

Abstract
Cancer is often associated with an increased risk of thrombotic events which are exacerbated by treatment with chemotherapeutics such as cyclosphosphamide (CP). Evidence suggests that thrombin can stimulate tumor progression via formation of fibrin and activation of protease-activated receptors (PARs) and platelets. We examined the effect of co-treatment with CP and dabigatran etexilate, a direct inhibitor of thrombin, using the murine orthotopic 4T1 tumor model. Mice receiving co-treatment with both low dose CP and dabigatran etexilate had significantly smaller mammary tumors and fewer lung metastases than mice treated with CP or dabigratran etexilate alone. Co-treatment with dabigatran etexilate and low dose CP also significantly decreased the number of arginase+Gr-1+CD11b+ myeloid derived suppressor cells as well as levels of TGF-β in spleens from tumor bearing mice. 4T1 tumors express procoagulant tissue factor (TF) and spontaneously release TF+ microparticles which are potent procoagulant factors that promote thrombin generation. Treatment with dabigatran etexilate alone prevented tumor-induced increases in circulating TF+ microparticles and also decreased the numbers of tumor-induced activated platelets by 40%. These results show that co-treatment with dabigatran etexilate and CP synergistically inhibits growth and metastasis of mammary tumors, suggesting that oral administration of the thrombin inhibitor dabigatran etexilate may be beneficial in not only preventing thrombotic events in cancer patients but also in treating malignant tumors themselves.
Keywords: breast cancer, metastasis, TGF-β, microparticles, Thrombin inhibitor
Abbreviations
- CP
cyclophosphamide
- MPs
microparticles
- PAR
protease-activated receptors
- PPP
platelet poor plasma
- PS
phosphatidylserine
- TAT
thrombin-antithrombin complex
- TF
tissue factor
- VTE
venous thromobembolism
- MDSC
myeloid derived suppressor cell
- Treg
regulatory T cell.
Introduction
The association between cancer and thrombosis has been known since 1865 when Armand Trousseau described the clinical association between venous thrombosis and cancer. Large epidemiological studies have shown that cancer patients have an elevated risk of developing a venous thromobembolism (VTE), defined as deep vein thrombosis and/or pulmonary embolism.1-3 VTE is one of the main causes of death during the progression of many types of cancer.4 Additionally, metastatic cancer has been associated with an increased risk for hemostatic complications.1 Cancer commonly induces abnormalities in one or more coagulation parameters, even without overt thrombotic events. Many hemostatic changes can be detected in cancer patients, such as plasma by-products of the clotting reactions (i.e. thrombin-antithrombin complex (TAT) or D-dimer) or high levels of procoagulant tissue factor (TF)-positive microparticles shed by tumor cells and platelets.5,6 Further compounding the risk for thrombotic complications, treatment with chemotherapeutic agents such as cyclophosphamide, fluorouracil-leucovorin, cisplatin, dexamethasone, vincristine, doxorubicin, and etoposide increases the incidence of symptomatic venous thromboembolism in cancer patients.7-10 The PROTECHT (PROphylaxis of ThromboEmbolism during CHemoTherapy) retrospective study confirmed chemotherapy to be an independent risk factor for thromboembolism in a wide cancer population.7,11 Although the mechanism is not well understood, the use of chemotherapeutic agents has pro-thrombotic side-effects associated with platelet activation, the release of nucleic acids, increased cellular exposure of phosphadidylserine (PS), and the elevation of TF+ microparticles.12,13
Circulating microparticles (100 – 1000 nm in size) originate from plasma membrane and are released by most cell types upon activation or when undergoing apoptosis, with platelets being the major sources of circulating microparticles in the blood.14 Microparticles may act as circulating signaling modules by activating receptors on target cells or by directly transferring part of their contents to recipient cells. Importantly, tumor cells express TF and spontaneously release TF+ microparticles into the blood.15 TF is a transmembrane receptor that binds plasma factor VII/VIIa and is the primary initiator of blood coagulation.16 TF+ microparticles are extremely potent pro-coagulant factors that can promote the conversion of prothrombin to thrombin. Accumulating reports suggest that these circulating TF+ microparticles contribute to the increased incidence of VTE seen in cancer patients.17,18
