

Abstract

The application of the bidentate, electron-rich bisphosphine ligand, 1,3-bis(dicyclohexyl)phosphine-propane (dcpp), in rhodium(I)-catalyzed intermolecular ketone hydroacylation is herein described. Isatins and α-keto amides are shown to undergo hydroacylation with a variety of non-chelating linear and branched aliphatic aldehydes. Also reported is the synthesis of new bidentate chiral phosphine ligands, and their application in hydroacylation is discussed.
Keywords: ketone, hydroacylation, rhodium, asymmetric catalysis, P ligands
While progress has been made in selective C–H oxidation via transition-metal catalyzed ketone hydroacylation,[1–4] the field is still young compared to that of the related ketone hydrosilylation,[5] olefin hydrogenation,[6] hydroformylation,[7] and hydroacylation[8] transformations. A complication that is common to both olefin and ketone hydroacylation arises from an energetically competent decarbonylation pathway that leads to deactivation of the transition metal catalyst, which has severely limited the efficacy and practicality of these methodologies in the past.[9] Our interest in this area has led to the development of systems bearing Lewis-basic heteroatoms to mitigate undesired decarbonylation in the intramolecular hydroacylation of ketones, and this strategy was applied to the enantioselective synthesis of a number of seven and eight-membered benzoxazepinones and benzoxazocinones.[10] We have also recently disclosed the first amide-directed asymmetric intermolecular ketone hydroacylation.[11] Herein, we describe alternative rhodium catalysts that enable the intermolecular hydroacylation of α-keto amides and isatins using a wider scope of substrates, including sterically-hindered α-branched aldehydes.
Due to the volume of successful intramolecular hydroacylations using Rh(I)-based catalysts[6,8,10–14] we sought to develop an analogous catalyst to enable a more general intermolecular transformation. To study this challenging reaction, α-keto amide 2a was chosen as the model ketone substrate, given its ability to chelate metal centers.[15,16] In the presence of a bisphosphine ligand, chelation of the 1,2-keto amide unit with concomitant oxidative addition of a simple aldehyde would prevent decarbonylation while directing insertion of the ketone into the Rh(III)-hydride (Figure 1).
Figure 1.

Reaction design with amide-bearing ketones.
While studying the ligand effect on the hydroacylation of α-keto amide 2a with hydrocinnamaldehyde 1a (2 equiv), we found that the transformation was sensitive to the bite-angle[17] and basicity of the bisphosphine ligand (Table 1). Catalysts derived from bis(diphenylphosphino)-type ligands L1–L5 resulted in low conversions to the desired product based on 1H NMR analysis of the reaction mixtures (entries 1–5). When the more electron-rich 1,3-bis(dicyclohexyl)phosphinopropane (dcpp) L8, which exhibits a bite-angle of 92.9° was used, the desired hydroacylation product 3aa was formed in 85% isolated yield in a chemoselective manner. Under the reaction conditions, no Tishchenko-type aldehyde homo-dimerization was observed. Deviations in the bite-angle inhibited reactivity (entries 7 and 9).
Table 1.
Ligand effects in intermolecular ketone hydroacylation.

| |||
|---|---|---|---|
| entry a | ligand | bite angle [°] | % conv. b |
| 1 | dppm L1 | 75.4 c | <5 |
| 2 | dppe L2 | 86.3 c | <5 |
| 3 | dppp L3 | 95.5 c | <5 |
| 4 | dppb L4 | 98.6 c | 25 |
| 5 | dpppent L5 | — d | 8 |
| 6 | dcpm L6 | 73.7 [18] | <5 |
| 7 | dcpe L7 | 84.4 [19] | 9 |
| 8 | dcpp L8 | 92.9 [20] | 85 (85)e |
| 9 | dcpb L9 | 97.1 [19] | 59 |
Conditions: 1a (2 equiv), 2a (1 equiv), [Rh(cod)2]BF4 (0.1 equiv), ligand (0.1 equiv), DCE, 70 °C, 16 h.
Conversion determined by 1H NMR analysis of the crude reaction mixture using 1,3,5-trimethylbenzene as an internal standard.
Bite angles calculated at the B3LYP/LACV3P** level with the ligand bound to a cationic Rh-center (ref. 10b).
