PBDE-100 binding residues and provides a useful tool to help explain and potentially predict both similarities and differences in compound selectivity across species (67). These interactions could also help experimentally define binding sites that are responsible for the adaptive evolution of P-gp into anthropogenic pollutants (68) and could prove applicable to the design of chemicals (69–71) with better potential for elimination by transporters. Indeed, with other transporter structures almost within reach, transporter interactions could soon represent an exciting new avenue for green chemistry.
MATERIALS AND METHODS
Chemicals
All OCPs, cyclosporine A (CsA), verapamil, DOX, Triton X-100, pesticides, and dimethyl sulfoxide (DMSO) were purchased from Sigma. All PCB and PBDE congeners were purchased from AccuStandard. With the exception of verapamil, which was dissolved in water, all stock solutions were prepared in DMSO and diluted to the final concentrations in reaction buffer (ATPase assay) or sterile water (yeast assay). The yeast nitrogen base without amino acids was purchased from Fluka, and the amino acid dropout supplement (−Leu) was obtained from Clontech. The final DMSO concentration in the ATPase and yeast cell assays did not exceed 2 and 0.5%, respectively.
Expression of mouse P-gp in DOX-sensitive yeast
Mouse P-gp (MmABCB1a) was cloned into Bam HI/Hind III restriction sites of the constitutive yeast expression vector p415GPD (Ampr LEU2 ARS/CEN) (72). The hypersensitized MATa S. cerevisiae strain lacking three ABC transporters (Δpdr5 Δsnq2 Δyor1) (27) was transformed with p415GPD plasmids containing MmABCB1a using the standard lithium acetate/polyethylene glycol method (73). The empty p415GPD plasmid was transformed as a control. Transformed cells were selected on synthetic complete dropout (SC-Leu) plates. A single colony from each transformation was picked and cultured overnight in SC-Leu media at 30°C for the spot assays. Overnight cultures were washed in sterile water and their concentrations were adjusted to an OD600 (optical density at 600 nm) of 0.1. To test the functional expression of mouse ABCB1 in yeast cells, fivefold serial dilutions of transformed cells were spotted on SC-Leu and SC-Leu + 10 μM DOX plates and incubated at 30°C for 36 and 48 hours, respectively. For the chemosensitization assay, 15 μl of the culture of p415GPD and p415GPD-MmABCB1a transformed cells was soaked in indicated concentrations of pollutants and spotted on SC-Leu and SC-Leu + 10 μM DOX plates. SC-Leu plates were incubated at 30°C for 40 hours and SC-Leu + DOX plates were incubated at 30°C for 48 to 72 hours.
Expression and purification of mouse P-gp protein
To generate purified mouse P-gp, we overexpressed both genes in Pichia pastoris and purified the protein using combined affinity tag and size exclusion chromatography (SEC). The expression and purification of mouse P-gp in P. pastoris were described previously (18, 25, 26). Briefly, P. pastoris transformed with yeast codon–optimized mouse P-gp (mouse ABCB1a, GenBank JF834158) harboring a C-terminal 6×His-tag was grown as 10-liter cultures in a Bioflow 415 bioreactor (New Brunswick Scientific), induced using slow methanol induction, and harvested. Cells were lysed at 40,000 psi by a single pass through a constant cell disrupter (TS-Series; Constant Systems Inc.), cell debris was separated by centrifugation at 12,500g, and membranes were isolated by centrifugation at 38,400g. Membranes were then solubilized and P-gp was purified using a Ni–nitrilotriacetic acid Superflow resin (Qiagen) via fast protein liquid chromatography (AEKTA, GE Life Sciences). The protein was concentrated (Centricon YM-100; Millipore), ultracentrifuged, and subjected to SEC (Superdex 200 16/60; GE Healthcare). The protein concentration was determined using the Micro BCA Protein Assay Kit (Pierce).
To evaluate the purity of the recombinant P-gp, 5 μg was separated by electrophoresis in a 7.5% SDS–polyacrylamide gel electrophoresis gel and subjected to immunoblotting using a 6×His epitope tag antibody (fig. S1A) (Pierce). The proteins were transferred to a polyvinylidene difluoride membrane by wet electroblotting (tank transfer) for 30 min at 100 V in transfer buffer [25 mM tris-base, 150 mM glycine, and 20% (v/v) methanol (pH 7.4)]. After the transfer, the membranes were blocked in 5% skim milk powder/1× TBST [0.1 M tris-base, 150 mM NaCl, and 0.05% Tween 20 (pH 7.4)] overnight at 23°C. After three washes with 1× TBST, the membranes were incubated for 2 hours with a 6×His epitope tag monoclonal antibody (mouse) in 5% skim milk powder/1× TBST at a ratio of 1:2000 (Pierce). Goat anti-mouse immunoglobulin G–horseradish peroxidase was used as secondary antibody in 5% skim milk powder/1× TBST at a ratio of 1:5000 for 1 hour at 23°C (Pierce). The proteins were visualized using the SuperSignal West Pico Chemiluminescent Substrate Kit (Pierce).
