

Abstract
Introduction
Platinum (Pt)-based antitumor agents remain important chemotherapeutic agents for treating many human malignancies. Elevated expression of the human high-affinity copper transporter 1 (hCtr1), resulting in enhanced Pt drug transport into cells, has been shown to be associated with improved treatment efficacy. Thus, targeting hCtr1 upregulation is an attractive strategy for improving the treatment efficacy of Pt-based cancer chemotherapy.
Area covered
Regulation of hCtr1 expression by cellular copper homeostasis is discussed. Association of elevated hCtr1 expression with intrinsic sensitivity of ovarian cancer to Pt drugs is presented. Mechanism of copper-lowering agents in enhancing hCtr1-mediated cis-diamminedichloroplatinum (II) (cisplatin, cDDP) transport is reviewed. Applications of copper chelation strategy in overcoming cDDP resistance through enhanced hCtr1 expression are evaluated.
Expert opinion
While both transcriptional and posttranslational mechanisms of hCtr1 regulation by cellular copper bioavailability have been proposed, detailed molecular insights into hCtr1 regulation by copper homeostasis remain needed. Recent clinical study using a copper-lowering agent in enhancing hCtr1-mediated drug transport has achieved incremental improvement in overcoming Pt drug resistance. Further improvements in identifying predictive measures in the subpopulation of patients that can benefit from the treatment are needed.
Keywords: cisplatin, copper homeostasis, high-affinity copper transporter 1, hCtr1, Sp1, copper chelators, platinum drug cancer chemotherapy
1. Introduction
The platinum (Pt)-based antitumor drugs, including cDDP, carboplatin, and oxaliplatin, have been the mainstay for treating a broad spectrum of human malignancies over the last four decades [1, 2]. cDDP has particular effectiveness in treating metastatic testicular cancers, where cure rates of greater than 90% can be achieved [3]. Pt drugs are also effective in treating ovarian cancers with response rates of about 70% [4]. Other tumor types that have been effective include head and neck, bladder, lung, breast, and malignant mesothelioma [1]. The primary lethality target of Pt drugs is DNA by forming intra-stranded and inter-stranded crosslinking [5]. It has been reported that the extent of DNA damage is directly correlated with cell lethality, which is also directly correlated with cellular Pt drug contents [6, 7]. Indeed, defective drug transport systems resulting in reduced Pt accumulation have long been recognized as an important mechanism of resistance [8–13]. Andrews and Howell [14] pointed out that almost all cDDP-resistant variants are associated with various degrees of reduced Pt levels in comparison with their cDDP-sensitive counterparts, implicating that drug transport is a widespread mechanism of cDDP resistance, underscoring the importance of understanding cDDP transport mechanisms for targeted therapy.
2. Mechanisms of cDDP transport
Early works reported that cDDP enters cells by means of passive diffusion and independently of protein carrier or channel/transporter, and that transport is unsaturable and un-competetable by Pt analogues [7, 8]. Import transporter for cDDP was identified by using yeast genetic approach as the high-affinity copper transporter 1 (Ctr1). Deletion of yCtr1 resulted in reduced cellular copper (Cu) and cDDP accumulation [15, 16]. This was confirmed in mouse embryonic fibroblasts with deleted mCtr1 alleles (mCtr1 (−/−)) which exhibited a similar phenotype [15]. However, Ctr1 apparently does not account for the total Cu and cDDP transport, because mCtr1(−/−) cells show only 60 – 70% reductions of Cu [17] and cDDP [18] transport capacities compared with those in mCtr1(+/+) counterparts. It has been suggested that divalent metal transporter 1 (Dmt1) may also transport Cu [19, 20], however, this remains controversial [21] and further investigations is needed.
2.1. Roles of copper transport systems in cDDP transport
Despite substantial differences in the biochemical properties of Cu ions and cDDP, the mechanisms of hCtr1-mediated transport of these two substrates are quite similar. Human Ctr1 (hCtr1) is a 190-amino acid protein of approximately 23 kDa with three transmembrane domains. Biochemical and structural analyses have shown that a functional hCtr1 is composed of three monomeric subunits forming a channel-like configuration [22]. The two highly conserved motifs Met-(X)n-Met (where X can any other amino acids except methionine), one located at the N-terminal extracellular domain and the other at the second transmembrane domain, are essential for Cu and cDDP transport [23]. X-ray fine absorption spectroscopy showed that two Cu+1 ions bind to the N-terminus of hCtr1, each coordinated by three sulfur atoms [24]. Deletion/mutation manipulations of these regions also result in impaired cDDP transport [23, 25, 26].
Using the N-terminal hCtr1 sequences as model peptides, it was suggested that prior to entering a cell, cDDP (molecular formula: cis-[PtCl2(NH3)2]) is activated by interacting with the extracellular methionine-clusters of hCtr1, resulting in release of the carrier ligand [27–29] and formation of the [Pt(Met)Cl(NH3)2]+1 intermediate which, like Cu+, has one positive charge. Traverses of Cu+1 and cDDP through hCtr1 induce conformational changes of trimeric hCtr1 as probed by a protein crosslinking approach [23]. Site-directed mutations of hCtr1 suggest that more than one molecule of Cu+1 or Pt are concurrently associated with one hCtr1 trimer during transport [23]. It has been proposed that the activated cDDP may use methionine-based intermolecular sulfur-sulfur exchange along the axis of the trimeric hCtr1 to facilitate its transport [30], much like the mechanisms underlying Cu+1 or Ag+1 transport by the CusA efflux pump in bacteria [31].
Once Cu+1 has traversed through the membrane, it is carried away by various Cu chaperones to various cuproenzymes in different cellular compartments: to superoxide dismutase 1 in the cytosol by CCS, to mitochondrial cytochrome C oxidase by Cox17, and to two trans-Golgi P1B-type ATPases (ATP7A and ATP7B) by Atox1 for loading on ceruloplasmin, a secretory protein for Cu elimination. Recent studies demonstrated that both CCS [32] and Atox1 [33] interact with hCtr1, suggesting a direct transfer of Cu+1 from hCtr1 to these carrier proteins (Figs. 1B & 1C). Bindings between cDDP and Atox1 [34–36] and between cDDP and Cox17 [37] have been demonstrated. Give the direct contact between these chaperones and hCtr1, cDDP may be similarly carried to its cellular destinations, but this remains to be demonstrated. Inside the cells, cDDP is transformed into [Pt(NH3)2Cl(OH2)]+ and [Pt(NH3)2(OH2)2]2+ because chloride ligand is slowly displaced by water [5]. The aqua ligands in these complexes are readily displaceable, allowing Pt atom to interact with cellular targets, notably the guanine residue of DNA [Pt(NH3)2Cl(Guanine-DNA)]+ [38] to elicit its cellular lethality mechanisms. Clearly, mechanisms of Pt drugs transport and their biotransformation resulting in formation of Pt-DNA adducts are complex and required further investigations.
Figure 1. The shared import and export mechanisms of Cu and platinum drugs.