Thrombin is the main effector protease of the coagulation cascade generated by the action of TF and other coagulation factors. Thrombin converts fibrinogen to fibrin which is deposited within the tumor vasculature, facilitating angiogenesis by providing a scaffold for new vessel formation.19 The fibrin matrix can also bind and sequester pro-tumor growth factors (i.e., insulin-like growth factor and vascular endothelial growth factor) and protect them from proteolytic degradation.20 Fibrin deposition favors metastatic progression by providing a favorable microenvironment for tumor cell adhesion and growth.21,22 In addition, thrombin is a potent activator of platelet adhesion, aggregation, and secretion. Platelet activation not only contributes to the hypercoagulation state in cancer patients but also promotes tumor growth, angiogenesis, and metastasis.22-24
Independent of clotting mechanisms, thrombin and TF participate in tumor progression via multiple mechanisms that result from interactions with specific receptors belonging to the family of protease-activated receptors (PAR) that are expressed by a variety of cell types including tumor cells, endothelial cells, vascular smooth muscle cells, macrophages and platelets.25,26 Thrombin cleaved PAR-1 stimulates growth factors, chemokines and the release of extracellular proteins that promote tumor cell proliferation and migration.27
Clinical studies have suggested that anticoagulation therapies, such as warfarin and low molecular weight heparins, interfere with metastatic cancer disease and increase cancer survival rates in addition to preventing hemostatic complications.28-31 However, warfarin requires frequent coagulation monitoring due to interactions with various foods and medications and low molecular weight heparins require daily subcutaneous injections, thus complicating their use in cancer patients.32 However dabigatran etexilate (Pradaxa®), a small molecule direct thrombin inhibitor, can be administered orally without the need to monitor anticoagulation status, providing stable, predictable inhibition of thrombin.33 Dabigatran etexilate is administrated as a prodrug which is rapidly converted to the active form of the drug (BIBR 953ZW) and achieves steady-state levels within 3 d.34
Because chemotherapy increases the development of a hypercoagulation state that has been shown to drive the progression of cancer, we tested the hypothesis that inhibition of thrombin with dabigatran etexilate will act synergistically with cyclophosphamide (CP), a common chemotherapeutic used to treat breast cancer, to inhibit tumor growth and metastasis. Using a murine orthotopic model of breast cancer, we show greater anti-tumor efficacy with dabigatran etexilate and CP co-treatment that is also accompanied by decreased numbers of circulating TF+ microparticles and platelet activation.
Results
Dabigatran etexilate and cyclophosphamide synergistically inhibit tumor growth and metastasis in vivo
Because chemotherapeutic agents have pro-thrombotic side-effects which can confer advantages to cancer cell growth, we evaluated the effect of thrombin inhibition with dabigatran etexilate in conjunction with low dose chemotherapeutic treatment using the 4T1 mammary adenocarcinoma model. Whereas we previously reported that treatment with dabigatran etexilate initiated before orthotopic injection of 4T1 tumor cells inhibited both the growth and metastasis of 4T1 mammary carcinomas,35 we tested the extent to which treatment with dabigatran and/or low dose cyclophosphamide might exert a therapeutic effect when treatment is initiated after appearance of a palpable primary tumor. Following orthotopic injection of 4T1 tumor cells in the mammary fat pad of Balb/c mice, treatment was initiated when the tumors were between 25–50 mm3. Mice were given 50 mg/kg CP once a week, with or without dabigatran etexilate administered by oral gavage (80 mg/kg twice daily, Monday-Friday, and 120 mg/kg once a day on weekends). Whereas the low dose of CP (50 mg/kg) alone resulted in a moderate inhibition of tumor growth, treatment with dabigatran etexilate alone had no effect on tumor growth when initiated 2 weeks after tumor cell injection when a primary tumor is already measurable. In contrast, there was a synergistic inhibitory effect on tumor growth in mice treated with both dabigatran etexilate and CP (Fig. 1A). Treatment with a lower dose of CP (25 mg/kg), which by itself had no effect on tumor growth, significantly inhibited tumor growth when mice were co-treated with dabigatran etexilate and CP (Fig. 1C).
Figure 1.