The bite angle for L5 has not been reported but is expected to be similar to that of L4 (ref. 13i).
Isolated yield.

With the identification of a catalyst that enables chemoselective cross-coupling between aldehyde 1a and α-keto amide 2a, we explored the scope (Table 2). A variety of aliphatic aldehydes underwent quantitative hydroacylation to the desired ester products. α-Keto amides bearing aryl-substituents were transformed into the corresponding α-acyloxyamides, with those containing more basic diethyl- and morpholino-amide directing groups undergoing hydroacylation in good to excellent yields. An isopropyl-substituted ketone was unreactive (not shown). Other incompatible coupling partners include aryl and alkenyl aldehydes, as well as a chiral aldehyde bearing an α-oxygen atom derived from oxidative cleavage of 1-O,2-O,5-O,6-O-diisopropylidene-mannitol.[21] While this new Rh(I)-catalyzed method accommodates unfunctionalized aliphatic aldehydes, the complementary NHC-[2] thiolate,[3] and selenide-catalyzed[4] intermolecular ketone hydroacylations achieve highest conversions to the Tishchenko products with aryl aldehydes.
Table 2.
Intermolecular hydroacylation of α-keto amides.

|
Conditions: 1 (2 equiv), 2 (1 equiv), [Rh(nbd)2]BF4 (0.05 or 0.1 equiv), dcpp (0.05 or 0.1 equiv), DCE, 70 °C.
Our group previously published an asymmetric intermolecular ketone hydroacylation which exerted only modest reactivity with isatin substrates.[11] However, we were pleased to find that the current [Rh(dcpp)]BF4 catalyst is highly active for the coupling of aliphatic aldehydes with N-substituted isatins (Table 3). Fluoro-, methyl-, and methoxy-substituted isatins were all viable coupling partners, and both N-methyl and N-benzyl isatins were well suited for this transformation.
Table 3.
Intermolecular hydroacylation of α-keto amides.

|
Conditions: 1c (2 equiv), 2 (1 equiv), [Rh(nbd)2]BF4 (0.05 or 0.1 equiv), dcpp (0.05 or 0.1 equiv), DCE, 70 °C.
Given the high activity of our dcpp-ligated Rh-catalyst, we sought to explore various chiral derivatives based on the dcpp scaffold. In addition to studying the reactivity of commercially-available (S,S)-BDPP (L10), which bears a similar bite-angle with respect to dcpp, a series of chiral variants bearing electron-rich phosphine donors were synthesized via a method reported by Mckinstry and Livinghouse.[22] Inspired by the work of Imamoto,[23] we also prepared C2-symmetric ligands that are chiral-at-phosphorus (L12–L14).
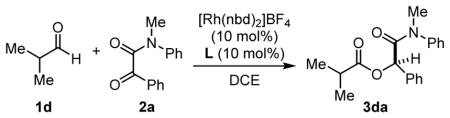
With the Rh-L10 catalyst, ester 3da was produced in 22% yield and 78% ee after 24 h at 30 °C, with the remainder of the mass balance being starting material (entry 1). No significant improvement in conversion was observed when the temperature was increased to 50 °C; however, the ee decreased to 46% (entry 2). When the analogous, more electron-rich L11 was employed as ligand, a substantial increase in yield was obtained (93%), albeit in nearly racemic form (entry 3). This observation supported our hypothesis that using bulkier, more electron-rich phosphines with cationic Rh(I) sources leads to highly active catalysts for ketone hydroacylation. We believe that BDPP’s planar phenyl substituents play a key role in inducing enantioselectivity.[24] The enantioselectivity was restored with P-stereogenic ligands. The catalyst derived from L12, which contains both P- and C- stereogenic centers, required elevated temperatures of 50 °C to achieve good yield and modest ee (entry 5, 86%, 37% ee). The diastereomeric ligand L13 was found to be slightly more reactive, achieving good conversion at 30 °C (67% yield), although with similar levels of enantioinduction (entry 6). Of this series of ligands, P-stereogenic L14 containing no chirality along the carbon backbone gave the best results, providing the hydroacylation product in 88% yield and 63% ee (entry 7).
