

Abstract
Edoxaban exposure‐response relationships from the phase III study evaluating edoxaban for prevention and treatment of venous thromboembolism (VTE) in patients with acute deep vein thrombosis (DVT) and/or pulmonary embolism (PE) were assessed by parametric time‐to‐event analysis. Statistical significant exposure‐response relationships were recurrent VTE with hazard ratio (HR) based on average edoxaban concentration at steady state (Cav) (HRCav) = 0.98 (i.e., change in the HR with every 1 ng/mL increase of Cav); the composite of recurrent DVT and nonfatal PE with HRCav = 0.99; and the composite of recurrent DVT, nonfatal PE, and all‐cause mortality HRCav = 0.98, and all death using maximal edoxaban concentration (Cmax) with HR (Cmax) = 0.99. No statistical significant exposure‐response relationships were found for clinically relevant bleeding or major adverse cardiovascular event. Results support the recommendation of once‐daily edoxaban 60 mg, and a reduced 30 mg dose in patients with moderate renal impairment, body weight ≤60 kg, or use of P‐glycoprotein inhibitors verapamil or quinidine.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THIS TOPIC? ☑ In the large‐scale phase III Hokusai‐VTE study, the nonvitamin K antagonist oral anticoagulant edoxaban was noninferior to warfarin in preventing recurrent VTE and caused statistically significant less bleeding. • WHAT QUESTION DID THIS STUDY ADDRESS? ☑ Potential relationships between edoxaban exposure and safety and efficacy endpoints in Hokusai‐VTE, associated risk factors influences, and the edoxaban efficacy/safety balance in patient subgroups were evaluated. • WHAT THIS STUDY ADDS TO OUR KNOWLEDGE ☑ Event risk for recurrent VTE; composite recurrent DVT and nonfatal PE; or composite recurrent DVT, nonfatal PE, and all‐cause mortality decreased with increasing average steady state edoxaban concentration. All‐cause mortality, but not clinically relevant bleeding or major adverse cardiovascular events, had statistically significant exposure‐response relationships. Identified risk factors were consistent with clinical knowledge. • HOW THIS MIGHT CHANGE CLINICAL PHARMACOLOGY AND THERAPEUTICS ☑ Recommendations of edoxaban 60 mg once daily for prevention and treatment of VTE in the general patient population and reduced‐dose edoxaban 30 mg in patients with moderate renal impairment, body weight ≤60 kg, or concomitant use of P‐glycoprotein inhibitors were supported.
Prevention of thromboembolic events is a significant global healthcare issue. Although vitamin K antagonist oral anticoagulants were used for several decades and are effective in preventing thromboembolic events, they have considerable limitations, such as delayed onset of anticoagulant action, a narrow therapeutic index requiring close laboratory monitoring, variable pharmacological response, and interactions with food.1 These limitations prompted clinical development of new nonvitamin K antagonist oral anticoagulants.2, 3 Edoxaban is a new nonvitamin K antagonist oral anticoagulant that directly inhibits activated Factor Xa, and dose‐dependently decreases thrombin generation.4
Edoxaban has an oral bioavailability of ∼62%, with maximum plasma concentration within 1 to 2 hours after oral administration.5 Plasma protein binding of edoxaban is relatively low, ranging from 40–59%.6 Edoxaban is eliminated via both renal excretion and liver metabolism pathways, with ∼50% of systemically absorbed drug excreted in urine.6 It is a P‐glycoprotein (P‐gp) substrate; hence, significant drug‐drug interactions are expected when edoxaban is used concurrently with strong P‐gp inhibitors.7, 8
Clinical efficacy and safety of edoxaban compared to warfarin were evaluated in a large‐scale phase III study, Hokusai‐VTE,9 for the prevention and treatment of venous thromboembolism in patients with acute deep vein thrombosis (DVT) and/or pulmonary embolism (PE). Patients received edoxaban 60 mg once daily or 30 mg once daily if they had moderate renal impairment (creatinine clearance 30–50 mL/min), body weight ≤60 kg, or concurrent treatment with potent P‐gp inhibitors, such as verapamil or quinidine. The study demonstrated that, after initial treatment with heparin, edoxaban once daily was noninferior to warfarin in preventing recurrent venous thromboembolism (VTE) but caused significantly less bleeding in a broad range of patients.9
Obtaining quantitative information regarding exposure‐response relationships and associated risk factors are critical for appropriate clinical management, dose selection, and reduction in patients receiving edoxaban therapy. Thus, the present analyses characterized potential relationships between edoxaban plasma exposure and efficacy or safety outcomes and evaluated potential influences of risk factors associated with these outcomes. Additionally, the efficacy/safety balance (clinical utility) of edoxaban was assessed overall and in patient subgroups receiving edoxaban 60 mg once daily and a reduced dose of edoxaban 30 mg once daily.
METHODS
Study design and data source
Details of the Hokusai‐VTE9 trial study design were described previously. Briefly, in this randomized, double‐blinded, noninferiority study, patients with DVT, PE, or both received initial heparin therapy (enoxaparin or unfractionated heparin) for at least 5 days, followed by edoxaban 60 mg once daily or warfarin. Edoxaban dose was reduced to 30 mg once daily in patients with body weight ≤60 kg, or creatinine clearance (CRCL) of 30 to 50 mL/min, or in patients with concomitant use of P‐gp inhibitors verapamil or quinidine. The warfarin dose was adjusted to maintain international normalized ratio between 2.0 and 3.0 based on clinical profiles and local practice guidelines or an authoritative dosing algorithm. Treatment duration was 3–12 months. Study protocol and amendments were approved by each participating center's ethics committee and the study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice Guidelines.
The primary efficacy endpoint was incidence of symptomatic recurrent VTE, defined as a composite of recurrent DVT, and fatal or nonfatal PE. The principal safety endpoint was incidence of clinically relevant bleeding, defined as a composite of major or clinically relevant nonmajor bleeding.
Pharmacokinetic data and exposure indices
Blood samples for edoxaban plasma concentration determination were collected from individual patients at predose and 1–3 hours postdose at the 3‐month visit, and any time during the 12‐month visit. One additional sample was collected at the time of an event (VTE, major or clinically relevant bleeding, or serious adverse event). Plasma edoxaban concentrations were measured by validated liquid chromatography‐tandem mass spectrometry assay with a lower limit of quantification of 0.764 ng/mL. The intra‐ and interassay precisions for edoxaban were ≤11.0% and ≤8.8%, respectively.5 Edoxaban has an active major metabolite, M4, but previous modeling work on renally impaired patients indicates that using M4 concentrations in exposure‐response analyses would not be of additional value. Hence, no metabolite data were used in this analysis.10
The pharmacokinetic (PK) exposure indices were obtained for individual patients based on an established reported population PK model.11 Area under the curve (AUC0‐24,ss), average concentration (Cav), peak plasma concentration (Cmax), and trough plasma concentration (Cmin) exposure indices were predicted for each occasion based on empirical Bayes estimates of PK parameters and interoccasion variability estimates from the PopPK model, as well as patients' specific dosing and covariate information. For patients with no available PK data (10% of the patients), PK exposure measures were imputed using typical PK parameter estimates and each patient's specific dosing and covariate information.