The yeast codon–optimized mouse P-gp used in this study has three N-glycosylation sites converted into glutamine (N83Q, N87Q, and N90Q) and a C-terminal His6-tag fusion. The calculated molecular mass of the resulting protein is ~142 kD, which was confirmed by matrix-assisted laser desorption/ionization mass spectrometry (MALDI MS) (fig. S1B). MALDI MS analysis was used with a time-of-flight mass detector on a Voyager Mass Spectrometer DE-STR (Applied Biosystems) and an effective ion path length of 2 m in the positive ion reflector mode. Sinapinic acid in 50% acetonitrile and 0.1% trifluoroacetic acid was used as matrix solution. Samples (1 mg/ml) were diluted 1:20 with the matrix solution and 1 μl was spotted onto the MALDI sample target plates and air-dried. Spectra were obtained in the mass range between 5000 and 200,000 daltons with 256 laser shots per spectrum. Accelerating voltage was 25,000 V with a grid voltage of 93% and a guide wire percentage of 0.3%. Spectra were recorded in delayed extraction with a delay time of 700 ns. Internal calibration was performed using bovine serum albumin standard (Sigma-Aldrich) with a calculated molecular mass of 66.5 kD. All data were analyzed using Voyager Data Explorer 4.0.0.0 (Applied Biosystems) and plotted using Origin 7.0 (OriginLab).
ATPase activity of purified mouse P-gp
To test for molecular interactions of pollutants with the P-gp transporters, we optimized an ATPase assay based on the malachite green method (28, 74). This assay is well established (29, 75), requiring only 1 to 2 μg of total protein per reaction for measurements of the ATPase activity of efflux transporters. Figure S1C shows the respective dose-response curves of mouse P-gp with the model ATPase stimulator verapamil and the model inhibitor CsA. Activation of mouse P-gp ATPase using verapamil resulted in a half maximal stimulation concentration of 9.4 ± 0.7 μM, which is in agreement with that found previously (76, 77). Verapamil-stimulated (100 μM) ATPase activity was inhibited by CsA with an IC50 of 1.3 ± 0.1 μM. Briefly, we used 2 μg of purified, solubilized mouse P-gp and added the protein to the wells of a chilled 96-well plate containing 60 μl of ATP-free reaction buffer [10 mM MgSO4, 0.05% (w/v) DDM, 1 mM tris(2-carboxyethyl)phosphine (TCEP), and Escherichia coli polar extract lipids (0.1 mg/ml) in 50 mM tris-Cl buffer (pH 7.5)] with serial dilutions of verapamil (control activator). To test inhibition, we used serial dilutions of CsA (control inhibitor) or pollutant compounds plus 100 μM verapamil. Then, 60 μl of ATP solution [5 mM Na-ATP, 10 mM MgSO4, 0.05% (w/v) DDM, 1 mM TCEP, and E. coli polar extract lipids (0.1 mg/ml) in 50 mM tris-Cl buffer (pH 7.5)] was added, mixed, and incubated for 3 min on ice. After incubation, the reaction mixtures in the 96-well polymerase chain reaction plate were transferred to a thermocycler and the reaction was started with the following cycling parameters: 3 s at 4°C, 5 min at 37°C, 15 s at 80°C (heat inactivation), and hold at 4°C. ATPase reactions (30 μl) were transferred to a 96-well enzyme-linked immunosorbent assay plate and the liberated inorganic phosphate was measured by adding 150 μl of an activated color development solution [17 mg of malachite green in 3.75 ml of Milli-Q H2O and 0.525 g of ammonium molybdate tetrahydrate in 12.5 ml of 4N HCl, activated with 0.02% (v/v) Triton X-100] in each sample well. The absorbance of each sample was immediately measured at 600 nm in a microplate reader (Spectramax M2) to minimize acid-catalyzed ATP hydrolysis in the color development solution (pH ~1 to 2). Control samples containing buffer and DMSO (CsA, pollutants) or H2O (verapamil) without added P-gp protein were subtracted as background values. Inorganic phosphate standards (KH2PO4) from 0.125 to 2 nmol served as controls. The ATPase activity data are given as mean ± SEM from three to six different measurements, and representative experiments are shown. For the calculation of IC50 values, the data were fitted to a Hill function: y = v1 + (v2 − v1) * xn/(kn + xn), where v1 and v2 are the initial and final reaction velocities, respectively, n is the Hill coefficient or the cooperativity of the dependence on x, and k is the concentration of the inhibitor (IC50) or the stimulator (EC50) that corresponds to 50% of the maximum (Michaelis constant). All calculations were performed using Origin software.
Reductive methylation and crystallization of mouse P-gp
We added freshly made borane dimethylamine (50 mM) and formaldehyde (100 mM) to purified mouse P-gp (~1 to 2 mg). The mixture was incubated (2 hours at 4°C), and the reaction was quenched with glycine, washed via concentration in SEC buffer [20 mM tris-HCl (pH 8.0), 20 mM NaCl, 0.01% LMNG (lauryl maltose neopentyl glycol), 0.01% sodium cholate, 0.2 mM TCEP, and 0.5 mM EDTA], and treated with 0.2 to 0.5 mM PBDE-100 dissolved in DMSO for an overnight incubation in the dark (12 to 16 hours at 4°C). The sample was then diluted 1:10 in SEC buffer, concentrated to ~12 to 15 mg/ml, and used for crystal trials. P-gp crystals were grown in 24-well Cryschem plates (Hampton Research) at a protein concentration of ~12 to 15 mg/ml using 4-μl sitting drops at a ratio of 1:1 protein/mother liquor [0.1 M Hepes, 50 mM lithium sulfate, 10 mM EDTA, and 24 to 29.5% (w/v) polyethylene glycol (PEG) 600 (pH 7.0 to 8.4)]. Crystals grown at 4°C typically appeared after 1 to 3 days and continued to grow to full size in approximately 2 weeks. Collected crystals were first cryoprotected by soaking in 0.1 M Hepes (at a pH identical to the crystal growth condition), 50 mM lithium sulfate, 10 mM EDTA, and 32% PEG 600. Collected crystals were typically ~650 μm × 400 μm × 300 μm.