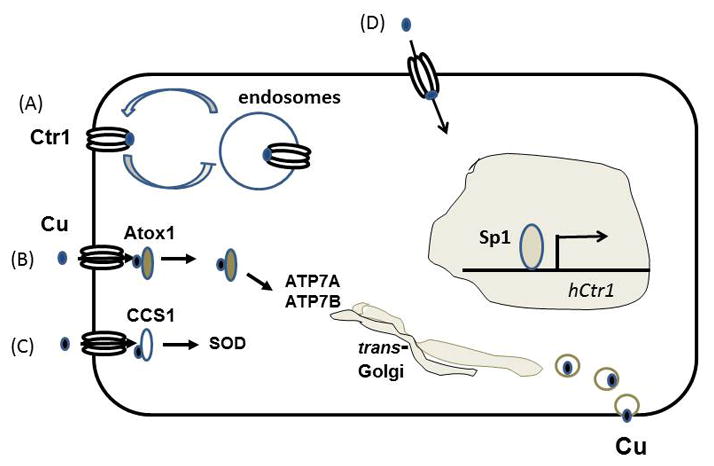
(A) hCtr1 transports Cu+1 by endocytosis, (B) The Cu chaperone Atox1 captures Cu by interacting with hCtr1. Cu is then transferred to ATP7A/ATP7B for eliminating by secretory vesicles. (C) The Cu chaperone CCS captures Cu by interacting with hCtr1. (D) hCtr1 may directly transport Cu into the cells.
Humans have a low-affinity Cu transporter, hCtr2 which has 143 amino acid residues that span through three transmembrane domains. In comparison with hCtr1, hCtr2 lacks the majority of the N-terminal ecto-sequence that is important for Cu+1 and cDDP transport but contains a M-X3-M sequence in the second transmembrane domain that is important for Cu transport. hCtr2 is located mainly in intracellular vesicles and has been proposed for Cu storage. mCtr2(−/−) mice display increased Cu accumulation in several organs compared with wild-type animals, demonstrating that mCtr2 plays a role in regulating systemic Cu distribution. At the cellular level, mCtr2(−/−) fibroblasts also elevated endosomal Cu accumulation. mCtr2 may interact with mCtr1 and causes formation of truncated Ctr1 lacking its ecto-domain containing Cu- and cDDP- binding motifs, but the mechanism of the proteolytic cleavage is not known [39]. Study also suggests that hCtr2 expression can also be modulated by Cu bioavailability and also plays a role in regulating cDDP sensitivity [40].
ATP7A and ATP7B are associated with Menke’s and Wilson’s diseases, respectively. Each contains eight transmembrane spannings and is organized into multiple functional domains involved in nucleotide-binding and catalytic ATP phosphorylation, trans-Golgi targeting and retention, and vesicle trafficking [41]. The N-termini of these ATPases contain six highly conserved CXXC motifs, each can bind one Cu+1. At low cellular Cu concentrations, Cu binding to these ATP7A and ATP7B induces transient catalytical phosphorylation thereby providing Cu+1 to cuproenzymes. At high Cu contents, Cu+1 binds to the metal-binding domain, inducing conformational changes and phosphorylation of several sites on the molecules by kinases. Hyper-phosphorylated ATPases then exit trans-Golgi and is present in endosomal/lysomal vesicles and trafficking to cell membrane where Cu is eliminated [41, 42]. Thus, both ATP7A and ATP7B play important roles in regulating cellular Cu homeostasis by eliminating excess Cu. The N-terminal CXXC motifs are also involved in Pt binding in cDDP treatments [43, 44]. However, whether the platination involves Atox1-bound cDDP is not clear. Moreover, whether elimination of cDDP by ATPases involves vesicle trafficking is not clear.
These observations collectively suggest that Cu+1 and cDDP share similar gross mechanisms in metal acquisition, distribution, and elimination, although differences in terms of transport kinetics and other parameters may exist.
2.2. Roles of copper transporter in cDDP sensitivity
Many studies have demonstrated that expression of hCtr1 and ATP7A/ATP7B affect cellular sensitivity of cDDP in cultured cell systems [45, 46]. In clinical settings, several studies have reported that expression levels of hCtr1 in tumors are significantly correlated with better treatment outcomes in terms of increased progression-free survival (PFS) time and overall survival (OS) times in patients treated with Pt-based therapeutics [47–50]. In some tumor types, elevated expression of hCtr1 is intrinsically sensitive to Pt-based treatment, suggesting that hCtr1 levels may be a predictive marker for effectiveness of Pt drug in cancer treatment. (see below). However, correlations between PFS or OS and ATP7 and ATP7B expression in ovarian cancer patients [50] and lung cancer patients were not as significant [47]. Moreover, expression of hCtr1, but not ATP7A or ATP7B, can be readily upregulated by manipulations of cellular Cu contents (see below). These results suggest that hCtr1 may be a valid target for improving Pt drug treatment efficacy through inducing its regulation for enhanced drug transport.
3. Regulation of cDDP sensitivity through modulation of hCtr1 expression
Copper is an essential micronutrient for cell survival but is poisonous when in excess. Therefore, cellular Cu levels have to be adequately regulated. Although the steady-state cellular Cu levels are regulated by the balancing acts among hCtr1, Cu chaperons, and Cu exporters, hCtr1 expression is regulated by acute Cu concentration changes. Under Cu-limited conditions, hCtr1 expression is upregulated, whereas under Cu-replete conditions, hCtr1 expression is repressed. Two major mechanisms have been described for hCtr1 regulation by Cu concentration variations as described below.
3.1 Regulation of hCtr1 by endocytosis
It has been reported that under Cu-replete conditions, hCtr1 is rapidly internalized by endocytosis where hCtr1 may [51] or may not be degraded [52]. In contrast, under Cu-deplete conditions, the internalized hCtr1 is recycled back to the plasma membrane [52]. This post-translational regulation model depicts that Cu and hCtr1 are mutually regulated and suggest that the sensing mechanism of Cu bioavailability that triggers hCtr1 endocytosis may reside on hCtr1 itself (Fig. 1A).
It must be noted that this two-component mutual regulation model was mainly carried out using anti-hCtr1 antibodies in cultured cells stably overexpressed hCtr1 by transfection of hCtr1 recombinants. The Ctr1 family is an evolutionally conserved group of proteins, and it has been difficult to obtain high affinity anti-hCtr1 antibodies for research. Polyclonal anti-hCtr1 antibodies produced by different laboratories using different portions of hCtr1 as antigens detected hCtr1 with different molecular masses by Western blotting, i.e. 28 kDa [53], 24 kDa [54], 35 kDa [55], 24 kDa and 36 kDa [56]. The 23- to 24-kDa proteins correspond to the unmodified hCtr1 monomer of 190 amino acid residues, whereas the 28-, 35- and 36-kDa proteins are considered as glycosylated hCtr1 [55, 57]. We produced a polyclonal anti-hCtr1 antibody using the N-terminal 50 amino acids ecto-peptide sequence as an antigen. This antibody recognizes the 23 kDa hCtr1 by Western blotting and stains cytoplasmic membrane localized hCtr1 in un-transfected cancer cells [50, 58]. It is unclear how the modified and unmodified hCtr1 may contribute to endocytosis under Cu stressed conditions.
Because of the difficulty of obtaining high quality anti-hCtr1 antibodies, current evidence showing Cu-induced hCtr1 endocytosis was mostly carried out using genetically engineered cell lines overproducing hCtr1 by transfection [41]. These hCtr1-overproducing cells therefore had been pre-loaded with Cu, causing reduction of endogenous hCtr1 levels [50, 59–61], much like cells that had been treated with Cu. As a consequence, these cells may have altered buffered bioavailable Cu capacities that control hCtr1 regulation under copper stressed conditions (see below).