Exposure‐response analysis
Efficacy endpoints included symptomatic recurrent VTE, a composite of recurrent DVT and nonfatal PE, and a composite of recurrent DVT, nonfatal PE, and all‐cause mortality. Safety endpoints analyzed were clinically relevant bleeding, all death, and major adverse clinical events (MACEs). All of these endpoints, and composite endpoints, were prespecified in both the statistical analysis and the exposure‐response analysis.
Prespecified risk factors for both efficacy and safety endpoints were age ≥75 years (AGE75), men/women (SEX), history of cancer (CAN), history of hepatic disease, history of renal disease, history of pulmonary disease (PUL), previous episode(s) of PE/DVT, recent active cancer (RCAN), recent surgery, trauma, or immobilization (SUR), use of estrogen‐containing drugs, presence of antiphospholipid antibodies, hyperhomocysteinemia, antithrombin deficiency, Factor V Leiden, protein C deficiency, protein S deficiency, prothrombin gene mutation, or concomitant use of aspirin (ASA). Additionally, history of dyslipidemia, prolonged sitting (≥4 hours), or history of cardiovascular disease were assessed as potential risk factors for efficacy endpoints. History of life‐threatening bleeding or history of cerebrovascular disease were assessed for the safety endpoints. However, PE/DVT, antithrombin deficiency, hyperhomocysteinemia, antiphospholipid antibodies, protein C deficiency, protein S deficiency, or prothrombin gene mutation were observed in fewer than 50 subjects receiving either warfarin or edoxaban and therefore were excluded from risk factor analysis. Warfarin was assumed to be in the therapeutic international normalized ratio range (i.e., warfarin patients were in the therapeutic international normalized ratio range in 63.5% of the warfarin treatment time9), hence, no warfarin‐specific covariates were investigated, and instead warfarin was treated as a “placebo arm” used to build the risk factor model.
For each endpoint, a parametric time‐to‐event (TTE) analysis was performed, with exponential, Weibull, or Gompertz distributed TTE. Generally, each endpoint risk factor model was first evaluated using data from all warfarin‐treated patients. After TTE data distribution exploration, an automated stepwise covariate model in Perl Speaks NONMEM PsN12, 13 (http://psn.sourceforge.net) identified potential statistically significant risk factors on the baseline hazard. A P ≤ 0.05 significance level was used for forward inclusion and backward deletion process. The selected warfarin risk factor model was subsequently applied to the edoxaban exposure‐response dataset, with re‐estimation of parameter values of risk factors. Dependent on fit improvement (i.e., a statistically significant drop in objective function value [OFV] at P ≤ 0.05), parameter estimates of the risk factors were either kept as the same as those in the warfarin risk factor model or modified per re‐estimation. Afterward, an exposure‐response relationship, including different PK exposure indices (AUC0‐24,ss,/Cav, Cmax, and Cmin) and linear or nonlinear exposure‐response relationships, was tested. The exposure‐response model further underwent a backward deletion process, with risk factor retention at a P ≤ 0.05 significance level, resulting in a final edoxaban model, including risk factors and an exposure‐response relationship.
Only time to first event, if occurring after first dose, was considered in the analysis. Time (days) to first event or censoring time was included, with censoring time set to date of common study end visit, patient's last assessment (or death), or the first of either 3 days after first study interruption or 3 days after final dose. Event time was set to the difference between the event day and the day subsequent to the first dose.
Hazard (h) was modeled over time (t) as:
where is the coefficient describing effect of risk factor and f() is the exposure‐response relationship (e.g., linear, power, Emax, or sigmoidal Emax type function); C(t) is the time‐varying PK exposure index; and is the baseline hazard. was modeled as for dichotomous risk factors and change from the median risk factor for continuous risk factors. was parameterized as:
where and are the scale and shape factor of the Weibull distribution, respectively, and γ fixed to 1 for an exponential distribution.
In the case of a Gompertz distribution, was parameterized as:
where and are the scale and shape factor of the Gompertz distribution, respectively.
Hazard ratios (HRs) were calculated to compare log‐linear risk factor effects on cumulative risk or event probability. For dichotomous risk factors, HRs represent change in risk for patients with the factor compared with patients without the factor. For continuous risk factors, the HR quantified change in risk/probability of an event for every unit of the risk factor. CRCL was investigated as an additional effect (sensitivity) on the exposure‐response parameter (i.e., proportional to EC50 in the Emax model or proportional to slope in a linear model). Methods used to calculate predicted event probability are described in the Supplementary Material.
Clinical utility index
The balance between efficacy and safety was calculated using:
where n is the number of endpoints, wi is weight for endpoint i and pi(…) represents event probability up to time T (i.e., the cumulative risk at time T) for corresponding endpoint i.
The efficacy vs. safety pair used in the clinical utility index (CUI) assessment was VTE vs. clinically relevant bleeding. Three different weights of 1:1, 2:1, and 1:2 were tested and cumulative risks were calculated at 1 year (365 days). The CUI was investigated over the range of PK exposures observed, and two dose subgroups (full dose of edoxaban 60 mg and reduced dose of edoxaban 30 mg).
Model selection was based on comparison of OFV between nested models, precision in parameter estimates, and scientific plausibility. A difference in OFV ≥3.84 is statistically significant at P ≤ 0.05 for 1 degree of freedom. Performance of TTE models was evaluated by visual predictive checks, the Kaplan–Meier curve of the observed data, plus a 95% prediction interval of simulated data from the TTE model with 100 replicates.
Dataset preparation, data exploration, graphical analyses, modeling, and simulations were performed using the software packages R (version 2.15.1, The R Foundation for Statistical Computing), NONMEM (version 7.2, ICON Development),14 Xpose (version 4.3.5, Uppsala University, Sweden),15 and PsN (version 3.5.8, Uppsala University, Sweden).12, 13
RESULTS
A total of 4,122 patients randomized to warfarin and receiving at least one dose were included in the risk factor dataset. A total of 4,118 patients randomized to edoxaban and receiving at least one dose were included in the edoxaban exposure‐response dataset. Approximately 17.8% of edoxaban‐treated patients received a 50% dose reduction, according to protocol‐defined criteria. Median time on study drug for warfarin and edoxaban was 250 and 248 days, respectively. Tables 1 and 2 summarize observed events related to efficacy and safety endpoints.