With [Rh(L14)]BF4 being the most promising catalyst for hydroacylation, we tested it with several other ketone substrates that had previously shown good reactivity with our [Rh(dcpp)]BF4 catalyst (Table 5). Isatins 2f and 2i were transformed with isobutyraldehyde (1d) in complete conversion, generating 3-acyloxy-oxindoles 3df and 3di in 86% and 87% yields, respectively (Table 5, entries 1 and 2). The enantioselectivities, however, were modest (40%). The coupling of α-ketomorpholine amide 2c and aldehyde 1d led to improved enantioselectivity (60%) in α-acyloxyamide 3dc, although the conversion was lower (36 % yield). In general, modifying the rhodium precursor with the new electron-rich ligands developed in this study gave rise to chiral rhodium catalysts that are superior in reactivity compared to the commercially-available BDPP-ligand L10.
Table 5.
Asymmetric hydroacylation of ketones using [Rh(L14)]BF4.

| |||||
|---|---|---|---|---|---|
| Entry | Keto Amide | Product | Time [h] | Yield [%] | ee [%] |
| 1 |
 2f |
 3df |
18 | 86 | 40 |
| 2 |
 2i |
 3di |
18 | 87 | 40 |
| 3 |
 2c |
 3dc |
45 | 36 | 60 |
Conditions: 1d (2 equiv), 2 (1 equiv), [Rh(nbd)2]BF4 (0.1 equiv), L14 (0.1 equiv), DCE, 30 °C.
We were able to successfully design a highly active catalyst system for the intermolecular hydroacylation of isatins and linear α-keto amides with simple aliphatic aldehydes. This protocol was enabled through the use of a cationic Rh(I) precursor and dcpp, a bulky, electron-rich bidentate phosphine. We have preliminary evidence that catalysts based on chiral variants of the dcpp ligand can indeed perform asymmetric intermolecular ketone hydroacylation for a variety of substrates. While commercially-available BDPP-derived Rh(I) gave the highest ee, the new P-stereogenic ligands synthesized in this study offered superior reactivity. These ligands are electron-rich, sterically-encumbered, and low in molecular weight, all of which are properties that are highly desirable for catalysis. We expect these ligands to be applicable to new reaction development beyond asymmetric hydroacylation.
Experimental Section
General Remarks
Commerical reagents were purchased from Sigma Aldrich, Strem, Alfa Aesar, and Acros and used without further purification. All reactions were carried out in a nitrogen-filled glovebox unless otherwise indicated. system. Solvents used in rhodium-catalyzed hydroacylations were first distilled over calcium hydride, degassed by three freeze-pump-thaw cycles and stored in a glove box. Other solvents were dried through two columns of activated alumina. Reactions were monitored using thin- layer chromatography (TLC) on EMD Silica Gel 60 F254 plates or by LC-MS. Visualization of the developed plateswas performed under UV light (254 nm) or KMnO4 stain. Column chromatography was performed with Silicycle Silia-P Flash Silica Gel using glass columns. Preparative-TLC was performed with 0.5 mm EMD Silica Gel 60 F254 plates. Organic solutions were concentrated under reduced pressure on a Büchi rotary evaporator. 1H and 13C NMR spectra were recorded on a Varian Mercury 300, Varian Mercury 400, or Bruker 400. NMR spectra were internally referenced to the residual solvent signal or TMS. Data for 1H NMR are reported as follows: chemical shift (δ ppm), multiplicity (s = singlet, d = doublet, dd = doublet of doublets, t = triplet, q = quartet, m = multiplet, br = broad), coupling constant (Hz), integration. Data for 13C NMR are reported in terms of chemical shift (δ ppm). High resolution mass spectra (HRMS) were obtained on a micromass 70S-250 spectrometer (EI) or an ABI/Sciex Qstar Mass Spectrometer (ESI). Enantiomeric excesses (ees) were ascertained on an Agilent 1200 Series HPLC using supercritical CO2 generated by an Aurora SFC
General Procedure for Catalytic Ketone Hydroacylation
In a nitrogen-filled glove box, 10 mol% ligand was dissolved in 100 μL of DCM and transferred to a vial containing 10 mol% [Rh(nbd)2]BF4, followed by an additional 100 μL DCM rinse, which was added to the catalyst mixture. The resulting pre-catalyst mixture was transferred to a 25 mL Schlenk tube equipped with a magnetic stir bar, followed by an additional 200 μL DCM rinse, which was added to the Schlenk tube. The tube was sealed and removed from the glovebox. The solution was degassed via two cycles of ‘freeze-pump-thaw’, after which the atmosphere was replaced with H2(g) and the reaction stirred at rt for 15 min. The solvent was then removed under reduced pressure. In the glovebox, 1.0 equiv ketone substrate was dissolved in 200 μL DCE, to which 2.0 equiv aldehyde was added. The resulting solution was transferred to the 25 mL Schlenk tube containing the catalyst, and the vial was rinsed with an additional 2×100 μL DCE, which was added to the Schlenk tube. The tube was sealed and heated to the indicated temperature for the appropriate time period. The crude reaction mixture was directly purified by preparative TLC.