3.2 Transcriptional regulation of hCtr1 by Cu bioavailability
Transcriptional regulation as an important mechanism of Ctr1 regulation has been well demonstrated in multiple organisms. In the yeast Saccaromyces cerevisiae, low Cu conditions activate the transcription factor Mac for upregulation of yCtr1 and yCtr3 and metal reductase Fre1, together encoding a high-affinity Cu import system [62]. At high Cu concentrations, Mac is inactive and unstable. Another transcription factor AceI is activated and induces the expression of Cu-chelating metallothionein encoded by CUP1 and CRS5 and the antioxidant superoxide dismutase encoded by SOD1 for detoxifying Cu overload [63]. In Drosophila under conditions of Cu starvation, Cu transporter dCtr1B expression is transcriptionally upregulated by metal-responsive transcription factor-1 (MTF-1). Strikingly, MTF-1 is a well-known metal-responsive transcriptional factor involved in upregulation of metallothionein genes for the detoxification of Cu and other heavy metals [64]. Thus, dMTF-1 responds to both excessive and limiting Cu conditions and regulates different genes accordingly. But how dMTF-1 senses these two different conditions and activates its target genes is not known. In Arabidopsis, transcription factor SQUAMOSA-promoter like binding protein 7 (SPL-7) upregulates the three Cu transporters, COPT1, COPT2 and COPT6 in response to Cu-limiting conditions [65–67].
We demonstrated that regulation of hCtr1 expression is controlled by transcription factor specific protein 1 (Sp1) which binds the GC boxes located at the Sp1 and hCtr1 promoter regions. Cu deficiency conditions, induced either by treatment with Cu chelators or by transfection with dominant-negative hCtr1 recombinant, upregulates Sp1 which in turn upregulates hCtr1 expression through promoter binding. In contrast, Cu sufficiency either by treating cells with CuSO4 or by overexpressing the wild-type hCtr1 recombinant, results in downregulation of Sp1 expression which in turn downregulates hCtr1 expression [46, 60]. These observations showed that human Cu homeostasis is controlled by the three-way mutually regulatory loop consisting of Cu, Sp1, and hCtr1 [46] (Fig. 2). This regulatory loop highlights the dynamic mechanism of Cu homeostatic regulation: changing any one component in the loop will affect the levels of the other two, resulting in a new homeostatic balance among all three. These findings have led to the development of using Cu chelators to upregulate hCtr1 expression, thereby enhancing cDDP transport capacity for its cell-killing activity (see below).
Figure 2. Schematics of Cu homeostasis regulation.
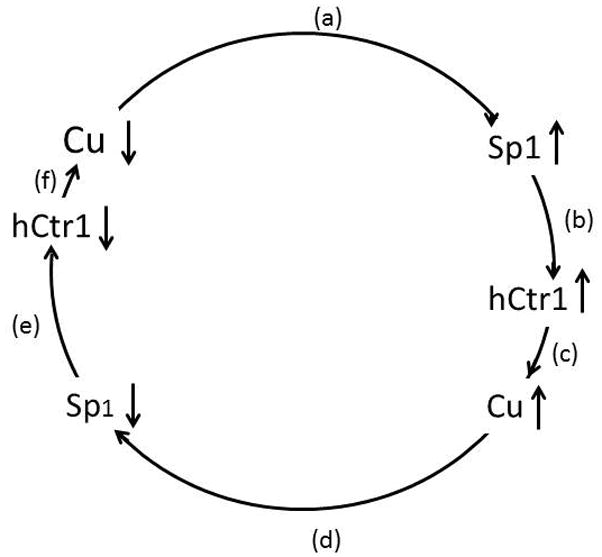
(a) Cu depletion upregulates Sp1. (b) Upregulated Sp1 increases hCtr1 expression, (c) Increased hCtr1 enhances Cu transport. (d) Increased Cu suppresses hCtr1 expression. (e) Reduced Sp1 down-regulates hCtr1, resulting in reduced Cu (f).
4. The sensing mechanisms of Cu bioavailability by Sp1
Many transcription factors mediate Ctr1 regulation through DNA binding therefore, these transcription factors may function as sensors of Cu bioavailability. For Ace1, excess Cu promotes its DNA binding [68]. In contrast, for Mac1 and SPL-7 [66], excess Cu inhibits their DNA-binding activities and corresponding promoter activities. These transcriptional regulators contain metal-binding motifs that respond to metal ions for their transcriptional activities. It appears that sensing of Cu bioavailability relies on Cu interactions between transcriptional regulators and the promoters of their target genes.
Sp1 is a ubiquitous transcription factor consisting of a DNA-binding domain that contains three zinc fingers (ZF) and a transactivation domain that contains two serine/threonine-rich and two glutamine-rich (Q-rich 1 and Q-rich 2) subdomains. Each ZF consists of Cys2·His2 that is coordinated by one Zn molecule. We previously demonstrated that both ZF and Q-rich 2 domains are essential for Sp1’s Cu homeostatic regulation [60]. Sp1 is constitutively bound by Zn ions because unliganded Sp1 is very unstable [69]. Excess Cu2+ competes the bound Zn2+, resulting in distortion of its DNA-binding activities and downregulation of target genes hCtr1 and Sp1. In contrast, Cu-limiting conditions enhance the stabilization of interactions between Sp1 and the Sp1 and hCtr1 promoters. These explanations are consistent with the ranked order of stability of metal complexes known as the Irving-Williams series [70] as follows: Cu+2 > Ni+2 > Co+2 > Zn+2 > Fe+2 > Mn+2 > Mg+2.
The effects of Cu+2, Zn+2, and Cu chelators (bathocuproine disulfonic acid (BCS)) on the stability of Sp1-DNA binding can be determined by using gel electrophoretic mobility shift (GEMS) assay which employs 32P-labeled double-stranded DNA probe with a Sp1 binding sequence [60] in the presence of increased concentrtions of Cu+2 or Zn+2 according to the published procedures [71]. This analysis revealed that Cu+2 ions indeed are more potent than Zn+2 in destabilization of the Sp1-DNA complex, consistent with the Irving and Williams’ series. In contrast, BCS increases Sp1-DNA complex stabilization (Z.D.L, and MTK, unpublished data).
Cupric ions (Cu+2) are at the top of the Irving-Williams series, suggesting that Cu+2 ions have a strong competitive edge over Zn+2 in binding to the same binding site [72]. Cuprous ions (Cu1+) are also highly competitive, and hence both Cu ions have the potential to displace less competitive metal ions from metellocomplexes [42, 73]. Cu ions in eukaryotic cytosol are predominantly in Cu+1 form, and is intracellularly trafficked via Cu1+-binding carrier proteins, because of mostly reduced environment. However, Cu+1 can be converted into Cu+2 by the Fenton reaction under oxidative stressed conditions, but Cu+2 can be converted back to Cu+1 by metal reductase (Fre1 from the yeast S. cerevisiae, which is homologous to the gp91Phox in the NADPH redox regulating complex [74]). Naturally occurred Cu is mostly in the form of Cu+2. It has to be converted to Cu+1 before transport by hCtr1.
We also found that the Q-rich 2 subdomain within the Sp1-TAD is also involved in Cu regulation of Sp1 and hCtr1 expression [60]. Cu can interact with the carboxylate group of glutamate (Q). The Q-rich 2 sequence interacts with TAFII110 in the TFIID, a core protein complex in the basal transcriptional machinery of RNA polymerase II [75, 76]. This domain is also involved in Sp1 self-multimerization resulting in DNA looping in the supra-activation of transcription [77, 78]. High concentrations of Cu may also disrupt Sp1 multimerization and its cross-talk with basal transcriptional machinery resulting in attenuation of Sp1’s transcription activity.