Table 1.
Observed events related to efficacy endpoints
| Treatment group | Recurrent VTE | Recurrent DVT + nonfatal PE | Recurrent DVT + nonfatal PE + all‐cause mortality | ||||
|---|---|---|---|---|---|---|---|
| Total no. of patients | No. of first events | % of events | No. of first events | % of events | No. of first events | % of events | |
| Warfarin | 4,122 | 80 | 1.94 | 71 | 1.72 | 100 | 2.43 |
| Edoxaban | |||||||
| 60 mg full dose | 3,385 | 53 | 1.57 | 46 | 1.36 | 66 | 1.95 |
| 30 mg reduced dose | 733 | 13 | 1.77 | 9 | 1.23 | 18 | 2.46 |
DVT, deep vein thrombosis; PE, pulmonary embolism; VTE, venous thromboembolism.
Table 2.
Observed events related to safety endpoints
| Treatment group | Clinically relevant bleeding | All death | MACE | ||||
|---|---|---|---|---|---|---|---|
| Total no. of patients | No. of first events | % of events | No. of first events | % of events | No. of first events | % of events | |
| Warfarin | 4,122 | 422 | 10.2 | 31 | 0.752 | 40 | 0.970 |
| Edoxaban | |||||||
| 60 mg full dose | 3,385 | 291 | 8.60 | 22 | 0.650 | 36 | 1.06 |
| 30 mg reduced dose | 733 | 58 | 7.91 | 9 | 1.23 | 12 | 1.64 |
MACE, major adverse cardiovascular events, defined as a composite of nonfatal myocardial infarction, nonfatal stroke, nonfatal systemic embolic events, and cardiovascular death.
Parameter estimates of final warfarin risk factor and edoxaban exposure‐response models for efficacy outcomes are presented in Table 3; corresponding model parameter estimates for safety outcomes are presented in Table 4. Overall, visual predictive check suggest each final endpoint model sufficiently predicted TTE data (Figure 1).
Table 3.
Final risk factor and exposure‐response models for efficacy endpoints
| Final warfarin risk factor model a | Final edoxaban exposure‐response model b | ||||
|---|---|---|---|---|---|
| Parameter | Estimate [90% CI] c | HR | Estimate [90% CI] c | HR | |
| Recurrent DVT | |||||
| λ [day−1] | 2.99·10−4 [2.18·10−4, 3.80·10−4] | ‐ | 2.62·10−6 [‐4.26·10−6, 9.50·10−6] | ‐ | |
| γ | −0.0137 [0.0169, −0.0105] | ‐ | 0.373 [0.297, 0.449] | ‐ | |
| βL,ER (Cav) [(ng/mL)−1] | ‐ | ‐ | −0.0218 [−0.0345, −0.00915] | 0.980 d | |
| Recurrent DVT + nonfatal PE | |||||
| λ [day−1] | 2.82·10−4 [2.02·10−4, 3.63·10−4] | ‐ | 2.302·10−7 [−6.83·10−7, 1.14·10−6] | ‐ | |
| γ | −0.0147 [−0.0182, −0.0112] | ‐ | 0.3576 [0.273, 0.443] | ‐ | |
| βL,ER (Cav) [(ng/mL)−1] | ‐ | ‐ | −0.01364 [−0.0303, 3.04·10−3] | 0.987 d | |
| βCRCL,ER | ‐ | ‐ | −0.00905 [−0.0244, 6.26·10−3] | ||
| Recurrent DVT + nonfatal PE + all‐cause mortality | |||||
| λ [day−1] | 3.65·10−4 [2.77·10−4, 4.53·10−4] | ‐ | 3.21·10−6 [−4.00·10−6, 1.04·10−5] | ‐ | |
| γ | −0.0133 [−0.0161, −0.0105] | ‐ | 0.398 [0.327, 0.469] | ‐ | |
| βL,ER (Cav) [(ng/mL)−1] | ‐ | ‐ | −0.0161 [−0.0276, −4.68·10−3] | 0.980 d | |
λ, scale factor of the exponential, Weibull, or Gompertz distribution; γ, shape factor of the Weibull or Gompertz distribution; β, coefficient describing risk factor; CI, confidence interval; CRCL, creatinine clearance; DVT, deep vein thrombosis; ER, exposure response; HR, hazard ratio; PE, pulmonary embolism; VTE, venous thromboembolism.
aBased on warfarin patients.
bBased on edoxaban patients.
cEstimates of risk factor effects parameterized as log hazard ratio, and CI obtained from the observed Fisher information matrix.
dDecrease in the hazard ratio with every 1 ng/mL of Cav of edoxaban.
Table 4.
Final risk factor and exposure‐response models for safety endpoints
| Final warfarin risk factor model a | Final edoxaban exposure‐response model b | ||||
|---|---|---|---|---|---|
| Parameter | Estimate [90% CI] c | HR | Estimate [90% CI] c | HR | |
| Clinically relevant bleeding | |||||
| λ [day−1] | 3.24·10−5 [1.83·10−5, 4.65·10−5] | ‐ | 3.05·10−5 [1.62·10−5, 4.48·10−5] | ‐ | |
| γ | 0.553 [0.511, 0.595] | ‐ | 0.629 [0.577, 0.681] | ‐ | |
| β AGE75 | 0.320 [0.110, 0.531] | 1.38 | 0.305 [0.076, 0.534] | 1.36 | |
| β ASA | 0.458 [0.244, 0.672] | 1.58 | 0.762 [0.547, 0.978] | 2.14 | |
| β CAN | 0.474 [0.216, 0.732] | 1.61 | 0.110 [−0.222, 0.442] | 1.12 | |
| β PUL | 0.465 [0.278, 0.653] | 1.59 | 0.168 [−0.0557, 0.392] | 1.18 | |
| β RCAN | 0.612 [0.206, 1.02] | 1.84 | 0.794 [0.309, 1.28] | 2.21 | |
| β SUR | 0.317 [0.123, 0.511] | 1.37 | 0.395 [0.186, 0.604] | 1.48 | |
| β SEX | 0.244 [0.0815, 0.407] | 1.28 | 0.678 [0.497, 0.859] | 1.97 | |
| All death | |||||
| λ [day−1] | 8.605·10−5 [4.703·10−5, 1.251·10−4] | ‐ | 3.15·10−5 [−6.37·10−6, 1.27·10−5] | ‐ | |
| γ | −0.01059 [−0.01500, −0.006177] | ‐ | 0.458 [0.327, 0.590] | ‐ | |
| βRCAN | 1.619 [0.6194, 2.619] | 5.048 | 1.49 [0.485, 2.49] | 4.42 | |
| βL,ER (Cmax) [(ng/mL)−1] | ‐ | ‐ | −0.00865 [−0.0139, −0.00337] | 0.991 d | |
| MACE | |||||
| λ [day−1] | 3.44·10−5 [2.22·10−5, 4.67·10−5] | ‐ | 4.16·10−6 [−2.435·10−6, 1.08·10−5] | ‐ | |
| γ | 0.685 [0.531, 0.840] | ‐ | |||
| βAGE75 | 0.9363 [0.327, 1.55] | 2.551 | 1.25 [0.747, 1.75] | 3.49 | |
| βCAN | 1.10 [0.484, 1.72] | 3.016 | ‐ | ‐ | |
| β SEX | −0.673 [−1.25, −0.0973] | 0.510 | ‐ | ‐ | |
λ, scale factor of the exponential, Weibull, or Gompertz distribution; γ, shape factor of the Weibull or Gompertz distribution; β, coefficient describing risk factor; AGE75, age ≥75 at baseline; ASA, concomitant use of aspirin or antiplatelet agent; CAN, history of cancer; CI, confidence interval; ER, exposure response; HR, hazard ratio; PUL, history of pulmonary disease; RCAN, recent active cancer; SEX, female gender; SUR, recent surgery, trauma, or immobilization.