X-ray data collection, structure determination, and refinement of the mouse P-gp/PBDE-100 cocrystal structure
X-ray diffraction data were collected at 100 K at the Canadian Light Source (08ID-1). X-ray fluorescence spectra were collected to verify the presence of bromine in P-gp crystals soaked with PBDE-100, and subsequent multiwavelength anomalous dispersion scans were conducted around the Br-K edge to maximize their anomalous signal contribution during data collection. All diffraction data were indexed and integrated with MOSFLM (78), processed with AIMLESS (79), and truncated with TRUNCATE within the CCP4 suite of programs (80). Phases were determined using the recently improved model of P-gp (26) as a search model in molecular replacement in Phaser (81). The resultant model underwent rigid-body and restrained positional refinement, with hydrogens applied in their riding positions, using PHENIX.REFINE (82) against a maximum likelihood target function with grouped B factors and secondary structure restraints. Rounds of refinement were interspersed with manual inspection and correction against Sigma-A–weighted electron density maps in Coot (83), and improvements to model geometry and stereochemistry were monitored using MolProbity (84). The side chains of residues proximal to PBDE-100 were modeled during the final rounds of refinement to avoid biasing their placement. Ligand description dictionaries were calculated using PHENIX.ELBOW (82), and the crystallographic position of PBDE-100 was validated using the anomalous scattering from the bromine atoms (Fig. 3C). The refined structure was judged to have excellent geometry as determined by MolProbity (84). The resulting refinement statistics are listed in table S1. Figures displayed in this paper were prepared using PyMOL (http://www.pymol.org). Atomic coordinates and structure factors for the cocrystal structure presented here were deposited with the Protein Data Bank (accession code 4XWK). Sequence alignments were performed using ClustalW2 Version 2.1 [European Molecular Biology Laboratory–European Bioinformatics Institute (EMBL-EBI)], and molecular interfaces were examined using PDBePISA (EMBL-EBI).
Tuna collection and pollutant data analysis
Yellowfin tuna (T. albacares) were collected from the GOM (28°29′N, 89°27′W). Tuna muscles were sent to AXYS (AXYS Analytical Services Ltd.) for analysis. Pollutants were measured according to the following EPA (U.S. Environmental Protection Agency) methods: for OCPs, 608, 625, 1625, 8081, and 8270; for PBDEs, 1614 and 625; and for PCBs, 1668 and 8270. For PFCs, methods developed by AXYS were used (AXYS MLA-041, AXYS MLA-042, AXYS MLA-043, and AXYS MLA-060). Analyses of pollutant data were performed according to previous studies (35, 85). Where pollutant values were below the detection limit, the values were treated as nondetectable (ND). Most blank measurements were at or below the detection limits and thus not subtracted from samples. In runs where blank values were above detection limits, only samples with values more than twice the blank value were reported (after subtraction of the blank value). The remaining values were treated as ND. The “ND” values were assigned zero when calculating the molar concentrations of POPs in the lipid fraction of the muscle tissue and for further statistical analysis, so as not to overestimate the level of pollutant in samples.
All tuna pollutant data were converted into micromolar lipid-normalized concentrations. Because the lipid composition of the tuna tissue samples was unknown, we assumed a lipid mass density of 1 for volume conversion (that is, 1 g of lipid is equal to 1 ml). To meet assumptions of normality and homogeneity of variance for statistical analyses, all data were log10-transformed. Zero values were excluded from the analyses. To avoid overestimating pollutant levels, one of nine fish initially collected from the GOM was treated as an outlier because of its high levels (more than two times the interquartile range) that did not appear to represent the concentration signature for the group. All data were analyzed and plotted using Origin software.
Vesicle inhibition assays and data analysis
Inhibition of the efflux of probe substrates into membrane vesicles expressing P-gp was carried out according to the manufacturer’s instructions, with some modifications (Sigma-Aldrich). Briefly, vesicle membrane suspensions were added to a 96-well plate stored on ice. Incubation media containing the single POP congener, the POP mixture, or verapamil as positive control were added to the plate and incubated for 15 min. Substrate solutions containing either Mg-ATP or Mg-AMP (adenosine monophosphate) and probe substrate (NMQ) were added to the plate for the designated time (3 min). The incubation was ended by the addition of the ice-cold wash mix. The sample solution was transferred to a filter plate and washed five times with wash mix. Plates were allowed to dry at room temperature for approximately 1 hour, after which a 50:50 methanol/water solution containing the internal standard was added to the filter plate wells and incubated for 15 min before it was transferred to a 96-well analytical plate. For each inhibitor, three assays were performed in duplicate. IC50 values were determined from the average percent inhibition values from each experiment ± SE of the measurement.