4.1. The cellular bioavailable Cu sensing capacity
It has been proposed that virtually all cellular Cu ions are tightly bound and that only a small fraction is “labile” or bioavailable for regulation [73]. The physiologically bioavailable Cu ion pools have to be kept within a window with lower and upper limits. The inter-regulatory loop of Cu-Sp1-hCtr1 in Cu homeostasis regulation (Fig. 2) is also consistent with this notion. Using fluorescent probes for AceI and Mac activations that sense opposite bioavailable Cu limits, it was demonstrated that the upper and lower limits of available Cu1+ concentrations in yeast are ~8.9 × 10−17 M and ~5.1 × 10−21 M, respectively [79]. In higher organisms, measurement of the buffered bioavailable Cu windows is more difficult. It was estimated that cellular buffered Cu+1 capacities are at about 10−15 M ranges whereas Zn+2 are at 10−12 M [72].
4.2. Cellular bioavailable Cu pool and hCtr1 regulation
The bioavailable buffered Cu pool, although still ill-defined in human cells, plays an important role in controlling hCtr1 regulation in response to Cu variations. The pool sizes may vary according to cell types at various physiological states. In addition, like cDDP, Cu can bind to many cellular constituents including small molecules such as amino acids, organic and inorganic ligands, and to the side-chain ligands on the surface of macromolecules such as cuproenzymes. Cu can also bind to sulfate-containing molecules such as glutathione (GSH) and metallothionein (MT) which are considered as the major Cu sink. Overexpression of GSH by elevated expression of γ-glutamylcysteine synthetase resulted in reduced bioavailable Cu pools, upregulation of hCtr1 and increased cellular cDDP sensitivity [80]. Cu can also displace Zn binding to MT and the released Zn may affect the transcription activities of ZF-containing transcription factors.
The capacities of the bioavailable Cu pool may also differ among different tissues/organs, reflecting in differential effectiveness for the induction of Ctr1 expression by copper-lowering agents. Intestinal epithelia are the main tissue involved in acquiring Cu from foodstuff in animals. Mice with intestinal epithelial ablation of both mCtr1 alleles (mCtr1int/int) show substantial variations in the severity of Cu reduction among different organs in the body as compared with their counterparts in mCtr1flox/flox mice [57]. Previously study showed that rats fed a Cu-deficient diet failed to show increased Ctr1 mRNA levels in livers and small intestines despite a substantial loss in Cu levels in these organs (69 ~ 89% reduction), compared with those in animals fed Cu-adequate diet [59]. These observations revealed that different organs have different responsivenesses of mCtr1 mRNA expression to Cu stress.
Elevated Cu levels have been frequently found in many human malignancies, including those of prostate, breast, colon, lung, and brain [81]. Cu can stimulate tumor cell growth and Cu chelation suppresses tumor growth in experimental animal tumor model [82]. Recent study demonstrated Cu is required for the oncogenic pathway elicited by the BRAFV600E mutation which occurs in approximately 50 % of malignant melanoma, by interacting with MEK1 which phosphorylates ERK1/2 growth signal [83]. These results demonstrated that the window of buffered Cu contents may differ between normal and neoplastic sources.
Sp1 is an essential enzyme that is ubiquitously expressed. Sp1 expression is transcriptionally self-regulated in the Cu homeostatic cycle [60]. More than 40 interacting proteins can associate with Sp1 and many of them are coregulators that either synergize or suppress Sp1’s transcription activity [84]. Moreover, posttranslational regulation such as that by glycosylation, acetylation, phosphorylation, SUMOylation, and ubiquitination can also affect its activity and stability [84]. Elevated expression of Sp1 are frequently found in tumors [85]. However, the roles of Sp1 in tumorigenesis are complex. For example, Sp1 upregulation is involved in Kras-induced lung tumorigenesis, however Sp1 expression is decreased during tumor progression, and reduced Sp1 levels is associated with lung tumor progression and metastasis [86]. Sp1 is a downstream effector of oncogenic Kras signaling. We found that expression of expression of Sp1 and hCtr1 is increased in the oncogenic Ras-transformed, SV40 T/t and telomerase reverse transcriptase-immortalized human fallopian tube epithelial (FTE) cells. FTE has been suggested as origin of ovarian cancers [87]. Furthermore, expression of hCtr1 and Sp1 can be induced in Kras-transformed FTE by Cu chelation but not in the non-transformed immortalized FTE cells cannot (L.Y., J. L, MTK, unpublished results). These findings are consistent with the notion that regulation of Sp1/hCtr1, and therefore Cu homeostasis, may differ between non-cancerous and cancerous cells.
5. Overcoming cDDP resistance by modulating hCtr1 expression
5.1. Preclinical studies
Induction of hCtr1 expression in cultured cells by Cu chelation occurs at about 30 minutes after treating with a Cu chelator, but takes 2 – 3 days to return to basal levels after Cu chelator is removed [50, 58]. Neither ATP7A nor ATP7B levels is changed under these conditions. Because of tight regulation of Sp1 and hCtr1 in the Cu homeostasis regulation loop, levels of regulation in regular cells are moderate. These moderate levels may also be contributed by the weak promoter of hCtr1, which is shared by another transcription unit encoding FK506 binding protein in the opposite direction located 201 nucleotides upstream from the transcription start site of hCtr1. This inter-genic sequence contains no enhancer-like sequence.
On the other hand, because cDDP-resistant variants often exhibit reduced hCtr1 levels, we demonstrated that these hCtr1low cells showed greater magnitudes of Sp1 and hCtr1 upregulation than hCtr1high cells by Cu chelation that are associated with greater re-sensitization to Pt drugs [50]. Moreover, cDDP by itself, like Ag+1 or Zn+2, can be considered as a Cu-lowering agent because it functions as a competitor of hCtr1-mediated Cu transport. cDDP upregulates Sp1 and hCtr1 expression, facilitating cDDP transport and cell killing capacity [58]. The preferential upregulation of hCtr1 levels in cDDP-resistant cells provides as a mechanistic basis of using Cu-lowering agents for overcoming cDDP resistance in clinical evaluation.
5.2 Clinical evaluation of Cu-lowering agents in overcoming Pt drug resistance
Cu-lowering agents such as trientine and D-pencillamine have been used for more than four decades for treating Cu toxicosis resulting from Cu accumulation in Wilson’s disease, a genetic disorder caused by defective ATP7B. A phase I exploratory study using carboplatin plus trientine in patients with advanced malignancies (n=55, 45 failed prior Pt drug treatment) has recently been completed at MD Anderson Cancer Center [88, 89]. These results demonstrated that (a) the combination of trientine with carboplatin was well-tolerated and associated with improved antitumor activity in a subset of Pt-refractory cancer patients who experienced substantial decreases of serum Cu levels, and (b) reduction of serum ceruloplasmin and/or Cu levels may be a prognostic marker for the treatment efficacy, i.e., patients who achieve rapid reduction in serum Cu levels are likely to respond better to the chemotherapy than those that did not show reduction. (c) No dose-limiting toxicity or treatment-related deaths were observed. The common grade 2 adverse events included anemia, thrombocytopenia, and neutropenia, fatique, nausea/vomiting, anorexia and neuropathy.
The response rate of the current Cu chelation therapy remains low (19%) and improvement is warrant from the following considerations: (a) This exploratory study included a heterogeneous cohort of patients with different tumor lineages. Most of the patients had unsuccessful prior Pt-based treatment. Because multiple mechanisms are known for cDDP resistance, it is imperative to determine whether the response to Cu chelation therapy in a subset of patients was related to reduced hCtr1 expression. (b) Moreover, different tumor types may have different buffered capacities for hCtr1 induction by Cu chelation as discussed above, much as cDDP shows favorable treatment efficacy for one type of tumor over others. The possible tumor type-related effectiveness of Cu-lowering agents in reducing Cu levels need to be investigated. (c) This current study measured serum Cu and ceruloplasmin levels as surrogate references for Cu chelation. However, the utility of these markers in monitoring tumors’ Cu levels remains to be critically evaluated..