aBased on warfarin patients.
bBased on edoxaban patients.
cEstimates of risk factor effects parameterized as log hazard ratio, and CI obtained from the observed Fisher information matrix.
dDecrease in the hazard ratio with every 1 ng/ml of Cmax of edoxaban.
Figure 1.
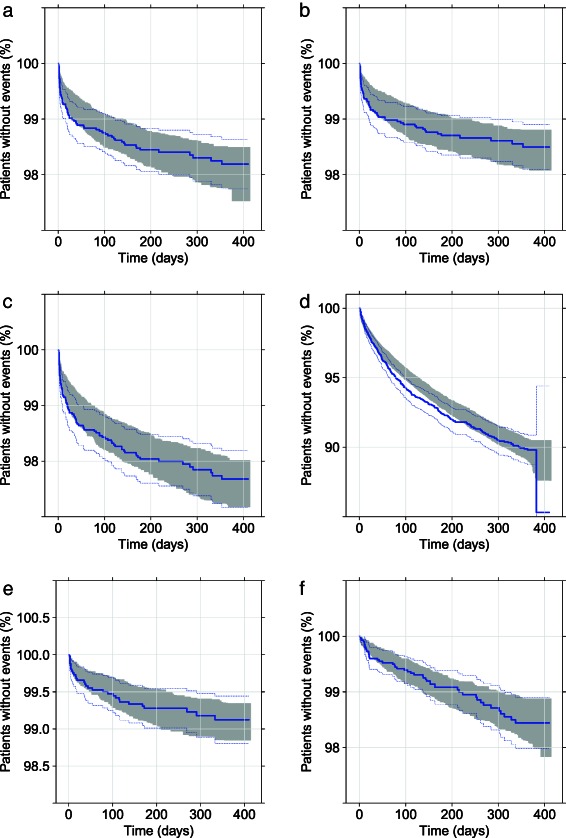
Visual predictive check of edoxaban final exposure‐response model (a) recurrent venous thromboembolism (VTE); (b) composite of deep vein thrombosis (DVT) and nonfatal pulmonary embolism (PE); (c) composite of DVT, nonfatal PE, and all‐cause mortality; (d) clinically relevant bleeding; (e) all death; and (f) major adverse clinical event (MACE). Solid line represents the Kaplan–Meier estimate of the observed data, dotted line represents the 95% confidence intervals of the observed data, and the shaded area represents the 95% prediction intervals using the final exposure‐response model for each endpoint.
Exposure‐efficacy relationship
A Gompertz distribution described TTE data well for the recurrent VTE warfarin risk factor model and provided a lower OFV compared with exponential and Weibull distributions (79.8 and 11.1 points, respectively). The final recurrent VTE warfarin risk factor model did not include any statistically significant risk factors (Table 3). When applying the final recurrent VTE risk factor model, including Gompertz distribution to only edoxaban data, the model fit to the early time points was unsatisfactory, and a Weibull distribution better described the data (∼24.2‐point drop in OFV). The final edoxaban‐recurrent VTE exposure‐response model included a linear relationship driven by average concentration over a dosing interval (Cav), with no statistically significant risk factors (Table 3). Predicted probability of recurrent VTE within 1 year decreased with increasing edoxaban Cav (Figure 2 a). In a typical edoxaban patient with edoxaban Cav of 36.7, 64.9, and 97.6 ng/mL (representing 2.5th, 50th, and 97.5th percentile of Cav in edoxaban‐treated patients), the predicted probability of recurrent VTE within 1 year was 3.31%, 1.80%, and 0.89%, respectively.
Figure 2.
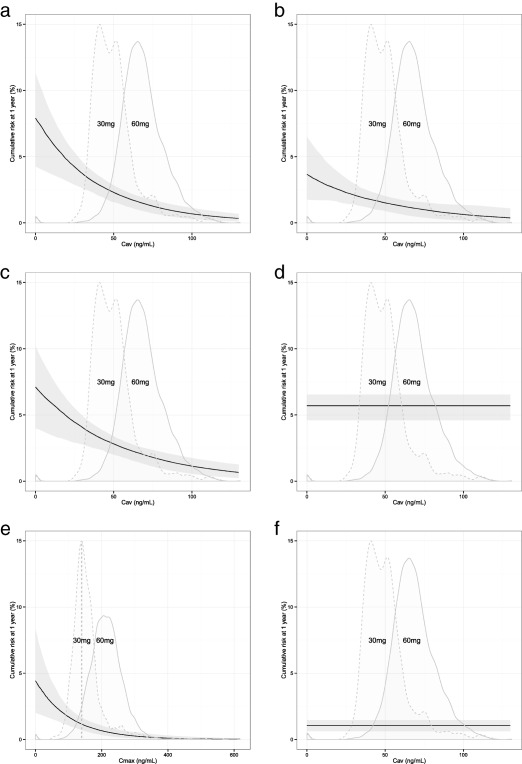
Probability of an event within 1 year/cumulative risk at 1 year vs. edoxaban exposure in a typical edoxaban patient with no risk factors (a) recurrent venous thromboembolism (VTE); (b) composite of deep vein thrombosis (DVT) and nonfatal pulmonary embolism (PE); (c) composite of DVT, nonfatal PE, and all‐cause mortality; (d) clinically relevant bleeding; (e) all death; and (f) major adverse clinical event (MACE). Solid line represents the median model prediction given the model parameters. Gray shaded area indicates the 90% confidence interval given the uncertainty in the model parameters. Distributions represent the predicted average concentration (Cav) or maximum/peak concentration (Cmax) in the full‐dose 60 mg (solid lines) and reduced‐dose 30 mg (dashed lines) groups. The distributions (densities) were adjusted to fit the graph for the purpose of illustration.