Analyst Instrument Control and Data Processing Software (AB SCIEX, version 1.6.1) was used to analyze the unlabeled probe substrate in P-gp vesicle assays (NMQ) for data collection and integration, which were then processed with Microsoft Excel 2007 (Microsoft). Calibration standards were used to calculate concentration on the basis of analyte/internal standard peak-area ratios with Analyst Instrument Control and Data Processing Software (AB SCIEX, version 1.6.1). A Shimadzu API 4000 mass spectrometer in positive mode (4500 V) was used in tandem with a Waters Atlantis (dC18, 5 μm, 100 × 2.1 mm) column (at 40°C) and a Luna C8 guard column (4.0 × 2.0 mm) for separation in a mobile phase of 0.2% formic acid in water and 0.2% formic acid in methanol and an injection volume of 1 μl. The mobile phase flow rate was 0.6 ml/min and the mass transitions used to identify NMQ were m/z (mass/charge ratio) = 339.1 and 339.3. Deuterated NMQ (d3-NMQ) was used as an internal standard and identified with mass transitions of m/z = 342.1 and 342.3.
Supplementary Material
Acknowledgments
We thank G. Moy for technical assistance, R. Doshi and I. L. Urbatsch for helpful discussions on ATPase assays, and R. J. Ferl and J. D. Rine for the yeast strain and the p415GPD plasmid. We thank B. Zgliczynski for providing the GOM map. We thank D. Epel, V. D. Vacquier, and J. Stegeman for discussions of earlier versions of the manuscript. Funding: This work was supported by the program on Oceans and Human Health through NIH grant ES021985 and NSF grant 1314480 to A.H. and G.C., by a WAITT Foundation grant to A.H. and S.S., and by a University of California San Diego Academic Senate Grant to A.H. A.P.M. was supported by a National Health and Medical Research Council CJ Martin Postdoctoral Research Fellowship. Author contributions: S.C.T.N. designed the research, conducted the assays, analyzed the data, and co-authored the manuscript. S.D.R. and A.P.M. conducted x-ray and protein purification work, and edited the manuscript. T.G. designed and performed the yeast assays, and edited the manuscript. L.T.B. collected tuna samples and analyzed the data. L.M.V. performed vesicle assays and analyzed the data. C.C. conducted protein purification work. G.L., S.S., and G.C. designed and supervised the research, and edited the manuscript. A.H. analyzed the data, designed the research, co-authored the manuscript, and supervised the project. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. The GOM map file was created using ArcGIS Desktop v10.3.1 and is licensed under the Educational Site License.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/4/e1600001/DC1
fig. S1. ATPase activity of purified, recombinant mouse P-gp.
fig. S2. Functional expression of mouse P-gp in yeast cells.
fig. S3. Comparison of PBDE-100 interacting residues.
fig. S4. Amino acid sequence alignment of human, mouse, zebrafish, and sea urchin P-gp.
fig. S5. Levels of POPs in wild-caught yellowfin tuna.
fig. S6. Individual POP congeners from the mixture inhibiting human P-gp.
table S1. Data collection and refinement statistics of the mouse P-gp/PBDE-100 cocrystal structure.
table S2. Metadata on yellowfin tuna specimens used in this study.
table S3. Physical and chemical properties of the 10 POP inhibitors.
REFERENCES AND NOTES
- 1.International Transporter Consortium Giacomini K. M., Huang S.-M., Tweedie D. J., Benet L. Z., Brouwer K. L. R., Chu X., Dahlin A., Evers R., Fischer V., Hillgren K. M., Hoffmaster K. A., Ishikawa T., Keppler D., Kim R. B., Lee C. A., Niemi M., Polli J. W., Sugiyama Y., Swaan P. W., Ware J. A., Wright S. H., Yee S. W., Zamek-Gliszczynski M. J., Zhang L., Membrane transporters in drug development. Nat. Rev. Drug Discov. 9, 215–236 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Galgani F., Cornwall R., Toomey B. H., Epel D. D., Interaction of environmental xenobiotics with a multixenobiotic defense mechanism in the bay mussel Mytilus galloprovincialis from the coast of California. Environ. Toxicol. Chem. 15, 325–331 (1996). [Google Scholar]
- 3.Bain L. J., McLachlan J. B., LeBlanc G. A., Structure-activity relationships for xenobiotic transport substrates and inhibitory ligands of P-glycoprotein. Environ. Health Perspect. 105, 812–818 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luckenbach T., Epel D., Nitromusk and polycyclic musk compounds as long-term inhibitors of cellular xenobiotic defense systems mediated by multidrug transporters. Environ. Health Perspect. 113, 17–24 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shabbir A., DiStasio S., Zhao J., Cardozo C. P., Wolff M. S., Caplan A. J., Differential effects of the organochlorine pesticide DDT and its metabolite p,p′-DDE on p-glycoprotein activity and expression. Toxicol. Appl. Pharmacol. 203, 91–98 (2005). [DOI] [PubMed] [Google Scholar]