6. Conclusion
The discovery that the Cu transporter systems also transport Pt drugs [15] has provided new insights into how modulation of cellular Cu levels can affect Pt drug sensitivity in cancer chemotherapy. Studies of the molecular bases of Cu homeostasis regulation by hCtr1 have resulted in an exploratory clinical investigation using a Cu-lowering agent to enhance Pt drugs transport. Although current results showed that about 20% of advanced cancer patients benefited from this treatment, however, this strategy remain attractive and the overall efficacy may be improved through better understanding the molecular mechanisms of hCtr1 regulation. Further studies are needed to improve the results of using Cu-chelation approach in improving the treatment efficacy of Pt drugs in cancer chemotherapy.
7. Expert Opinion
Since its approval by FDA in 1978, cDDP has been a highly prescribed antineoplastic agent and has become a pillar of cancer chemotherapy in treating many types of human malignancies. cDDP can be used in front-line therapy as well as in savage therapy after failure with targeted agents. cDDP resistance has been an important problem associated with cDDP failure because once resistance develops, other effective treatment options are limited. Mechanisms of cDDP resistance are multifactorial due to multiple cellular targets in its mode of toxicities [90, 91]. Over the years, substantial efforts have been devoted in developing effective agents to circumvent Pt drug resistance. Classical studies have been concentrated on DNA damages induced by cDDP, identifying genes and pathways that repair DNA damages and developing small molecules that target these pathways. Along this line, olaparib, rucaparib, niraparib, and BMN673, inhibitors of poly (ADP-ribose) polymerase targeting the repair system of cDDP-induced DNA damages [92], have shown promise for improving cDDP therapy although its clinical utility requires further studies [93].
Platinum drug transporters have been an important area of Pt research for the last decade. However, translating this research into clinical benefit remains a largely uncharted domain. The observations that vast majority of cDDP resistant variants are associated with reduced drug accumulation underscore the likelihood that cDDP transport systems are important targets for overcoming cDDP resistance. Of particular attractive is hCtr1 which controls the import of cDDP and carboplatin and to much reduced extent oxaliplatin drugs, particularly, its expression is correlated with clinical sensitivity of Pt drugs [47–50]. The exploratory clinical trial using a copper-lowering agent in overcoming cDDP resistance represents the first-in-human study and has obtained an incremental success. Current results showed about 20% of Pt-refractory patients with advanced malignancies experienced response to Pt drug in combination with Cu chelator treatment. Admittedly, clinical investigations targeting hCtr1 upregulation using Cu chelation strategy remain at their infancy, although Cu-lowering agents have been used for treating other indications for a long time. This strategy, upon further development, would likely to become a low cost treatment option. Another strength of this strategy lies on the differential sensitivity of hCtr1 induction by Cu chelation between drug-resistant tumor cells and normal cells, providing a favorable “therapeutic index” in cancer chemotherapy. Given the complexity of Cu homeostasis in controlling hCtr1 expression, more work is imperative. Another area that is equally important is the exploration of drug retention mechanisms regulated by ATP7A and ATP7B. Specifically, agents that can suppress activities and/or expression levels of these drug exporters are likely to be useful. This area of research remains understudied.
Since Pt transport mechanisms are intrinsically tied into the Cu homeostasis system, better understanding on global regulation mechanisms of Cu metabolism under various challenges will be needed to identify interacting pathway networking that affects the bioavailability of cellular Pt drugs. Moreover, global analyses of transcriptional and post-transcriptional regulators, epigenetic modifiers, and interferences of small molecules including metal ions such as Zn+2 and Fe+2 [94] that would likely regulate the expression of these transporters will need to be integrated into the core regulators described here. These studies may provide an integrated therapeutic rationale for improving the overall treatment efficacy of Pt drugs in cancer chemotherapy.
List of Abbreviations used in the manuscript
- ATP7A and ATP7B
two trans-Golgi PIB-type ATPases
- Atox1 and CCS
two copper chaperones
- BCS
bathocuproine disulfic acid
- cDDP
cis-diamminedichloroplatinum (II), cisplatin
- Ctr1
high-affinity copper transporter
- Cu
copper
- GEMS
gel electrophoretic mobility shift
- MTF-1
metal-responsive transcription factor
- MT
metallothionein
- Q-rich
glutamine-rich
- SCLC
small-cell-lung cancer
- Sp1
specific protein 1
- SPL-7
SQUAMOSA-promoter like-binding protein 7
- ZF
zinc finger
Footnotes
Financial and competing interest’s disclosure
The authors have been supported by grants from the National Science Council, Taiwan (NSC102-2628-B-006-014-MY3) (HHWC), and RO1 CA149260 (MTK, NS, and LGF) and R01 CA152197 (MTK) and CA16672 (MD Anderson Core) from the National Cancer Institute. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. We thank Michael Worley (Scientific Publications, MD Anderson Cancer Center) for editing the manuscript.
Bibliography
- 1.Kelland L. The resurgence of platinum-based cancer chemotherapy. Nature reviews Cancer. 2007;7(8):573–84. doi: 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]
- 2.Muggia F. Platinum compounds 30 years after the introduction of cisplatin: implications for the treatment of ovarian cancer. Gynecologic oncology. 2009;112(1):275–81. doi: 10.1016/j.ygyno.2008.09.034. [DOI] [PubMed] [Google Scholar]
- 3.Einhorn LH. Curing metastatic testicular cancer. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(7):4592–5. doi: 10.1073/pnas.072067999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bast RC, Jr, Hennessy B, Mills GB. The biology of ovarian cancer: new opportunities for translation. Nature reviews Cancer. 2009;9(6):415–28. doi: 10.1038/nrc2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang D, Lippard SJ. Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov. 2005;4(4):307–20. doi: 10.1038/nrd1691. [DOI] [PubMed] [Google Scholar]
- 6.Kim ES, Lee JJ, He G, et al. Tissue platinum concentration and tumor response in non-small-cell lung cancer. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2012;30(27):3345–52. doi: 10.1200/JCO.2011.40.8120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hall MD, Okabe M, Shen DW, et al. The role of cellular accumulation in determining sensitivity to platinum-based chemotherapy. Annual review of pharmacology and toxicology. 2008;48:495–535. doi: 10.1146/annurev.pharmtox.48.080907.180426. [DOI] [PubMed] [Google Scholar]
- 8.Gately DP, Howell SB. Cellular accumulation of the anticancer agent cisplatin: a review. British journal of cancer. 1993;67(6):1171–6. doi: 10.1038/bjc.1993.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oldenburg J, Begg AC, van Vugt MJ, et al. Characterization of resistance mechanisms to cis-diamminedichloroplatinum(II) in three sublines of the CC531 colon adenocarcinoma cell line in vitro. Cancer Res. 1994;54(2):487–93. [PubMed] [Google Scholar]