A Gompertz distribution described the TTE of a composite of recurrent DVT and nonfatal PE (DVT + nonfatal PE) well in warfarin‐treated patients, and provided a lower OFV compared with exponential and Weibull distributions (ΔOFV = −77.6 and −11.6, respectively). No statistically significant risk factor was included in the final DVT + nonfatal PE risk factor model for warfarin (Table 3). A Weibull distribution was a statistically significantly better fit to the edoxaban data, and reduced OFV by 19.7 points compared with the Gompertz distribution. Edoxaban Cav (and implicitly AUC0‐24,ss) were statistically significant using linear relationships descriptors between exposure and DVT + nonfatal PE TTE. Additionally, CRCL had a statistically significant influence on the sensitivity of the exposure‐response relationship (ΔOFV = −4.13). The final DVT + nonfatal PE exposure‐response model was a Weibull distribution with a linear relationship driven by Cav, including CRCL as an additional sensitivity factor on the slope of the linear relationship (Table 3). Figure 2 b shows the predicted risk of DVT + nonfatal PE with relation to edoxaban Cav. In a typical edoxaban patient with edoxaban Cav of 36.7, 64.9, and 97.6 ng/mL (representing the 2.5th, 50th, and 97.5th percentile of Cav in edoxaban‐treated patients), predicted probability of a composite event of recurrent DVT and nonfatal PE within 1 year was 2.09%, 1.43%, and 0.92%, respectively.
Final risk factor and exposure‐response models selected for the composite outcome of recurrent DVT, nonfatal PE, and all‐cause mortality (DVT + nonfatal PE + all‐cause mortality) were similar to models developed for recurrent VTE TTE (Table 3). The risk of DVT + nonfatal PE + all‐cause mortality decreased with increasing edoxaban Cav (Figure 2 c). In a typical edoxaban patient with edoxaban Cav of 36.7, 64.9, and 97.6 ng/mL (representing the 2.5th, 50th, and 97.5th percentile of Cav in edoxaban‐treated patients), predicted probability of DVT + nonfatal PE + all‐cause mortality within 1 year was 3.70%, 2.36%, and 1.40%, respectively.
Exposure‐safety relationship
A Weibull distribution provided a lower OFV (ΔOFV = −217) compared with the exponential distribution for the TTE of clinically relevant bleeding in warfarin‐treated patients. Seven statistically significant risk factors were identified: age ≥75 years (AGE75); female gender (SEX); concomitant use of aspirin or antiplatelet agent (ASA); history of cancer (CAN); history of pulmonary disease (PUL); recent active cancer (RCAN); and recent surgery, trauma, or immobilization (SUR) (Table 4).
No PK exposure indices were statistically significant descriptors of TTE of clinically relevant bleeding in edoxaban‐treated patients. All risk factors identified in the warfarin risk factor model were statistically significant when applied to edoxaban patient data. The predicted probability of clinically relevant bleeding within 1 year was 5.74% in a typical edoxaban patient without any risk factors. Patients with AGE75 had a 36% higher risk of a clinically relevant bleeding event than those aged <75 years; female patients had a 97% higher risk compared with male patients. Concomitant use of ASA, CAN, PUL, RCAN, and SUR increased the risk by 114%, 12%, 18%, 121%, and 48%, respectively (Table 4).
A Gompertz distribution described well the TTE of all death in warfarin‐treated patients and provided a lower OFV compared with exponential and Weibull distributions (ΔOFV = −20.1 and ΔOFV = −2.19 points, respectively). The risk factor RCAN was statistically significant and included in the final warfarin model (Table 4).
At early time points, a Weibull distribution fit edoxaban patient data better than the Gompertz distribution, decreasing OFV by 5.31 points. Among the PK exposure metrics evaluated, the Cmax was a statistically significant descriptor of TTE of all death for a linear exposure‐response relationship. The risk factor RCAN remained statistically significant in edoxaban patients. The final exposure‐response model for edoxaban patients was a Weibull distribution with a linear exposure‐response relationship driven by Cmax, including RCAN as a risk factor (Table 4). Risk of an all‐death event decreased with increasing edoxaban Cmax (Figure 2 e). In a typical edoxaban patient with edoxaban Cmax of 107, 203, and 311 ng/mL (representing the 2.5th, 50th, and 97.5th percentile of Cmax, respectively, in edoxaban‐treated patients) and no other risk factors, predicted probability of an all‐death event within 1 year was 1.76%, 0.78%, 0.30%, respectively. The predicted risk was 442% higher in a patient with RCAN compared to a similar patient without recent active cancer.
Weibull or exponential distributions produced similar OFVs (OFVs differ by 1.70) when applied to major adverse cardiovascular events (defined as a composite of nonfatal myocardial infarction, nonfatal stroke, nonfatal systemic embolic events, and cardiovascular death [MACE]), TTE, and warfarin patient data. An exponential distribution was selected to describe TTE of MACE because it had one fewer parameter than the Weibull distribution. Risk factors AGE75, SEX, and CAN were statistically significant and included in the final warfarin risk factor model (Table 4).
An exponential distribution did not satisfactorily fit edoxaban patient data. A Weibull distribution better described the TTE of MACE data in edoxaban‐treated patients (9.01 points lower OFV). No PK exposure indices were statistically significant descriptors of MACE TTE. Among the three risk factors identified in the warfarin risk factor model, only AGE75 remained statistically significant using edoxaban patient data. The predicted probability of MACE within 1 year was 1.17% in edoxaban‐treated patients with AGE75, and increased by 349% in patients with AGE75 (Table 4).
CUI assessment
CUI was assessed to balance safety and efficacy using the efficacy endpoint recurrent VTE and the safety endpoint clinically relevant bleeding. CUI was calculated using the final exposure‐response model for recurrent VTE, including a linear relationship with edoxaban Cav. As no statistically significant exposure‐response relationship was identified in the final edoxaban clinically relevant bleeding model, a linear relationship with Cav was used in the CUI evaluation. The re‐estimated model with a linear Cav relationship is presented in Supplementary Table S1 and Supplementary Figure S1.
Figure 3 a shows the CUI prediction in a typical edoxaban patient with no risk factors, assuming three different approaches for weighting efficacy and safety event probability. Higher efficacy weight (i.e., changing weight from 1:1 to 2:1) rewarded higher PK exposures (i.e., higher dose), whereas higher safety weight (i.e., changing weight from 1:1 to 1:2) favored lower PK exposures (i.e., lower dose). Overall, CUIs were lower for the 60 mg dose group compared with the 30 mg dose‐reduced group for all three weights. Figure 3 b shows the CUI prediction in a typical edoxaban patient with different risk factors, assuming a 1:1 weight. As expected, CUI values varied among different risk factors. Overall event risk (VTE and clinically relevant bleeding) was pronounced in patients with RCAN, concomitant use of ASA, or who were women. However, the CUI curve shape appeared similar, and edoxaban Cav corresponding to CUI nadir did not vary greatly among various risk factors.