- 6.Sreeramulu K., Liu R., Sharom F. J., Interaction of insecticides with mammalian P-glycoprotein and their effect on its transport function. Biochim. Biophys. Acta 1768, 1750–1757 (2007). [DOI] [PubMed] [Google Scholar]
- 7.Stevenson C. N., MacManus-Spencer L. A., Luckenbach T., Luthy R. G., Epel D., New perspectives on perfluorochemical ecotoxicology: Inhibition and induction of an efflux transporter in the marine mussel, Mytilus californianus. Environ. Sci. Technol. 40, 5580–5585 (2006). [DOI] [PubMed] [Google Scholar]
- 8.Anselmo H. M. R., van den Berg J. H. J., Rietjens I. M. C. M., Murk A. J., Inhibition of cellular efflux pumps involved in multi xenobiotic resistance (MXR) in echinoid larvae as a possible mode of action for increased ecotoxicological risk of mixtures. Ecotoxicology 21, 2276–2287 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Bircsak K. M., Richardson J. R., Aleksunes L. M., Inhibition of human MDR1 and BCRP transporter ATPase activity by organochlorine and pyrethroid insecticides. J. Biochem. Mol. Toxicol. 27, 157–164 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dankers A. C. A., Roelofs M. J. E., Piersma A. H., Sweep F. C. G. J., Russel F. G. M., van den Berg M., van Duursen M. B. M., Masereeuw R., Endocrine disruptors differentially target ATP-binding cassette transporters in the blood-testis barrier and affect leydig cell testosterone secretion in vitro. Toxicol. Sci. 136, 382–391 (2013). [DOI] [PubMed] [Google Scholar]
- 11.Smital T., Luckenbach T., Sauerborn R., Hamdoun A. M., Vega R. L., Epel D., Emerging contaminants—Pesticides, PPCPs, microbial degradation products and natural substances as inhibitors of multixenobiotic defense in aquatic organisms. Mutat. Res. 552, 101–117 (2004). [DOI] [PubMed] [Google Scholar]
- 12.Buss D. S., Callaghan A., Interaction of pesticides with p-glycoprotein and other ABC proteins: A survey of the possible importance to insecticide, herbicide and fungicide resistance. Pestic. Biochem. Phys. 90, 141–153 (2008). [Google Scholar]
- 13.Juliano R. L., Ling V., A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim. Biophys. Acta 455, 152–162 (1976). [DOI] [PubMed] [Google Scholar]
- 14.Ambudkar S. V., Dey S., Hrycyna C. A., Ramachandra M., Pastan I., Gottesman M. M., Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu. Rev. Pharmacol. Toxicol. 39, 361–398 (1999). [DOI] [PubMed] [Google Scholar]
- 15.Dean M., Annilo T., Evolution of the ATP-binding cassette (ABC) transporter superfamily in vertebrates. Annu. Rev. Genomics Hum. Genet. 6, 123–142 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Leslie E. M., Deeley R. G., Cole S. P. C., Multidrug resistance proteins: Role of P-glycoprotein, MRP1, MRP2, and BCRP (ABCG2) in tissue defense. Toxicol. Appl. Pharmacol. 204, 216–237 (2005). [DOI] [PubMed] [Google Scholar]
- 17.Gottesman M. M., Fojo T., Bates S. E., Multidrug resistance in cancer: Role of ATP–dependent transporters. Nat. Rev. Cancer 2, 48–58 (2002). [DOI] [PubMed] [Google Scholar]
- 18.Aller S. G., Yu J., Ward A., Weng Y., Chittaboina S., Zhuo R., Harrell P. M., Trinh Y. T., Zhang Q., Urbatsch I. L., Chang G., Structures of P-glycoproteins reveals a molecular basis for poly-specific drug binding. Science 323, 1718–1722 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Borst P., Elferink R. O., Mammalian ABC transporters in health and disease. Annu. Rev. Biochem. 71, 537–592 (2002). [DOI] [PubMed] [Google Scholar]
- 20.Martin C., Berridge G., Higgins C. F., Mistry P., Charlton P., Callaghan R., Communication between multiple drug binding sites on P-glycoprotein. Mol. Pharmacol. 58, 624–632 (2000). [DOI] [PubMed] [Google Scholar]
- 21.Ferreira R. J., Ferreira M.-J. U., dos Santos D. J. V. A., Molecular docking characterizes substrate-binding sites and efflux modulation mechanisms within P-glycoprotein. J. Chem. Inf. Model. 53, 1747–1760 (2013). [DOI] [PubMed] [Google Scholar]
- 22.A. Sturm, H. Segner, in Biochemistry and Molecular Biology of Fishes, T. P. Mommsen, T. W. Moon, Eds. (Elsevier B.V., Amsterdam, Netherlands, 2005), vol. 6, pp. 495–533. [Google Scholar]
- 23.Epel D., Luckenbach T., Stevenson C. N., MacManus-Spencer L. A., Hamdoun A., Smital T., Efflux transporters: Newly appreciated roles in protection against pollutants. Environ. Sci. Technol. 42, 3914–3920 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Döring B., Petzinger E., Phase 0 and phase III transport in various organs: Combined concept of phases in xenobiotic transport and metabolism. Drug Metab. Rev. 46, 261–282 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Ward A. B., Szewczyk P., Grimard V., Lee C.-W., Martinez L., Doshi R., Caya A., Villaluz M., Pardon E., Cregger C., Swartz D. J., Falson P. G., Urbatsch I. L., Govaerts C., Steyaert J., Chang G., Structures of P-glycoprotein reveal its conformational flexibility and an epitope on the nucleotide-binding domain. Proc. Natl. Acad. Sci. U.S.A. 110, 13386–13391 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Szewczyk P., Tao H., McGrath A. P., Villaluz M., Rees S. D., Lee S. C., Doshi R., Urbatsch I. L., Zhang Q., Chang G., Snapshots of ligand entry, malleable binding and induced helical movement in P-glycoprotein. Acta Crystallogr. D Biol. Crystallogr. 71, 732–741 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jeong H., Herskowitz I., Kroetz D. L., Rine J., Function-altering SNPs in the human multidrug transporter gene ABCB1 identified using a Saccharomyces-based assay. PLOS Genet. 3, e39 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van Veldhoven P. P., Mannaerts G. P., Inorganic and organic phosphate measurements in the nanomolar range. Anal. Biochem. 161, 45–48 (1987). [DOI] [PubMed] [Google Scholar]