- 10.Song IS, Savaraj N, Siddik ZH, et al. Role of human copper transporter Ctr1 in the transport of platinum-based antitumor agents in cisplatin-sensitive and cisplatin-resistant cells. Molecular cancer therapeutics. 2004;3(12):1543–9. [PubMed] [Google Scholar]
- 11.Teicher BA, Holden SA, Herman TS, et al. Characteristics of five human tumor cell lines and sublines resistant to cis-diamminedichloroplatinum(II) Int J Cancer. 1991;47(2):252–60. doi: 10.1002/ijc.2910470214. [DOI] [PubMed] [Google Scholar]
- 12.Twentyman PR, Wright KA, Mistry P, et al. Sensitivity to novel platinum compounds of panels of human lung cancer cell lines with acquired and inherent resistance to cisplatin. Cancer Res. 1992;52(20):5674–80. [PubMed] [Google Scholar]
- 13.Waud WR, Leopold WR, Elliott WL, et al. Antitumor activity of ethyl 5-amino-1,2-dihydro-2-methyl-3-phenyl-pyrido [3,4-b]pyrazin-7-ylcarbamate, 2-hydroxyethanesulfonate, hydrate (NSC 370147) against selected tumor systems in culture and in mice. Cancer Res. 1990;50(11):3239–44. [PubMed] [Google Scholar]
- 14.Andrews PA, Howell SB. Cellular pharmacology of cisplatin: perspectives on mechanisms of acquired resistance. Cancer cells. 1990;2(2):35–43. [PubMed] [Google Scholar]
- 15••.Ishida S, Lee J, Thiele DJ, et al. Uptake of the anticancer drug cisplatin mediated by the copper transporter Ctr1 in yeast and mammals. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(22):14298–302. doi: 10.1073/pnas.162491399. This work was the groundbreaking article demonstrating that Ctr1 is a cDDP transporter. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin X, Okuda T, Holzer A, et al. The copper transporter CTR1 regulates cisplatin uptake in Saccharomyces cerevisiae. Mol Pharmacol. 2002;62(5):1154–9. doi: 10.1124/mol.62.5.1154. [DOI] [PubMed] [Google Scholar]
- 17.Lee J, Petris MJ, Thiele DJ. Characterization of mouse embryonic cells deficient in the ctr1 high affinity copper transporter. Identification of a Ctr1-independent copper transport system. J Biol Chem. 2002;277(43):40253–9. doi: 10.1074/jbc.M208002200. [DOI] [PubMed] [Google Scholar]
- 18.Holzer AK, Manorek GH, Howell SB. Contribution of the major copper influx transporter CTR1 to the cellular accumulation of cisplatin, carboplatin, and oxaliplatin. Mol Pharmacol. 2006;70(4):1390–4. doi: 10.1124/mol.106.022624. [DOI] [PubMed] [Google Scholar]
- 19.Arredondo M, Mendiburo MJ, Flores S, et al. Mouse divalent metal transporter 1 is a copper transporter in HEK293 cells. Biometals: an international journal on the role of metal ions in biology, biochemistry, and medicine. 2014;27(1):115–23. doi: 10.1007/s10534-013-9691-6. [DOI] [PubMed] [Google Scholar]
- 20.Jiang L, Garrick MD, Garrick LM, et al. Divalent metal transporter 1 (Dmt1) mediates copper transport in the duodenum of iron-deficient rats and when overexpressed in iron-deprived HEK-293 cells. The Journal of nutrition. 2013;143(12):1927–33. doi: 10.3945/jn.113.181867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Illing AC, Shawki A, Cunningham CL, et al. Substrate profile and metal-ion selectivity of human divalent metal-ion transporter-1. J Biol Chem. 2012;287(36):30485–96. doi: 10.1074/jbc.M112.364208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pope CR, Flores AG, Kaplan JH, Unger VM. Structure and function of copper uptake transporters. Current topics in membranes. 2012;69:97–112. doi: 10.1016/B978-0-12-394390-3.00004-5. [DOI] [PubMed] [Google Scholar]
- 23•.Liang ZD, Stockton D, Savaraj N, et al. Mechanistic comparison of human high-affinity copper transporter 1-mediated transport between copper ion and cisplatin. Mol Pharmacol. 2009;76(4):843–53. doi: 10.1124/mol.109.056416. This paper demonstrates that cDDP and Cu treatments induce conformational changes of hCtr1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24••.De Feo CJ, Aller SG, Siluvai GS, et al. Three-dimensional structure of the human copper transporter hCTR1. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(11):4237–42. doi: 10.1073/pnas.0810286106. This paper describes the hCtr1 conformation using cryoelectronmicroscopy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Larson CA, Adams PL, Blair BG, et al. The role of the methionines and histidines in the transmembrane domain of mammalian copper transporter 1 in the cellular accumulation of cisplatin. Mol Pharmacol. 2010;78(3):333–9. doi: 10.1124/mol.110.064766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Larson CA, Adams PL, Jandial DD, et al. The role of the N-terminus of mammalian copper transporter 1 in the cellular accumulation of cisplatin. Biochemical pharmacology. 2010;80(4):448–54. doi: 10.1016/j.bcp.2010.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sze CM, Khairallah GN, Xiao Z, Wedd AG, et al. Interaction of cisplatin and analogues with a Met-rich protein site. J Biol Inorg Chem. 2009;14(2):163–5. doi: 10.1007/s00775-008-0452-x. [DOI] [PubMed] [Google Scholar]
- 28.Crider SE, Holbrook RJ, Franz KJ. Coordination of platinum therapeutic agents to met-rich motifs of human copper transport protein1. Metallomics. 2010;2(1):74–83. doi: 10.1039/b916899k. [DOI] [PubMed] [Google Scholar]
- 29.Wu Z, Liu Q, Liang X, Yang X, et al. Reactivity of platinum-based antitumor drugs towards a Met- and His-rich 20mer peptide corresponding to the N-terminal domain of human copper transporter 1. J Biol Inorg Chem. 2009;14(8):1313–23. doi: 10.1007/s00775-009-0576-7. [DOI] [PubMed] [Google Scholar]
- 30.Wang X, Du X, Li H, et al. The effect of the extracellular domain of human copper transporter (hCTR1) on cisplatin activation. Angew Chem Int Ed Engl. 2011;50(12):2706–11. doi: 10.1002/anie.201006739. [DOI] [PubMed] [Google Scholar]
- 31.Long F, Su CC, Zimmermann MT, et al. Crystal structures of the CusA efflux pump suggest methionine-mediated metal transport. Nature. 2010;467(7314):484–8. doi: 10.1038/nature09395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pope CR, De Feo CJ, Unger VM. Cellular distribution of copper to superoxide dismutase involves scaffolding by membranes. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(51):20491–6. doi: 10.1073/pnas.1309820110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Flores AG, Unger VM. Atox1 contains positive residues that mediate membrane association and aid subsequent copper loading. The Journal of membrane biology. 2013;246(12):903–13. doi: 10.1007/s00232-013-9592-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arnesano F, Banci L, Bertini I, et al. Probing the interaction of cisplatin with the human copper chaperone Atox1 by solution and in-cell NMR spectroscopy. Journal of the American Chemical Society. 2011;133(45):18361–9. doi: 10.1021/ja207346p. [DOI] [PubMed] [Google Scholar]
- 35.Boal AK, Rosenzweig AC. Crystal structures of cisplatin bound to a human copper chaperone. Journal of the American Chemical Society. 2009;131(40):14196–7. doi: 10.1021/ja906363t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Calandrini V, Nguyen TH, Arnesano F, et al. Structural biology of cisplatin complexes with cellular targets: the adduct with human copper chaperone atox1 in aqueous solution. Chemistry. 2014;20(37):11719–25. doi: 10.1002/chem.201402834. [DOI] [PubMed] [Google Scholar]
- 37.Zhao L, Cheng Q, Wang Z, et al. Cisplatin binds to human copper chaperone Cox17: the mechanistic implication of drug delivery to mitochondria. Chemical communications. 2014;50(20):2667–9. doi: 10.1039/c3cc48847k. [DOI] [PubMed] [Google Scholar]