Supplementary Material
Table 4.
Catalysis using chiral dcpp-inspired ligands.
| |||||
|---|---|---|---|---|---|
| Entry | L | Temp [°C] | Time [h] | Yield [%] | ee [%] |
| 1 |
 L10 |
30 | 24 | 22 | 78 |
| 2 | 50 | 24 | 26 | 46 | |
|
| |||||
| 3 |
 (S,S)-L11 |
30 | 45 | 93 | 4 |
|
| |||||
| 4 |
 (SP,SP)-(2R,4R)-L12 |
30 | 24 | 19 | 52 |
| 5 | 50 | 24 | 86 | 37 | |
|
| |||||
| 6 |
 (SP,SP)-(2S,4S)-L13 |
30 | 20 | 67 | 38 |
|
| |||||
| 7 |
(SP,SP)-L14 |
30 | 24 | 88 | 63 |
Acknowledgments
Funding provided by UC Irvine and the National Institutes of Health (GM105938). K. G. M. K. and L. E. L. acknowledge NSERC of Canada for graduate scholarships.
Footnotes
Dedicated to Prof. Stephen L. Buchwald on the occasion of his 60th birthday.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/adsc.201######.
References
- 1.For recent reviews, see: Huang H, Yang L. Chem Rev. 2015 doi: 10.1021/cr500610p.Tsakos M, Schaffert ES, Clement LL, Villadsen NL, Poulsen TB. Nat Prod Rep. 2015;32:605–632. doi: 10.1039/c4np00106k.Liu B, Hu F, Shi BF. ACS Catal. 2015;5:1863–1881.
- 2.For NHC-catalyzed Tishchenko processes, see: Chan A, Scheidt KA. J Am Chem Soc. 2006;128:4558–4559. doi: 10.1021/ja060833a.Sreenivasulu M, Kumar KA, Reddy KS, Kumar KS, Kumar PR, Kumar KB, Chandrasekhar KB, Pal M. Tetrahedron Lett. 2011;52:727–732.Du D, Lu Y, Jin J, Tang W, Lu T. Tetrahedron. 2011;67:7557–7562.
- 3.a) Cronin L, Manoni F, O’Connor CJ, Connon SJ. Angew Chem Int Ed. 2010;49:3045–3048. doi: 10.1002/anie.200907167. [DOI] [PubMed] [Google Scholar]; b) O’Connor CJ, Mononi F, Curran SP, Connon SJ. New J Chem. 2011;35:551–553. [Google Scholar]
- 4.Curran SP, Connon SJ. Org Lett. 2012;14:1074–1077. doi: 10.1021/ol203439g. [DOI] [PubMed] [Google Scholar]
- 5.For a recent review, see: Riener K, Högerl MP, Gigler P, Kühn FE. ACS Catal. 2012;2:613–621.