- 29.Henkel R. D., VandeBerg J. L., Walsh R. A., A microassay for ATPase. Anal. Biochem. 169, 312–318 (1988). [DOI] [PubMed] [Google Scholar]
- 30.Sarkadi B., Price E. M., Boucher R. C., Germann U. A., Scarborough G. A., Expression of the human multidrug resistance cDNA in insect cells generates a high activity drug-stimulated membrane ATPase. J. Biol. Chem. 267, 4854–4858 (1992). [PubMed] [Google Scholar]
- 31.Doige C. A., Yu X., Sharom F. J., ATPase activity of partially purified P-glycoprotein from multidrug-resistant Chinese hamster ovary cells. Biochim. Biophys. Acta 1109, 149–160 (1992). [DOI] [PubMed] [Google Scholar]
- 32.Tampal N. M., Robertson L. W., Srinivasan C., Ludewig G., Polychlorinated biphenyls are not substrates for the multidrug resistance transporter-1. Toxicol. Appl. Pharmacol. 187, 168–177 (2003). [DOI] [PubMed] [Google Scholar]
- 33.Lowe C. J., Clarke D. N., Medeiros D. M., Rokhsar D. S., Gerhart J., The deuterostome context of chordate origins. Nature 520, 456–465 (2015). [DOI] [PubMed] [Google Scholar]
- 34.Lohmann R., Jurado E., Pilson M. E. Q., Dachs J., Oceanic deep water formation as a sink of persistent organic pollutants. Geophys. Res. Lett. 33, L12607 (2006). [Google Scholar]
- 35.Schecter A., Päpke O., Harris T. R., Tung K. C., Musumba A., Olson J., Birnbaum L., Polybrominated diphenyl ether (PBDE) levels in an expanded market basket survey of U.S. food and estimated PBDE dietary intake by age and sex. Environ. Health Perspect. 114, 1515–1520 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schecter A., Colacino J., Haffner D., Patel K., Opel M., Päpke O., Birnbaum L., Perfluorinated compounds, polychlorinated biphenyls, and organochlorine pesticide contamination in composite food samples from Dallas, Texas, USA. Environ. Health Perspect. 118, 796–802 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Loke M., Geslani C., Takenaka B., Leung P., An overview of seafood consumption and supply sources: Hawai’i versus U.S. Econ. Issues 22, 1–9 (2012). [Google Scholar]
- 38.ISSF Tuna Stock Status Update, 2015: Status of the world fisheries for tuna (ISSF Technical Report 2015-03A, International Seafood Sustainability Foundation, Washington, 2015). [Google Scholar]
- 39.Lin J. H., Yamazaki M., Role of P-glycoprotein in pharmacokinetics: Clinical implications. Clin. Pharmacokinet. 42, 59–98 (2003). [DOI] [PubMed] [Google Scholar]
- 40.Dietrich C. G., Geier A., Oude Elferink R. P. J., ABC of oral bioavailability: Transporters as gatekeepers in the gut. Gut 52, 1788–1795 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hites R. A., Foran J. A., Carpenter D. O., Hamilton M. C., Knuth B. A., Schwager S. J., Global assessment of organic contaminants in farmed salmon. Science 303, 226–229 (2004). [DOI] [PubMed] [Google Scholar]
- 42.Johnson T. N., Tanner M. S., Taylor C. J., Tucker G. T., Enterocytic CYP3A4 in a paediatric population: Developmental changes and the effect of coeliac disease and cystic fibrosis. Br. J. Clin. Pharmacol. 51, 451–460 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Johnson T. N., Thomson M., Intestinal metabolism and transport of drugs in children: The effects of age and disease. J. Pediatr. Gastroenterol. Nutr. 47, 3–10 (2008). [DOI] [PubMed] [Google Scholar]
- 44.Smith D., Worldwide trends in DDT levels in human breast milk. Int. J. Epidemiol. 28, 179–188 (1999). [DOI] [PubMed] [Google Scholar]
- 45.Bouwman H., Kylin H., Sereda B., Bornman R., High levels of DDT in breast milk: Intake, risk, lactation duration, and involvement of gender. Environ. Pollut. 170, 63–70 (2012). [DOI] [PubMed] [Google Scholar]
- 46.Crone T. J., Tolstoy M., Magnitude of the 2010 Gulf of Mexico oil leak. Science 330, 634 (2010). [DOI] [PubMed] [Google Scholar]
- 47.Reddy C. M., Arey J. S., Seewald J. S., Sylva S. P., Lemkau K. L., Nelson R. K., Carmichael C. A., McIntyre C. P., Fenwick J., Ventura G. T., Van Mooy B. A. S., Camilli R., Composition and fate of gas and oil released to the water column during the Deepwater Horizon oil spill. Proc. Natl. Acad. Sci. U.S.A. 109, 20229–20234 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lampen A., Ebert B., Stumkat L., Jacob J., Seidel A., Induction of gene expression of xenobiotic metabolism enzymes and ABC-transport proteins by PAH and a reconstituted PAH mixture in human Caco-2 cells. Biochim. Biophys. Acta 1681, 38–46 (2004). [DOI] [PubMed] [Google Scholar]