- 38.Zwelling LA, Kohn KW. Mechanism of action of cis-dichlorodiammineplatinum(II) Cancer treatment reports. 1979;63(9–10):1439–44. [PubMed] [Google Scholar]
- 39••.Ohrvik H, Nose Y, Wood LK, et al. Ctr2 regulates biogenesis of a cleaved form of mammalian Ctr1 metal transporter lacking the copper- and cisplatin-binding ecto-domain. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(46):E4279–88. doi: 10.1073/pnas.1311749110. This work describes the cross-talk between Ctr1 and Ctr2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang CP, Fofana M, Chan J, et al. Copper transporter 2 regulates intracellular copper and sensitivity to cisplatin. Metallomics: integrated biometal science. 2014;6(3):654–61. doi: 10.1039/c3mt00331k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hasan NM, Lutsenko S. Regulation of copper transporters in human cells. Current topics in membranes. 2012;69:137–61. doi: 10.1016/B978-0-12-394390-3.00006-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Polishchuk EV, Concilli M, Iacobacci S, et al. Wilson disease protein ATP7B utilizes lysosomal exocytosis to maintain copper homeostasis. Developmental cell. 2014;29(6):686–700. doi: 10.1016/j.devcel.2014.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Safaei R, Adams PL, Mathews RA, et al. The role of metal binding and phosphorylation domains in the regulation of cisplatin-induced trafficking of ATP7B. Metallomics: integrated biometal science. 2013;5(8):964–72. doi: 10.1039/c3mt00131h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tadini-Buoninsegni F, Bartolommei G, Moncelli MR, et al. Translocation of platinum anticancer drugs by human copper ATPases ATP7A and ATP7B. Angewandte Chemie. 2014;53(5):1297–301. doi: 10.1002/anie.201307718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45••.Kuo MT, Chen HH, Song IS, et al. The roles of copper transporters in cisplatin resistance. Cancer Metastasis Rev. 2007;26(1):71–83. doi: 10.1007/s10555-007-9045-3. This review article presents the three-way mutual regulation mechanism of Cu-Sp1-hCtr1 homeostasis. [DOI] [PubMed] [Google Scholar]
- 46.Kuo MT, Fu S, Savaraj N, Chen HH. Role of the human high-affinity copper transporter in copper homeostasis regulation and cisplatin sensitivity in cancer chemotherapy. Cancer Res. 2012;72(18):4616–21. doi: 10.1158/0008-5472.CAN-12-0888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen HH, Yan JJ, Chen WC, et al. Predictive and prognostic value of human copper transporter 1 (hCtr1) in patients with stage III non-small-cell lung cancer receiving first-line platinum-based doublet chemotherapy. Lung cancer. 2012;75(2):228–34. doi: 10.1016/j.lungcan.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ishida S, McCormick F, Smith-McCune K, et al. Enhancing tumor-specific uptake of the anticancer drug cisplatin with a copper chelator. Cancer cell. 2010;17(6):574–83. doi: 10.1016/j.ccr.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim ES, Tang X, Peterson DR, et al. Copper transporter CTR1 expression and tissue platinum concentration in non-small cell lung cancer. Lung cancer. 2014;85(1):88–93. doi: 10.1016/j.lungcan.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50•.Liang ZD, Long Y, Tsai WB, et al. Mechanistic basis for overcoming platinum resistance using copper chelating agents. Molecular cancer therapeutics. 2012;11(11):2483–94. doi: 10.1158/1535-7163.MCT-12-0580. This paper presents evidence showing differential upregulation of hCtr1 by Cu-chelating agents between cDDP-resistant and cDDP-sensitive cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guo Y, Smith K, Lee J, et al. Identification of methionine-rich clusters that regulate copper-stimulated endocytosis of the human Ctr1 copper transporter. J Biol Chem. 2004;279(17):17428–33. doi: 10.1074/jbc.M401493200. [DOI] [PubMed] [Google Scholar]
- 52.Molloy SA, Kaplan JH. Copper-dependent recycling of hCTR1, the human high affinity copper transporter. J Biol Chem. 2009;284(43):29704–13. doi: 10.1074/jbc.M109.000166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Klomp AE, Tops BB, Van Denberg IE, et al. Biochemical characterization and subcellular localization of human copper transporter 1 (hCTR1) The Biochemical journal. 2002;364(Pt 2):497–505. doi: 10.1042/BJ20011803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kuo YM, Gybina AA, Pyatskowit JW, et al. Copper transport protein (Ctr1) levels in mice are tissue specific and dependent on copper status. The Journal of nutrition. 2006;136(1):21–6. doi: 10.1093/jn/136.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Maryon EB, Molloy SA, Kaplan JH. O-linked glycosylation at threonine 27 protects the copper transporter hCTR1 from proteolytic cleavage in mammalian cells. J Biol Chem. 2007;282(28):20376–87. doi: 10.1074/jbc.M701806200. [DOI] [PubMed] [Google Scholar]
- 56.Nose Y, Wood LK, Kim BE, et al. Ctr1 is an apical copper transporter in mammalian intestinal epithelial cells in vivo that is controlled at the level of protein stability. J Biol Chem. 2010;285(42):32385–92. doi: 10.1074/jbc.M110.143826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nose Y, Kim BE, Thiele DJ. Ctr1 drives intestinal copper absorption and is essential for growth, iron metabolism, and neonatal cardiac function. Cell metabolism. 2006;4(3):235–44. doi: 10.1016/j.cmet.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 58.Liang ZD, Long Y, Chen HH, et al. Regulation of the high-affinity copper transporter (hCtr1) expression by cisplatin and heavy metals. Journal of biological inorganic chemistry: JBIC: a publication of the Society of Biological Inorganic Chemistry. 2014;19(1):17–27. doi: 10.1007/s00775-013-1051-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59•.Lee J, Prohaska JR, Dagenais SL, et al. Isolation of a murine copper transporter gene, tissue specific expression and functional complementation of a yeast copper transport mutant. Gene. 2000;254(1–2):87–96. doi: 10.1016/s0378-1119(00)00287-0. This paper describes the insensitivity of murine Ctr1 upregulation in normal tissues in mice fed Cu-deficient diet. [DOI] [PubMed] [Google Scholar]
- 60.Liang ZD, Tsai WB, Lee MY, et al. Specificity protein 1 (sp1) oscillation is involved in copper homeostasis maintenance by regulating human high-affinity copper transporter 1 expression. Mol Pharmacol. 2012;81(3):455–64. doi: 10.1124/mol.111.076422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61••.Song IS, Chen HH, Aiba I, et al. Transcription factor Sp1 plays an important role in the regulation of copper homeostasis in mammalian cells. Mol Pharmacol. 2008;74(3):705–13. doi: 10.1124/mol.108.046771. This is an important article demonstrating the role of Sp1 in hCtr1 transcriptional regulation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rutherford JC, Bird AJ. Metal-responsive transcription factors that regulate iron, zinc, and copper homeostasis in eukaryotic cells. Eukaryot Cell. 2004;3(1):1–13. doi: 10.1128/EC.3.1.1-13.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thiele DJ. ACE1 regulates expression of the Saccharomyces cerevisiae metallothionein gene. Molecular and cellular biology. 1988;8(7):2745–52. doi: 10.1128/mcb.8.7.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Selvaraj A, Balamurugan K, Yepiskoposyan H, et al. Metal-responsive transcription factor (MTF-1) handles both extremes, copper load and copper starvation, by activating different genes. Genes & development. 2005;19(8):891–6. doi: 10.1101/gad.1301805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Penarrubia L, Andres-Colas N, Moreno J, et al. Regulation of copper transport in Arabidopsis thaliana: a biochemical oscillator? Journal of biological inorganic chemistry: JBIC: a publication of the Society of Biological Inorganic Chemistry. 15(1):29–36. doi: 10.1007/s00775-009-0591-8. [DOI] [PubMed] [Google Scholar]