- 6.a) Evans AP, editor. Modern Rhodium-Catalyzed Organic Reactions. Wiley-VCH; Weinheim: 2005. [Google Scholar]; b) Tang W, Zhang X. Chem Rev. 2003;103:3029–3070. doi: 10.1021/cr020049i. [DOI] [PubMed] [Google Scholar]; c) Xie JH, Zhu SF, Zhou QL. Chem Soc Rev. 2012;41:4126–4139. doi: 10.1039/c2cs35007f. [DOI] [PubMed] [Google Scholar]; d) Roseblade SJ, Pfaltz A. Acc Chem Res. 2007;40:1402–1411. doi: 10.1021/ar700113g. [DOI] [PubMed] [Google Scholar]
- 7.a) van Leeuwen PWNM, Claver C, editors. Rhodium Catalyzed Hydroformylation. Kluwer Academic Publishers; Dordrecht, The Netherlands: 2000. [Google Scholar]; b) Claver C, Diéguez M, Pàmies O, Castillón S. Top Organomet Chem. 2006;18:35–64. [Google Scholar]
- 8.Willis MC. Chem Rev. 2010;110:725–748. doi: 10.1021/cr900096x. [DOI] [PubMed] [Google Scholar]
- 9.For examples of hydroacylation reactions demonstrating a competitive decarbonylation pathway, see: Sakai K, Ide J, Oda O, Nakamura N. Tetrahedron Lett. 1972;13:1287–1290.Milstein D. J Chem Soc, Chem Commun. 1982:1357–1358.Milstein D. Organometallics. 1982;1:1549–1551.Bergens SH, Fairlie DP, Bosnich B. Organometallics. 1990;9:566–571.
- 10.a) Shen Z, Khan HA, Dong VM. J Am Chem Soc. 2008;130:2916–2917. doi: 10.1021/ja7109025. [DOI] [PubMed] [Google Scholar]; b) Shen Z, Dornan PK, Khan HA, Woo TK, Dong VM. J Am Chem Soc. 2009;131:1077–1091. doi: 10.1021/ja806758m. [DOI] [PubMed] [Google Scholar]; c) Khan HA, Kou KGM, Dong VM. Chem Sci. 2011;2:407–410. [Google Scholar]
- 11.Kou KGM, Le DN, Dong VM. J Am Chem Soc. 2014;136:9471–9476. doi: 10.1021/ja504296x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.For intermolecular Rh-catalyzed hydroacylation of alkenes using salicylaldehydes, see: Imai M, Tanaka M, Tanaka K, Yamamoto Y, Imai-Ogata N, Shimowatari M, Nagumo S, Kawahara N, Suemune H. J Org Chem. 2004;69:1144–1150. doi: 10.1021/jo035395u.Tanaka K, Tanaka M, Suemune H. Tetrahedron Lett. 2005;46:6053–6056.Stemmler RT, Bolm C. Adv Synth Catal. 2007;349:1185–1198.Inui Y, Tanaka M, Imai M, Tanaka K, Suemune H. Chem Pharm Bull. 2009;57:1158–1160. doi: 10.1248/cpb.57.1158.Coulter MM, Kou KGM, Galligan B, Dong VM. J Am Chem Soc. 2010;132:16330–16333. doi: 10.1021/ja107198e.Phan DHT, Kou KGM, Dong VM. J Am Chem Soc. 2010;132:16354–16355. doi: 10.1021/ja107738a.Murphy SK, Petrone DA, Coulter MM, Dong VM. Org Lett. 2011;13:6216–6219. doi: 10.1021/ol202663p.Zhang HJ, Bolm C. Org Lett. 2011;13:3900–3903. doi: 10.1021/ol201431c.Murphy SK, Coulter MM, Dong VM. Chem Sci. 2012;3:355–358.von Delius M, Le CM, Dong VM. J Am Chem Soc. 2012;134:15022–15032. doi: 10.1021/ja305593y.
- 13.For intermolecular Rh-catalyzed hydroacylation of alkenes using β-sulfur aldehydes, see: Willis MC, McNally SJ, Beswick PJ. Angew Chem Int Ed. 2004;43:340–343. doi: 10.1002/anie.200352751.Willis MC, Randell-Sly HE, Woodward RL, Currie GS. Org Lett. 2005;7:2249–2251. doi: 10.1021/ol050638l.Moxham GL, Randell-Sly HE, Brayshaw SK, Woodward RL, Weller AS, Willis MC. Angew Chem Int Ed. 2006;45:7618–7622. doi: 10.1002/anie.200603133.Willis MC, Randell-Sly HE, Woodward RL, McNally SJ, Currie GS. J Org Chem. 2006;71:5291–5297. doi: 10.1021/jo060582o.Moxham GL, Randell-Sly H, Brayshaw SK, Weller AS, Willis MC. Chem Eur J. 2008;14:8383–8397. doi: 10.1002/chem.200800738.Osborne JD, Randell-Sly HE, Currie GS, Cowley AR, Willis MC. J Am Chem Soc. 2008;130:17232–17233. doi: 10.1021/ja8069133.Randell-Sly HE, Osborne JD, Woodward RL, Currie GS, Willis MC. Tetrahedron. 2009;65:5110–5117.González-Rodríguez C, Parsons SR, Thompson AL, Willis MC. Chem Eur J. 2010;16:10950–10954. doi: 10.1002/chem.201001748.Pawley RJ, Moxham GL, Dallanegra R, Chaplin AB, Brayshaw SK, Weller AS, Willis MC. Organometallics. 2010;29:1717–1728.González-Rodríguez C, Pawley RJ, Chaplin AB, Thompson AL, Weller AS, Willis MC. Angew Chem Int Ed. 2011;50:5134–5138. doi: 10.1002/anie.201100956.