- 49.Vaché C., Camares O., De Graeve F., Dastugue B., Meiniel A., Vaury C., Pellier S., LEoz-Garziandia E., Bamdad M., Drosophila melanogaster P-glycoprotein: A membrane detoxification system toward polycyclic aromatic hydrocarbon pollutants. Environ. Toxicol. Chem. 25, 572–580 (2006). [DOI] [PubMed] [Google Scholar]
- 50.Hamdoun A. M., Griffin F. J., Cherr G. N., Tolerance to biodegraded crude oil in marine invertebrate embryos and larvae is associated with expression of a multixenobiotic resistance transporter. Aquat. Toxicol. 61, 127–140 (2002). [DOI] [PubMed] [Google Scholar]
- 51.Valton E., Amblard C., Wawrzyniak I., Penault-Llorca F., Bamdad M., P-gp expression in brown trout erythrocytes: Evidence of a detoxification mechanism in fish erythrocytes. Sci. Rep. 3, 3422 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Incardona J. P., Gardner L. D., Linbo T. L., Brown T. L., Esbaugh A. J., Mager E. M., Stieglitz J. D., French B. L., Labenia J. S., Laetz C. A., Tagal M., Sloan C. A., Elizur A., Benetti D. D., Grosell M., Block B. A., Scholz N. L., Deepwater Horizon crude oil impacts the developing hearts of large predatory pelagic fish. Proc. Natl. Acad. Sci. U.S.A. 111, E1510–E1518 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brette F., Machado B., Cros C., Incardona J. P., Scholz N. L., Block B. A., Crude oil impairs cardiac excitation-contraction coupling in fish. Science 343, 772–776 (2014). [DOI] [PubMed] [Google Scholar]
- 54.Mull C. G., Lyons K., Blasius M. E., Winkler C., O’Sullivan J. B., Lowe C. G., Evidence of maternal offloading of organic contaminants in white sharks (Carcharodon carcharias). PLOS One 8, e62886 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lyons K., Adams D. H., Maternal offloading of organochlorine contaminants in the yolk-sac placental scalloped hammerhead shark (Sphyrna lewini). Ecotoxicology 24, 553–562 (2015). [DOI] [PubMed] [Google Scholar]
- 56.Mackay D., Fraser A., Bioaccumulation of persistent organic chemicals: Mechanisms and models. Environ. Pollut. 110, 375–391 (2000). [DOI] [PubMed] [Google Scholar]
- 57.Cole B. J., Hamdoun A., Epel D., Cost, effectiveness and environmental relevance of multidrug transporters in sea urchin embryos. J. Exp. Biol. 216, 3896–3905 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Eytan G. D., Regev R., Oren G., Assaraf Y. G., The role of passive transbilayer drug movement in multidrug resistance and its modulation. J. Biol. Chem. 271, 12897–12902 (1996). [DOI] [PubMed] [Google Scholar]
- 59.Regev R., Katzir H., Yeheskely-Hayon D., Eytan G. D., Modulation of P-glycoprotein-mediated multidrug resistance by acceleration of passive drug permeation across the plasma membrane. FEBS J. 274, 6204–6214 (2007). [DOI] [PubMed] [Google Scholar]
- 60.Raub T. J., P-glycoprotein recognition of substrates and circumvention through rational drug design. Mol. Pharm. 3, 3–25 (2006). [DOI] [PubMed] [Google Scholar]
- 61.Chufan E. E., Kapoor K., Ambudkar S. V., Drug–protein hydrogen bonds govern the inhibition of the ATP hydrolysis of the multidrug transporter P-glycoprotein. Biochem. Pharmacol. 101, 40–53 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Emond C., Sanders J. M., Wikoff D., Birnbaum L. S., Proposed mechanistic description of dose-dependent BDE-47 urinary elimination in mice using a physiologically based pharmacokinetic model. Toxicol. Appl. Pharmacol. 273, 335–344 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.van Herwaarden A. E., Schinkel A. H., The function of breast cancer resistance protein in epithelial barriers, stem cells and milk secretion of drugs and xenotoxins. Trends Pharmacol. Sci. 27, 10–16 (2006). [DOI] [PubMed] [Google Scholar]
- 64.Chhabra R. S., Intestinal absorption and metabolism of xenobiotics. Environ. Health Perspect. 33, 61–69 (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kurelec B., A new type of hazardous chemical: The chemosensitizers of multixenobiotic resistance. Environ. Health Perspect. 105 (Suppl. 4), 855–860 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Montanari F., Ecker G. F., Prediction of drug–ABC-transporter interaction—Recent advances and future challenges. Adv. Drug Deliv. Rev. 86, 17–26 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gökirmak T., Campanale J. P., Shipp L. E., Moy G. W., Tao H., Hamdoun A., Localization and substrate selectivity of sea urchin multidrug (MDR) efflux transporters. J. Biol. Chem. 287, 43876–43883 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wirgin I., Roy N. K., Loftus M., Chambers R. C., Franks D. G., Hahn M. E., Mechanistic basis of resistance to PCBs in Atlantic tomcod from the Hudson River. Science 331, 1322–1325 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Anastas P., Eghbali N., Green chemistry: Principles and practice. Chem. Soc. Rev. 39, 301–312 (2010). [DOI] [PubMed] [Google Scholar]