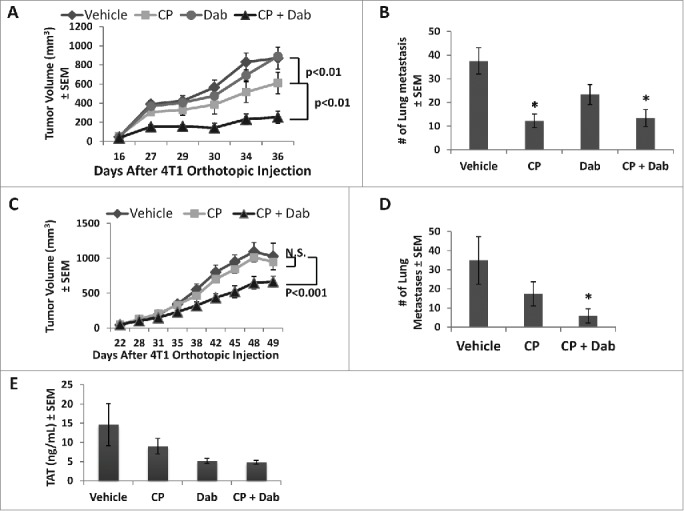
Inhibitory effects of CP and dabigatran etexilate co-treatment on 4T1 tumor growth, metastases, and thrombin antithrombin (TAT) complex. Balb/c mice were orthotopically injected with 2 × 104 4T1 cells in the inguinal mammary fat pad. When the primary tumors were between 25–50 mm3 in size, treatment was initiated with i.p. injection of cyclophosphamide (CP) at 50 mg/kg (A, B) or 25 mg/kg (C, D, E) once weekly with or without dabigatran etexilate (Dab) administration by oral gavage (80 mg/kg twice daily, Mon-Fri, and 120 mg/kg once a day on weekends). Mice were sacrificed 24 hrs after last CP treatment. (A, C) Graph shows tumor growth rate and volume in mice bearing 4T1 tumors under different treatments (n = 5–9 mice/treatment group; mean volume per time point ± SEM). (B, D) Lungs were perfused with India ink to visualize surface lung metastatic nodules of mice treated with 50 mg/kg (B) or 25 mg/kg (D) of CP. (E) Platelet poor plasma (PPP) was used to determine levels of TAT by ELISA assay. * = p < 0.05 compared to control vehicle-treated tumor bearing mice.
4T1 tumor cells metastasize efficiently to multiple organs affected in human breast cancer, particularly to the lungs.36 By 4 weeks following the injection of 4T1 cells, lung metastases were visible in most animals with no or very few macroscopic metastases observed in other tissues. Treatment with CP (50 mg/kg) with or without dabigatran etexilate significantly lowered the number of metastatic lung nodules compared to that in untreated tumor bearing mice (Fig. 1B). However, only the co-treatment group saw significant reductions in metastatic lung nodules when the lower dose of CP (25 mg/kg) was used (Fig. 1D).
To characterize the effect of treatment on in vivo procoagulant activity, we measured circulating TATs. TAT complexes are formed by the neutralization of thrombin by antithrombin III and have been used as a surrogate measure of thrombin generation.37 Treatment with dabigatran etexilate with or without CP lowered the levels of circulating TAT complexes compared to that seen in control vehicle-treated tumor-bearing mice (Fig. 1E). In general, TAT levels correlated with tumor size, suggesting that differences in effects of treatment with dabigatran etexilate on tumor growth are associated with differential procoagulant activity in the mice.
Dabigatran etexilate treatment inhibits the generation of tissue factor (TF) positive microparticles in vivo
Exposure of anionic phospholipids, such as PS, on the surface of cells and microparticles is associated with increased TF activity.38 Studies have implicated the expression of procoagulant molecules such as TF and microparticles in the development of systemic coagulopathies in cancer patients 17,18 and possibly tumor progression.39 It has also been hypothesized that chemotherapy may cause an increased shedding of TF+ microparticles, which are potent initiators of the coagulation cascade as activators of thrombin, from apoptotic endothelial, tumor cells or activated platelets.13 To investigate this, we exposed 4T1 tumor cells to the cytotoxic chemotherapeutic doxorubicin. Treatment doses of doxorubicin were chosen to parallel plasma concentrations achieved in patients.15 Cells exposed to doxorubicin released 3 times as many TF+ microparticles as untreated cells (Fig. 2A). The majority (>90 %) of microparticles shed from 4T1 cells expressed the tumor epithelial marker CD326 (data not shown). To investigate the effect of chemotherapy on circulating TF+ microparticles in vivo, non-tumor bearing mice were i.p. injected with a single dose of 200 mg/kg CP and sacrificed at 2, 6 and 24 h post injection. Blood was drawn from the vena cava and microparticles in PPP were analyzed. Levels of TF+ microparticles increased with administration of CP, peaking at 6 h post injection (data not shown). TF+ microparticles remained elevated at 24 h post injection, but at reduced levels compared to 6 h post injection. To determine if treatment with dabigatran etexilate could ameliorate this response, non-tumor bearing mice were fed dabigatran chow (10 mg dabigatran etexilate/g) ad lib for 2 d prior to i.p. injection of CP. At 6 h after CP injection, circulating TF+ microparticles were increased almost fold10- in mice injected with CP compared to untreated mice (Fig. 2B). However, treatment with dabigatran etexilate prevented the CP induction of circulating TF+ microparticles.