Figure 3.
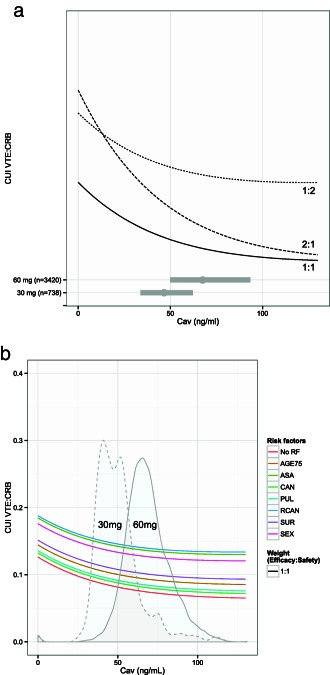
Clinical utility index (CUI) prediction based on cumulative risk of recurrent venous thromboembolism (VTE) and clinically relevant bleeding at 1 year. (a) In a typical edoxaban patient with no risk factors and for three clinical weights of 1:1, 2:1, and 1:2. Horizontal bars represent the 90% prediction interval of edoxaban average concentration (Cav) in patients with 60 mg or 30 mg. The point represents the median edoxaban Cav exposure. (b) In a typical edoxaban patient stratified by one risk factor and for the clinical weight of 1:1. The figure is stratified by no risk factors (No RF); AGE75, age ≥ 75 at baseline; ASA, concomitant use of aspirin or antiplatelet agent; CAN, history of cancer; PUL, history of pulmonary disease; RCAN, recent active cancer; SEX, female gender; or SUR, recent surgery, trauma, or immobilization. The distributions represent the predicted edoxaban Cav in 60 mg (solid lines) and 30 mg (dashed lines) subgroups. The distributions (densities) were adjusted to fit the graph for purpose of illustration.
DISCUSSION
A statistically significant exposure‐response relationship was identified for all three efficacy endpoints—recurrent VTE; a composite of recurrent DVT and nonfatal PE; or a composite of recurrent DVT, nonfatal PE, and all‐cause mortality. The predicted probability of an event related to these endpoints decreased with increasing edoxaban Cav. However, no evaluated risk factor was statistically significant, likely due to low numbers of patients and events in each risk factor subgroup.
No statistically significant exposure‐response relationship was found for clinically relevant bleeding in this analysis, although a number of risk factors were statistically significant. Not identifying an exposure‐response relationship may have several causes out of which a narrow PK exposure range may be likely because a statistically significant exposure‐response relationship for major bleeding was found in nonvalvular atrial fibrillation patients treated with edoxaban (the ENGAGE study, with two dose regimens; 60 mg with dose adjustments to 30 mg and 30 mg with dose adjustments to 15 mg).16 However, other differences between the ENGAGE and Hokusai‐VTE studies, like patient populations, sample size, frequency of events, study length, and study interruptions, may also play a role for the disagreement in identifying an exposure‐response relationship.
Based on the final model, the predicted probability of a clinically relevant bleeding event within 1 year was 5.73% in a typical edoxaban patient without any risk factors. Predicted risk was markedly increased—by 97%, 121%, and 114%, respectively—in female patients, patients with RCAN, or patients with concomitant ASA. Additionally, patients with AGE≥75 had a 36% higher risk of clinically relevant bleeding than those aged <75 years. Factors CAN, PUL, and recent surgery increased risk by 12%, 18%, and 48%, respectively. Identified risk factors were generally consistent with current knowledge. Several VTE observational studies using warfarin or nonvitamin K antagonist oral anticoagulants reported SEX is an independent predictor of bleeding.17, 18, 19 Major bleeding risk in anticoagulated patients was 2.5% per year in those aged >80 years compared with 0.9% per year in younger patients.17, 20 In a phase II study, coadministration of low‐dose ASA (100 mg), high‐dose ASA (325 mg), and naproxen with edoxaban increased bleeding time.21 Patients with cancer also experience high risk of anticoagulant‐associated major bleeding.22, 23
Similar to the TTE of clinically relevant bleeding, no statistically significant exposure‐response relationship was identified for MACE. This warrants cautious interpretation and can be attributed to various factors. For example, a narrow PK exposure range may limit identification of a statistically significant exposure‐response relationship. A very strong correlation between the event probability (i.e., clinically relevant bleeding or MACE) and risk factor may also mask a statistically significant relationship between event probability and PK exposure.
Evaluation of CUIs should consider relative clinical weights of probabilities of efficacy and safety outcomes. For a safety emphasis, the nadir benefit/risk CUI will tend to favor lower exposures. For an efficacy emphasis, the nadir benefit/risk CUI will tend to favor higher exposures. The weights of efficacy to safety of 1:1, 2:1, and 1:2 used in this analysis were subjective and intended to provide scenarios for further interpretation using clinical judgment. In this analysis, the evaluated CUI favored exposures in the upper end of the observed distribution, especially in the dose adjusted patients, because the identified exposure‐response relationship for clinically relevant bleeding was flat and the exposure‐response relationship for recurrent VTE was steeper in the 30 mg exposure region compared to the 60 mg exposure region. Thus, based solely on this analysis/evidence, a higher dose would be advocated/envisaged. However, the primary statistical analysis showed satisfactory outcome for the dose regimen in the VTE patient population.9 The studied dose regimen (edoxaban 60 mg dose adjusted to 30 mg) showed superiority vs. warfarin for clinically relevant bleeding and noninferiority for recurrent VTE in the primary statistical analysis. In addition, the subgroups analyses showed a similar efficacy between warfarin and edoxaban while significantly less bleeding using edoxaban in patients with and without dose adjustments.9 As discussed above, there is uncertainty in the flat exposure‐response relationship observed for clinically relevant bleeding, thus explaining the difference to the primary analysis. Additionally, other clinical factors not included in the exposure‐response models will impact the choice of dose.