- 14.For intermolecular Rh-catalyzed hydroacylation of alkenes using non-chelating aldehydes, see: Roy AH, Lenges CP, Brookhart M. J Am Chem Soc. 2007;129:2082–2093. doi: 10.1021/ja066509x.Tanaka K, Shibata Y, Suda T, Hagiwara Y, Hirano M. Org Lett. 2007;9:1215–1218. doi: 10.1021/ol070153s.Shibata Y, Tanaka K. J Am Chem Soc. 2009;131:12552–12553. doi: 10.1021/ja905908z.Murphy SK, Bruch A, Dong VM. Angew Chem Int Ed. 2014;53:2455–2459. doi: 10.1002/anie.201309987.Murphy SK, Bruch A, Dong VM. Chem Sci. 2015;6:174–180. doi: 10.1039/c4sc02026j.
- 15.Khalil MMH, Al-Seif FA. J Coord Chem. 2007;60:1191–1201. [Google Scholar]
- 16.Khalil MMH, Al-Seif FA. Res Lett Inorg Chem. 2008;2008:1–4. [Google Scholar]
- 17.For discussions on how bite angles affect reactivity, see: van Leeuwen PWNM, Kamer PCJ, Reek JNH. Pure Appl Chem. 1999;71:1443–1452.Birkholz (née Gensow) M-N, Freixa Z, van Leeuwen PWNM. Chem Soc Rev. 2009;38:1099–1118. doi: 10.1039/b806211k.
- 18.Leitner W, Bühl M, Fornika R, Six C, Baumann W, Dinjus E, Kessler M, Krüger C, Rufińska A. Organometallics. 1999;18:1196–1206. [Google Scholar]
- 19.Kohrt C, Baumann W, Spannenberg A, Drexler H-J, Gridnev ID, Heller D. Chem—Eur J. 2013;19:7443–7451. doi: 10.1002/chem.201204336. The X-ray crystallography data (CCDC-913096, [Rh(dcpe)(mac)]BF4) and (CCDC-913098, [Rh(dcpb)(cod)]BF4) were obtained from The Cambridge Crystallographic Data Centre. [DOI] [PubMed] [Google Scholar]
- 20.Six C, Gabor B, Görls H, Mynott R, Philipps P, Leitner W. Organometallics. 1999;18:3316–3326. This bite angle is obtained from an X-ray crystal structure of [Ru(dcpp)(η3-C6H8)(η3-C6H8)] [Google Scholar]
- 21.Daumas M, Vo-Quang Y, Vo-Quang L, Le Goffic F. Synthesis. 1989:64–65. Systems that involve directing elements on both coupling partners (i.e., a double-chelation approach) have been found to be unreactive when using catalysts modified with bidentate phosphine ligands (see ref. 12e) [Google Scholar]
- 22.McKinstry L, Livinghouse T. Tetrahedron. 1995;51:7655–7666. [Google Scholar]
- 23.a) Oohara N, Imamoto T. Bull Chem Soc Jpn. 2002;75:1359–1365. [Google Scholar]; b) Imamoto T, Tamura K, Zhang Z, Horiuchi Y, Sugiya M, Yoshida K, Yanagisawa A, Gridnev ID. J Am Chem Soc. 2012;134:1754–1769. doi: 10.1021/ja209700j. [DOI] [PubMed] [Google Scholar]
- 24.Fryzuk MD, Bosnich B. J Am Chem Soc. 1977;99:6262–6267. doi: 10.1021/ja00461a014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.