- 66.Yamasaki H, Hayashi M, Fukazawa M, et al. SQUAMOSA Promoter Binding Protein-Like7 Is a Central Regulator for Copper Homeostasis in Arabidopsis. Plant Cell. 2009;21(1):347–61. doi: 10.1105/tpc.108.060137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Garcia-Molina A, Xing S, Huijser P. Functional characterisation of Arabidopsis SPL7 conserved protein domains suggests novel regulatory mechanisms in the Cu deficiency response. BMC plant biology. 2014;14:231. doi: 10.1186/s12870-014-0231-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Furst P, Hu S, Hackett R, et al. Copper activates metallothionein gene transcription by altering the conformation of a specific DNA binding protein. Cell. 1988;55(4):705–17. doi: 10.1016/0092-8674(88)90229-2. [DOI] [PubMed] [Google Scholar]
- 69.Bittel DC, Smirnova IV, Andrews GK. Functional heterogeneity in the zinc fingers of metalloregulatory protein metal response element-binding transcription factor-1. J Biol Chem. 2000;275(47):37194–201. doi: 10.1074/jbc.M003863200. [DOI] [PubMed] [Google Scholar]
- 70.Irving H, Williams RJ. Order of stability of metal complexes. Nature. 1948;162:746–47. [Google Scholar]
- 71.Aiba I, Hossain A, Kuo MT. Elevated GSH level increases cadmium resistance through down-regulation of Sp1-dependent expression of the cadmium transporter ZIP8. Mol Pharmacol. 2008;74(3):823–33. doi: 10.1124/mol.108.046862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Foster AW, Osman D, Robinson NJ. Metal preferences and metallation. J Biol Chem. 2014;289(41):28095–103. doi: 10.1074/jbc.R114.588145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Davis AV, O’Halloran TV. A place for thioether chemistry in cellular copper ion recognition and trafficking. Nature chemical biology. 2008;4(3):148–51. doi: 10.1038/nchembio0308-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shinyashiki M, Pan CJ, Lopez BE, et al. Inhibition of the yeast metal reductase heme protein fre1 by nitric oxide (NO): a model for inhibition of NADPH oxidase by NO. Free radical biology & medicine. 2004;37(5):713–23. doi: 10.1016/j.freeradbiomed.2004.05.031. [DOI] [PubMed] [Google Scholar]
- 75.Gill G, Pascal E, Tseng ZH, Tjian R. A glutamine-rich hydrophobic patch in transcription factor Sp1 contacts the dTAFII110 component of the Drosophila TFIID complex and mediates transcriptional activation. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(1):192–6. doi: 10.1073/pnas.91.1.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liu WL, Coleman RA, Ma E, et al. Structures of three distinct activator-TFIID complexes. Genes & development. 2009;23(13):1510–21. doi: 10.1101/gad.1790709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mastrangelo IA, Courey AJ, Wall JS, et al. DNA looping and Sp1 multimer links: a mechanism for transcriptional synergism and enhancement. Proceedings of the National Academy of Sciences of the United States of America. 1991;88(13):5670–4. doi: 10.1073/pnas.88.13.5670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Su W, Jackson S, Tjian R, et al. DNA looping between sites for transcriptional activation: self-association of DNA-bound Sp1. Genes & development. 1991;5(5):820–6. doi: 10.1101/gad.5.5.820. [DOI] [PubMed] [Google Scholar]
- 79••.Wegner SV, Sun F, Hernandez N, et al. The tightly regulated copper window in yeast. Chemical communications. 2011;47(9):2571–3. doi: 10.1039/c0cc04292g. This paper describes experimental evidence of buffered Cu regulation window. [DOI] [PubMed] [Google Scholar]
- 80••.Chen HH, Song IS, Hossain A, et al. Elevated glutathione levels confer cellular sensitization to cisplatin toxicity by up-regulation of copper transporter hCtr1. Mol Pharmacol. 2008;74(3):697–704. doi: 10.1124/mol.108.047969. The work describes overexpression of GSH enhances cDDP sensitivity through upregulation of hCtr1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tisato F, Marzano C, Porchia M, et al. Copper in diseases and treatments, and copper-based anticancer strategies. Medicinal research reviews. 2010;30(4):708–49. doi: 10.1002/med.20174. [DOI] [PubMed] [Google Scholar]
- 82.Ishida S, Andreux P, Poitry-Yamate C, et al. Bioavailable copper modulates oxidative phosphorylation and growth of tumors. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(48):19507–12. doi: 10.1073/pnas.1318431110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83••.Brady DC, Crowe MS, Turski ML, et al. Copper is required for oncogenic BRAF signalling and tumorigenesis. Nature. 2014;509(7501):492–6. doi: 10.1038/nature13180. This paper demonstrates the role of Cu in oncogenic behavior of BRAF-mutant melaonoma. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Beishline K, Azizkhan-Clifford J. Sp1 and the ‘hallmarks of cancer’. The FEBS journal. 2015;282(2):224–58. doi: 10.1111/febs.13148. [DOI] [PubMed] [Google Scholar]
- 85.Safe S, Abdelrahim M. Sp transcription factor family and its role in cancer. Eur J Cancer. 2005;41(16):2438–48. doi: 10.1016/j.ejca.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 86.Hsu TI, Wang MC, Chen SY, et al. Sp1 expression regulates lung tumor progression. Oncogene. 2012;31(35):3973–88. doi: 10.1038/onc.2011.568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shan W, Mercado-Uribe I, Zhang J, et al. Mucinous adenocarcinoma developed from human fallopian tube epithelial cells through defined genetic modifications. Cell cycle. 2012;11(11):2107–13. doi: 10.4161/cc.20544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88•.Fu S, Hou MM, Wheler J, et al. Exploratory study of carboplatin plus the copper-lowering agent trientine in patients with advanced malignancies. Investigational new drugs. 2014;32(3):465–72. doi: 10.1007/s10637-013-0051-8. This is the first-in-human study of Cu chelation and Pt drug cancer chemotherapy. [DOI] [PubMed] [Google Scholar]
- 89.Fu S, Naing A, Fu C, et al. Overcoming platinum resistance through the use of a copper-lowering agent. Molecular cancer therapeutics. 2012;11(6):1221–5. doi: 10.1158/1535-7163.MCT-11-0864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Stewart DJ. Mechanisms of resistance to cisplatin and carboplatin. Crit Rev Oncol Hematol. 2007;63(1):12–31. doi: 10.1016/j.critrevonc.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 91.Maccio A, Madeddu C. Cisplatin: an old drug with a newfound efficacy -- from mechanisms of action to cytotoxicity. Expert opinion on pharmacotherapy. 2013;14(13):1839–57. doi: 10.1517/14656566.2013.813934. [DOI] [PubMed] [Google Scholar]
- 92.Lord CJ, Tutt AN, Ashworth A. Synthetic Lethality and Cancer Therapy: Lessons Learned from the Development of PARP Inhibitors. Annual review of medicine. 2015;66:455–70. doi: 10.1146/annurev-med-050913-022545. [DOI] [PubMed] [Google Scholar]
- 93.Marchetti C, Imperiale L, Gasparri ML, et al. Olaparib, PARP1 inhibitor in ovarian cancer. Expert opinion on investigational drugs. 2012;21(10):1575–84. doi: 10.1517/13543784.2012.707189. [DOI] [PubMed] [Google Scholar]
- 94.Robinson NJ, Winge DR. Copper metallochaperones. Annual review of biochemistry. 2010;79:537–62. doi: 10.1146/annurev-biochem-030409-143539. [DOI] [PMC free article] [PubMed] [Google Scholar]