In Hokusai‐VTE, ∼18% of patients received a 50% dose reduction according to protocol‐defined criteria (i.e., moderate renal impairment, body weight ≤60 kg, or concurrent treatment with potent P‐gp inhibitors); the majority of those patients had moderate renal impairment.9 As a very small number of patients received a reduced edoxaban dose for the other criteria, the current analysis was conducted for the entire reduced‐dose group. Patients receiving reduced‐dose edoxaban 30 mg once daily had ∼30% lower Cav compared with patients receiving full‐dose edoxaban 60 mg once daily (median Cav: 47 ng/mL vs. 67 ng/mL).11 The observed clinically relevant bleeding rate was lower in the 30 mg reduced‐dose group than the 60 mg dose group (7.91% vs. 8.60%), although no statistically significant exposure‐response relationship was identified. The rate of recurrent VTE was slightly higher in the 30 mg reduced‐dose group compared with the 60 mg dose group (1.77% vs. 1.57%), but efficacy was retained. As reported previously, both doses were noninferior to warfarin for prevention and treatment of VTE, and had statistically significant lower rates of bleeding than warfarin, according to intent‐to‐treat analysis.9 Further, within the edoxaban Cav range observed in Hokusai‐VTE, predicted CUI curves were generally flat toward increased or decreased Cav, suggesting marginal change in overall event risk (VTE and clinically relevant bleeding) with a change in edoxaban Cav, given the weighing of efficacy:safety of 1:1, 1:2, or 2:1. Therefore, modeling results aligned with clinical observations and support the recommendation of edoxaban 60 mg once daily for prevention and treatment of VTE in the general patient population, and a 50% dose reduction for patients with moderate renal impairment, body weight ≤60 kg, or concurrent treatment with the potent P‐gp inhibitors verapamil or quinidine. An alternative to the label dosing would be to identify a target exposure and adjust doses based on PK covariates and/or risk factors. This approach might be advantageous but at the same time problematic in how to identify the target exposure. As for CUI, clinical judgment in balancing safety and efficacy might also be debatable. Given all together, we believe that, although perhaps not optimal for all patients, the studied doses are supported in this work. This is also confirmed by the statistically analysis that showed noninferiority for efficacy and superiority for safety.9
These results also provide useful information for appropriate clinical management of patient subgroups receiving edoxaban therapy. Risk factors identified for the principal safety endpoint (clinically relevant bleeding) were generally consistent with current knowledge or clinical reports. Based on CUI assessment, edoxaban Cav corresponding to CUI nadir does not vary greatly among various risk factors, indicating a limited need for dose reduction according to risk factors. However, CUI values vary among different risk factors, and overall event risk (VTE and clinically relevant bleeding) were pronounced in patients with RCAN, concomitant use of ASA, or who were women. This information may be useful in treating and monitoring patients with specific risk factors.
Because of the large dataset and long run times, simultaneous estimation of PK and TTE was not performed. Instead, individual PK exposure indices (i.e., Cmax, minimum or trough concentration at steady state [Cmin], AUC0‐24, and Cav at steady state) were obtained based on individual PK parameters from a population PK analysis and used in subsequent exposure‐response modeling. Due to high η‐shrinkage (shrinkage toward typical parameters) in some population PK parameters, all predictions may have shrunken toward the population or typical predictions, and associated variability might be underestimated.11 However, this is expected to exert limited bias on the exposure‐response analysis, because individual PK parameter estimates vary with covariate effects (i.e., dose, age, body weight, renal function, and concomitant use of P‐gp inhibitors), which were accounted for in deriving individual PK exposure indices. Exposure indices that are dependent on absorption and volume of distribution (e.g., Cmax) might further be biased due to inaccurate sampling or dosing history when sampled close to an event. However, the numbers of samples close to events are relatively low compared to the total amount of samples. Moreover, Cmax has been shown in previous phase II edoxaban studies to be correlated with bleeding events which is another rationale for investigating this exposure index despite high potential shrinkage.
Warfarin patients were used as a placebo arm in this analysis with the argument that the warfarin patients were dose‐adjusted to have an international normalized ratio within two to three. The warfarin patients were in this international normalized ratio range 60% of the time. However, we believe that the impact of not being in the range 100% of the treated time is minor because the warfarin models were only used to characterize risk factors and the base line hazard was re‐evaluated in the edoxaban arm. Moreover, the risk factor model was reassessed as a final step in the exposure‐response model development. Hence, any treatment effects that were masked by nonoptimal warfarin treatment are likely to be captured in these reassessments during the edoxaban arm model development.
CRCL was investigated as a sensitivity parameter on the exposure‐response relationship because it was indicated that rivaroxaban did have steeper PK/pharmacodynamic relationship between exposure and bleeding for renal impairment patients than accounted for in the exposure. However, even though CRCL was found as statistically significant for the endpoint recurrent DVT + nonfatal PE, the effect is quite modest compared to the Cav effect and was not found for the other efficacy endpoints.
In summary, TTE data related to Hokusai‐VTE efficacy and safety outcomes were described well overall by corresponding exposure‐response models. Statistically significant exposure‐response relationships were identified for all three efficacy endpoints—recurrent VTE; a composite of recurrent DVT and nonfatal PE; or a composite of recurrent DVT, nonfatal PE, and all‐cause mortality. A statistically significant exposure‐response relationship was also identified for the safety endpoint all death, but not clinically relevant bleeding or MACE. Exposure‐response analysis results, together with CUI assessment, support recommendations of edoxaban 60 mg once daily for prevention and treatment of VTE in the general patient population, and reduced doses of 30 mg in patients with moderate renal impairment, body weight ≤60 kg, or concurrent treatment with potent P‐gp inhibitors.
Study Highlights
WHAT IS THE CURRENT KNOWLEDGE ON THIS TOPIC?
In the large‐scale phase III Hokusai‐VTE study, the nonvitamin K antagonist oral anticoagulant edoxaban was noninferior to warfarin in preventing recurrent VTE and caused statistically significant less bleeding.
WHAT QUESTION DID THIS STUDY ADDRESS?
Potential relationships between edoxaban exposure and safety and efficacy endpoints in Hokusai‐VTE, associated risk factors influences, and the edoxaban efficacy/safety balance in patient subgroups were evaluated.
WHAT THIS STUDY ADDS TO OUR KNOWLEDGE
Event risk for recurrent VTE; composite recurrent DVT and nonfatal PE; or composite recurrent DVT, nonfatal PE, and all‐cause mortality decreased with increasing average steady state edoxaban concentration. All‐cause mortality, but not clinically relevant bleeding or major adverse cardiovascular events, had statistically significant exposure‐response relationships. Identified risk factors were consistent with clinical knowledge.
HOW THIS MIGHT CHANGE CLINICAL PHARMACOLOGY AND THERAPEUTICS
Recommendations of edoxaban 60 mg once daily for prevention and treatment of VTE in the general patient population and reduced‐dose edoxaban 30 mg in patients with moderate renal impairment, body weight ≤60 kg, or concomitant use of P‐glycoprotein inhibitors were supported.
Supporting information
Supporting Information S01
Supporting Information S02
Acknowledgments
Editorial support was provided by Terri Schochet, PhD, of AlphaBioCom, LLC, King of Prussia, PA, and funded by Daiichi Sankyo.