Figure 2.

Effect of CP and dabigatran etexilate co-treatment on circulating microparticles (MPs). 4T1 cells in culture were treated with 1µg/mL of doxorubicin for 24 hours and compared to untreated cells. Conditioned culture media was analyzed by flow cytometry for TF+ MPs (A). Non-tumor bearing mice were i.p. injected with 200 mg/kg of CP with or without Dabigatran chow (10 mg dabigatran etexilate/g food pellet) ad lib for 2 d prior to CP injection. Mice were sacrificed 6 h after CP injection. TF+ MPs in PPP were quantified by flow cytometry (B). Number of MPs in PPP isolated from 4T1 tumor bearing mice (C). Percentage of MPs positive for TF in PPP isolated from 4T1 tumor bearing mice (D). Cell origin of TF+ MPs stained with anti-CD41 (platelets), anti-CD45 (leukocytes), anti-CD31 (endothelial cells) and anti-CD326 (epithelial cells) antibodies in PPP isolated from 4T1 tumor bearing mice (E). Cell origin of total MPs in PPP isolated from 4T1 tumor bearing mice (F). Percentage of total MPs of platelet origin in PPP isolated from 4T1 tumor bearing mice (G). n = 4–8 mice per group. * = p < 0.05, # = p< 0.01 compared to control vehicle-treated tumor bearing mice.
Studies have shown that the numbers of circulating microparticles is increased in cancer patients compared to healthy individuals.40 Similarly, untreated mice with 4T1 tumors showed a fold12- increase in numbers of circulating microparticles compared to nontumor-bearing mice (Fig. 2C). Treatment with low dose CP (25 mg/kg) did not significantly affect microparticle numbers compared to vehicle-treated, tumor-bearing mice. Administration of dabigatran extexilate with or without CP prevented the tumor-induction of circulating microparticles resulting in numbers of microparticles similar to that in non-tumor bearing normal mice (Fig. 2C).
Since circulating TF+ microparticles are elevated in patients with advanced breast cancer,18 we also investigated the effect of treatment with dabigatran etexilate with and without CP on the percentage of microparticles that were positive for TF. Control vehicle-treated mice with 4T1 tumors not only had more circulating microparticles compared to normal mice, but a much higher percentage of those microparticles were positive for TF (58% versus 4%) (Fig. 2D). Treatment of tumor bearing mice with low dose CP did not significantly affect the percentage of TF+ microparticles. Administration of dabigatran etexilate alone resulted in significant fold5- reduction in percentage of TF+ microparticles compared to tumor-bearing control mice while co-treatment with dabigatran etexilate and CP reduced the percentage of TF+ microparticles to that of non-tumor bearing normal mice (Fig. 2D).
The composition of the microparticles depends on their cell origin, with microparticles from different cells containing different profiles of proteins, lipids and nucleic acids. Circulating microparticles from control vehicle-treated tumor-bearing mice were stained for CD41 (platelets), CD45 (leukocytes), CD31 (endothelial cells) and CD326 (4T1 epithelial cells) to determine the microparticle cell origin. Circulating TF+ microparticles in 4T1 tumor bearing mice were primarily of platelet and tumor epithelial origin, with equal numbers staining for CD41 and CD326 (Fig. 2E). Due to the low number of circulating TF+ microparticles in tumor bearing mice in the dabigatran ± CP treatment groups, we were unable to reliably determine their cellular origin. Total circulating microparticles in 4T1 tumor bearing control mice were primarily of platelet origin with 72.3 ± 6.2% of the microparticles staining positively for CD41 (Fig. 2F). Treatment with CP alone did not alter the percentage of circulating microparticles originating from platelets; however, treatment with dabigatran etexilate with or without CP caused a dramatic reduction to 32.7% ± 7.7% and 36.3% ± 4.4%, respectively (Fig. 2G). There were no significant effects of treatment with dabigatran etexilate with or without CP on CD45, CD31 and CD326 positive microparticles (data not shown).