- 70.Zimmerman J. B., Anastas P. T., Toward substitution with no regrets. Science 347, 1198–1199 (2015). [DOI] [PubMed] [Google Scholar]
- 71.A Framework to Guide Selection of Chemical Alternatives (National Research Council, The National Academies Press, Washington, DC, 2014), 334 pp. [PubMed] [Google Scholar]
- 72.Mumberg D., Müller R., Funk M., Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 156, 119–122 (1995). [DOI] [PubMed] [Google Scholar]
- 73.R. D. Gietz, in Methods in Molecular Biology, W. Xiao, Ed. (Humana Press, Clifton, NJ, 2014), vol. 1205, pp. 1–12. [Google Scholar]
- 74.Urbatsch I. L., Al-Shawi M. K., Senior A. E., Characterization of the ATPase activity of purified Chinese hamster P-glycoprotein. Biochemistry 33, 7069–7076 (1994). [DOI] [PubMed] [Google Scholar]
- 75.Venter H., Velamakanni S., Balakrishnan L., van Veen H. W., On the energy-dependence of Hoechst 33342 transport by the ABC transporter LmrA. Biochem. Pharmacol. 75, 866–874 (2008). [DOI] [PubMed] [Google Scholar]
- 76.Swartz D. J., Weber J., Urbatsch I. L., P-glycoprotein is fully active after multiple tryptophan substitutions. Biochim. Biophys. Acta 1828, 1159–1168 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bai J., Swartz D. J., Protasevich I. I., Brouillette C. G., Harrell P. M., Hildebrandt E., Gasser B., Mattanovich D., Ward A., Chang G., Urbatsch I. L., A gene optimization strategy that enhances production of fully functional P-glycoprotein in Pichia pastoris. PLOS One 6, e22577 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Battye T. G. G., Kontogiannis L., Johnson O., Powell H. R., Leslie A. G. W., iMOSFLM: A new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 67, 271–281 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.P. Evans, in Acta Crystallographica Section D: Biological Crystallography (International Union of Crystallography, Chester, UK, 2006), vol. 62, pp. 72–82. [Google Scholar]
- 80.Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G. W., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S., Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J., Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H., PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Emsley P., Lohkamp B., Scott W. G., Cowtan K., Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chen V. B., Arendall W. B. III, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C., MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hites R. A., Foran J. A., Schwager S. J., Knuth B. A., Hamilton M. C., Carpenter D. O., Global assessment of polybrominated diphenyl ethers in farmed and wild salmon. Environ. Sci. Technol. 38, 4945–4949 (2004). [DOI] [PubMed] [Google Scholar]
- 86.Johansen H. R., Alexander J., Rossland O. J., Planting S., Løvik M., Gaarder P. I., Gdynia W., Bjerve K. S., Becher G., PCDDs, PCDFs, and PCBs in human blood in relation to consumption of crabs from a contaminated Fjord area in Norway. Environ. Health Perspect. 104, 756–764 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bedi J. S., Gill J. P. S., Kaur P., Sharma A., Aulakh R. S., Evaluation of pesticide residues in human blood samples from Punjab (India). Vet. World 8, 66–71 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fourth National Report on Human Exposure to Environmental Chemicals (Centers for Disease Control and Prevention, Atlanta, GA, 2009), 530 pp. [Google Scholar]
- 89.Mackay D., Mascarenhas R., Shiu W. Y., Aqueous solubility of polychlorinated biphenyls. Chemosphere 9, 257–264 (1980). [Google Scholar]
- 90.ATSDR, Toxicological profile for aldrin/dieldrin. Agency Toxic Subst. Dis. Regist., 1–354 (2002). [PubMed]
- 91.ATSDR, Toxicological profile for endrin. Agency Toxic Subst. Dis. Regist., 1–227 (1996). [PubMed]
- 92.ATSDR, Toxicological profile for DDT, DDE, and DDD. Agency Toxic Subst. Dis. Regist., 1–497 (2002). [PubMed]
- 93.ATSDR, Toxicological profile for polychlorinated biphenyls (PCBs). Agency Toxic Subst. Dis. Regist., 1–948 (2000). [PubMed]
- 94.ATSDR, Toxicological profile for polybrominated diphenyl ethers (PBDEs). Agency Toxic Subst. Dis. Regist., 1–532 (2015). [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/4/e1600001/DC1
fig. S1. ATPase activity of purified, recombinant mouse P-gp.
fig. S2. Functional expression of mouse P-gp in yeast cells.
fig. S3. Comparison of PBDE-100 interacting residues.
fig. S4. Amino acid sequence alignment of human, mouse, zebrafish, and sea urchin P-gp.
fig. S5. Levels of POPs in wild-caught yellowfin tuna.
fig. S6. Individual POP congeners from the mixture inhibiting human P-gp.
table S1. Data collection and refinement statistics of the mouse P-gp/PBDE-100 cocrystal structure.
table S2. Metadata on yellowfin tuna specimens used in this study.
table S3. Physical and chemical properties of the 10 POP inhibitors.