Conflict of Interest
The study and analysis were sponsored by Daiichi Sankyo Pharma Development. Mats Karlsson and Ulrika Simonsson have obtained consultancy fees from Daiichi Sankyo Pharma Development. Raymond Miller and Ophelia Yin are employees of Daiichi Sankyo Pharma Development. The other authors have no conflicts of interest to declare.
Author Contributions
J.N., O.Q.P.Y., and U.S. wrote the manuscript. J.N. performed the research. J.N., K.E.K., S.J., R.M., M.O.K., and U.S. analyzed the data.
References
- 1. Verhamme, P. & Bounameaux, H. Direct oral anticoagulants for acute venous thromboembolism: closing the circle? Circulation 129, 725–727 (2014). [DOI] [PubMed] [Google Scholar]
- 2. Ahrens, I. , Lip, G.Y. & Peter, K. New oral anticoagulant drugs in cardiovascular disease. Thromb. Haemost. 104, 49–60 (2010). [DOI] [PubMed] [Google Scholar]
- 3. Ansell, J. et al Pharmacology and management of the vitamin K antagonists: American College of Chest Physicians Evidence‐Based Clinical Practice Guidelines (8th Edition). Chest 133(6 Suppl), 160S–198S (2008). [DOI] [PubMed] [Google Scholar]
- 4. Furugohri, T. et al DU‐176b, a potent and orally active factor Xa inhibitor: in vitro and in vivo pharmacological profiles. J. Thromb. Haemost. 6, 1542–1549 (2008). [DOI] [PubMed] [Google Scholar]
- 5. Matsushima, N. , Lee, F. , Sato, T. , Weiss, D. & Mendell, J. Bioavailability and safety of the factor Xa inhibitor edoxaban and the effects of quinidine in healthy subjects. Clin. Pharmacol. Drug. Dev. 2, 358–366 (2013). [DOI] [PubMed] [Google Scholar]
- 6. Bathala, M.S. , Masumoto, H. , Oguma, T. , He, L. , Lowrie, C. & Mendell, J. Pharmacokinetics, biotransformation, and mass balance of edoxaban, a selective, direct factor Xa inhibitor, in humans. Drug Metab. Dispos. 40, 2250–2255 (2012). [DOI] [PubMed] [Google Scholar]
- 7. Mikkaichi, T. et al Edoxaban transport via P‐glycoprotein is a key factor for the drug's disposition. Drug Metab. Dispos. 42, 520–528 (2014). [DOI] [PubMed] [Google Scholar]
- 8. Mendell, J. et al Drug‐drug interaction studies of cardiovascular drugs involving P‐glycoprotein, an efflux transporter, on the pharmacokinetics of edoxaban, an oral factor Xa inhibitor. Am. J. Cardiovasc. Drugs 13, 331–342 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hokusai‐VTE Investigators et al Edoxaban versus warfarin for the treatment of symptomatic venous thromboembolism. N. Engl. J. Med. 369, 1406–1415 (2013). [DOI] [PubMed] [Google Scholar]
- 10. Jönsson, S. , Simonsson, U.S. , Miller, R. & Karlsson, M.O. Population pharmacokinetics of edoxaban and its main metabolite in a dedicated renal impairment study. J. Clin. Pharmacol. 55, 1268–1279 (2015). [DOI] [PubMed] [Google Scholar]
- 11. Niebecker, R. et al Population pharmacokinetics of edoxaban in patients with symptomatic deep‐vein thrombosis and/or pulmonary embolism – The Hokusai‐VTE phase 3 study. Br. J. Clin. Pharmacol. 80, 1374–1387 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lindbom, L. , Pihlgren, P. & Jonsson, E.N. PsN‐Toolkit–a collection of computer intensive statistical methods for non‐linear mixed effect modeling using NONMEM. Comput. Methods Programs Biomed. 79, 241–257 (2005). [DOI] [PubMed] [Google Scholar]
- 13. Lindbom, L. , Ribbing, J. & Jonsson, E.N. Perl‐speaks‐NONMEM (PsN)–a Perl module for NONMEM related programming. Comput. Methods Programs Biomed. 75, 85–94 (2004). [DOI] [PubMed] [Google Scholar]
- 14. Beal, S.L. & Sheiner, L.B. NONMEM Users' Guide. NONMEM Project Group, University of California at San Francisco, San Francisco, CA: (1998). [Google Scholar]
- 15. Jonsson, E.N. & Karlsson, M.O. Xpose–an S‐PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput. Methods Programs Biomed. 58, 51–64 (1999). [DOI] [PubMed] [Google Scholar]
- 16. Ruff, C.T. et al Association between edoxaban dose, concentration, anti‐Factor Xa activity, and outcomes: an analysis of data from the randomised, double‐blind ENGAGE AF‐TIMI 48 trial. Lancet 385, 2288–2295 (2015). [DOI] [PubMed] [Google Scholar]
- 17. Kuijer, P.M. , Hutten, B.A. , Prins, M.H. & Büller, H.R. Prediction of the risk of bleeding during anticoagulant treatment for venous thromboembolism. Arch. Intern. Med. 159, 457–460 (1999). [DOI] [PubMed] [Google Scholar]
- 18. Alotaibi G.S., Almodaimegh H., McMurtry M.S. & Wu, C. Do women bleed more than men when prescribed novel oral anticoagulants for venous thromboembolism? A sex‐based meta‐analysis. Thromb. Res. 132, 185–189 (2013). [DOI] [PubMed] [Google Scholar]
- 19. Bauersachs, R.M. Use of anticoagulants in elderly patients. Thromb. Res. 129, 107–115 (2012). [DOI] [PubMed] [Google Scholar]
- 20. Geldhof, V. , Vandenbriele, C. , Verhamme, P. & Vanassche, T. Venous thromboembolism in the elderly: efficacy and safety in non‐VKA oral anticoagulants. Thromb. J. 12, 21–30 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mendell, J. , Lee, F. , Chen, S. , Worland, V. , Shi, M. & Samama, M.M. The effects of the antiplatelet agents, aspirin and naproxen, on pharmacokinetics and pharmacodynamics of the anticoagulant edoxaban, a direct factor Xa inhibitor. J. Cardiovasc. Pharmacol. 62, 212–221 (2013). [DOI] [PubMed] [Google Scholar]
- 22. Prandoni, P. et al Recurrent venous thromboembolism and bleeding complications during anticoagulant treatment in patients with cancer and venous thrombosis. Blood 100, 3484–3488 (2002). [DOI] [PubMed] [Google Scholar]
- 23. Ruíz–Giménez, N. et al Predictive variables for major bleeding events in patients presenting with documented acute venous thromboembolism. Findings from the RIETE Registry. Thromb. Haemost. 100, 26–31 (2008). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information S01
Supporting Information S02