Dabigatran etexilate reduces platelet activation in 4T1 tumor bearing mice
Another potent initiator of the clotting cascade is activated platelets, which can be activated by thrombin. Red blood cells in the blood of 4T1 tumor bearing mice were lysed by ammonium oxalate and the platelets counted. The number of platelets per µl of blood was similar in all groups (Fig. 3A). Platelets were then assessed for activation by flow cytometric analysis after staining for CD41, a platelet marker, and CD62p (p-selectin) which is expressed upon platelet activation. The number of activated platelets was dramatically increased in 4T1 tumor bearing control mice compared to non-tumor bearing normal mice, increasing from 15,858 ± 596 activated platelets per µl of blood in nontumor bearing mice to 68,401 ± 8,208 activated platelets per µl of blood in untreated 4T1 tumor bearing control mice (Fig. 3B). Treatment with dabigatran etexilate with or without CP significantly reduced the number of activated platelets by similar amounts (Fig. 3B). Treatment with CP alone also reduced the number of activated platelets in tumor bearing mice even though, at this dose, CP had no effect on tumor growth, metastasis or microparticle generation, indicating that while low dose CP has negligible effects on the tumor it still may have physiological effects in the animals.
Figure 3.
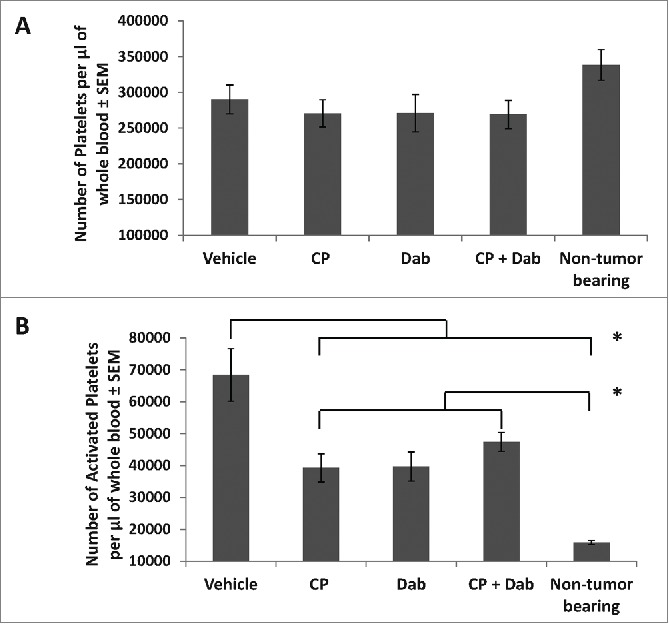
Dabigatran etexilate reduces platelet activation in tumor-bearing mice. Balb/c mice were orthotopically injected with 2 × 104 4T1 cells in the inguinal mammary fat pad. When the primary tumors were between 25–50 mm3 in size, treatment was initiated with i.p. injection of CP at 15 mg/kg once weekly with or without dabigatran (administration by oral gavage 80 mg/kg twice daily, Mon-Fri, and dabigatran chow (10 mg dabigatran etexilate/g food pellet) on the weekends). Mice were sacrificed 24 hrs after last CP treatment. Blood was collected and treated with ammonium oxalate to lyse red blood cells. Platelets were counted manually using a hemocytometer (A). Activated platelets were identified by flow cytometry (B). n = 7–8 mice per group. * = p < 0.05 compared to indicated group.
Dabigatran etexilate reduces a subpopulation of splenic myeloid-derived suppressor cells
As previously reported,41 4T1 tumor-bearing mice have significantly larger spleens than non-tumor bearing mice (Fig. 4A). This splenomegaly is caused by a dramatic leukemoid reaction resulting in a massive granulocytic infiltration in the spleen. Treatment with either CP or dabigatran etexilate decreased the tumor induced splenomegaly but co-treatment with both dabigatran etexilate and CP maintained spleen weights comparable to that of non-tumor bearing mice (Fig. 4A). Spleens from 4T1 tumor bearing mice were further analyzed for inflammatory infiltrates by flow cytometry analysis. Following treatment with dabigatran etexilate and CP there were no significant differences in total CD45+ immune cells in viable, disaggregated spleens (data not shown). Treatment with either CP or dabigatran etexilate resulted in only minor differences in CD45+ leukocyte subpopulations in the spleens of tumor bearing mice; however, co-treatment with CP and dabigatran etexilate significantly altered their distribution (Fig. 4B). Co-treatment with CP and dabigatran etexilate decreased the population of arginase+ CD11b+ Gr-1+ cells which are markers for myeloid derived suppressor cells (MDSC), a major immunosuppressive cell population found in many tumors (Fig. 4B). Since TGF-β plays a crucial role in the generation, accumulation, and immunosuppressive effects of immune suppressive cells including MDSCs and regulatory T cells (Tregs), we examined the effect of dabigatran etexilate and/or CP on protein levels of TGF-β in the spleen of 4T1 tumor-bearing mice. Whereas treatment with only CP or dabigatran did not significantly decrease splenic TGF-β levels, co-treatment with both CP and dabigatran etexilate reduced TGF-β levels in tumor bearing mice to those found in spleens from nontumor-bearing mice (Fig. 4C).
Figure 4.

Dabigatran etexilate reduces 4T1 tumor-induced splenomegaly, immunosuppressive cell population, and TGF-β. Balb/c mice were orthotopically injected with 2 × 104 4T1 cells in the inguinal mammary fat pad. When the primary tumors were between 25–50 mm3 in size, treatment was initiated with i.p. injection of CP at 15 mg/kg once weekly with or without dabigatran (administration by oral gavage 80 mg/kg twice daily, Mon-Fri, and dabigatran chow (10 mg dabigatran etexilate/g food pellet) on the weekends). Mice were sacrificed 24 hrs after last CP treatment. Spleens were excised and (A) weighed, (B) analyzed for the percentage of arginase+ Gr-1+ CD11b+ cells in the population of CD45+ splenocytes by flow cytometry, and (C) assayed for levels of TGF-β protein by Cytokine Bead Array and flow cytometry. n = 7–8 mice per group. * = p < 0.05; # = p< 0.01 compared to control vehicle-treated tumor bearing mice or indicated group.
Discussion
Thrombotic complications are one of the primary causes of death in patients with advanced stages of cancer,42 and the pro-thrombotic microenvironment also promotes tumor growth and metastasis.19 Although chemotherapy is a mainstay of treatment for many types of cancer, it can also greatly increase the risk of systemic coagulopathies in cancer patients. Consequently, we hypothesized that inhibiting thrombin may increase the anti-tumor efficacy of a chemotherapeutic agent that promotes a pro-thrombotic state. Our results show, for the first time, that co-treatment with CP and dabigatran etexilate, an orally administered direct thrombin inhibitor, synergistically inhibits mammary tumor growth and metastasis in vivo.
The pathogenesis of blood coagulation in cancer is complex, but one key element is the expression of TF on the tumor cell surface and its active release as TF+ microparticles.43-45 Microparticles released by tumor cells have exposed PS on their surface, which in conjunction with TF, are potent initiators of the conversion of prothrombin to thrombin. As has been reported in human cancer patients,46-49 we observed a significant increase in the number of circulating TF+ microparticles in 4T1 tumor-bearing mice compared with non-tumor bearing mice. Importantly, thrombin inhibition with dabigatran etexilate treatment completely prevented the tumor-induced increase in circulating TF+ microparticles.
We and others 15 have shown that treatment with chemotherapeutic agents increases the number of shed TF+ microparticles from tumor cells in vitro as well as circulating TF+ microparticles in nontumor-bearing mice treated with cyclophosphamide. However, no increase in circulating TF+ microparticles was seen in 4T1 tumor bearing mice administered low dose cyclophosphamide. Similarly, no increase in circulating TF+ microparticles was observed in plasma samples from early-stage breast cancer patients drawn either 2 or 8 d after the start of chemotherapy.50 It is possible that an earlier time point is needed to observe a chemotherapy-induction of circulating TF+ microparticles since we observed a sharp increase in circulating TF+ microparticles within hours following a single injection of CP in non-tumor bearing mice. Alternatively, chemotherapeutic agents may promote a procoagulant state via other mechanisms such as tumor cell lysis and the subsequent release of cell-free DNA that can induce thrombosis.51 Animal studies have also shown that cyclophosphamide and acrolein, a by-product of cyclophosphamide metabolism, are potent pro-oxidative stressors that upregulate procoagulant pathways that generate thrombin while impairing endogenous anticoagulant pathways such as activated protein C.52
Thrombin has been shown to directly promote tumor growth and metastasis by many different mechanisms (Fig. 5). For instance, thrombin activity leads to fibrin deposition in the tumor vasculature which can provide a scaffold for new vessel formation and support angiogenesis. This fibrin matrix can also bind to pro-tumorigenic growth factors, such as VEGF and IGF-1, and protect them from proteolytic degradation.20 Fibrin deposition has also been shown to facilitate metastasis by stabilizing tumor cells as they adhere to the endothelium.20 Thrombin also promotes tumor growth and metastasis by activating the G-coupled 7 transmembrane protease-activated receptor (PAR-1) by cleaving the receptor's N-terminal end, which folds back on the second extracellular loop and activates the cell. PAR-1 activation stimulates many different signaling cascades that have been implicated in tumor growth, survival, angiogenesis, and metastasis.53,54 Thrombin promotes angiogenesis by inducing VEGF expression in tumor cells and by upregulating the VEGF receptors, VEGFR2 and angiopoietin-2 in endothelial cells.55-57
Figure 5.
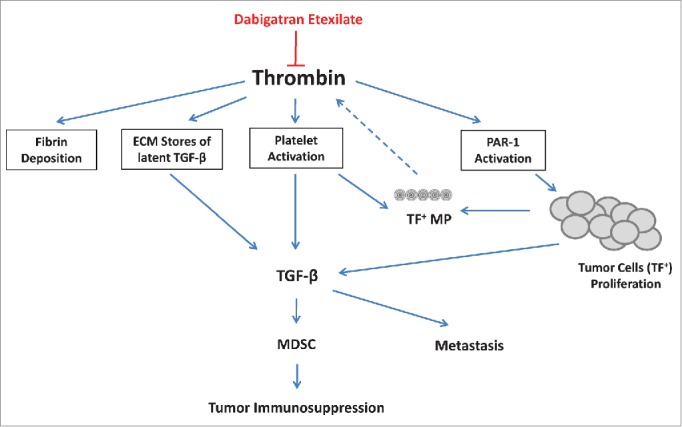
Inhibition of cancer progression by dabigatran etexilate. Thrombin has many activities that can support cancer progression and metastasis including fibrin deposition, PAR-1 activation (stimulating tumor cell proliferation), and platelet activation. Thrombin can induce TGF-β both via the release of TGF-β from activated platelets and via the release of latent TGF-β from extracellular matrix stores. Tumor cells also produce large amounts of TGF-β which strongly impacts the generation and expansion of myeloid derived suppresser cells (MDSC) that contribute to an immunosuppressive microenvironment. Dabigatran etexilate directly inhibits thrombin, potentially disrupting many of these protumorigenic pathways.
Thrombin is also a potent activator of platelets, which enhance metastasis via multiple mechanisms. Activated platelets interact with tumor cells during their transit through the circulation, forming tumor-platelet emboli, which can impair the ability of the immune system to identify and eliminate malignant cells.58 Activated platelets secrete growth factors, chemokines, and cytokines including TGF-β, PDGF, VEGF, angiopoietin-1, and lysophosphatidic acid.19 Platelet-derived TGF-β inhibits natural killer cell activity by down-regulating the activating immunoreceptor natural killer group 2, member D (NKG2D) on natural killer cells.59 TGF-β secreted from activated platelets combined with direct contact with tumor cells also activates TGF-β/Smad and NFκB pathways in tumor cells resulting in their transition to an invasive epithelial-mesenchymal-like phenotype and promoting metastasis.60
Treatment with both dabigatran etexilate and CP not only synergistically inhibited tumor growth but also inhibited the splenomegaly and accumulation of arginase+ Gr-1+ CD11b+ MDSCs characteristically seen in 4T1 tumor-bearing mice. There is a good correlation between tumor progression and accumulation of MDSCs in many types of cancer.61 MDSCs are key mediators of immunosuppression in cancer, and chronic inflammation has been shown to enhance the accumulation of MDSC and to increase their suppression of T cells.62 Thrombin has been demonstrated to drive inflammatory responses in many pathologies including cancer.63 In addition to proinflammatory cytokines such as IL-1β, IL-6, and PGE2, other tumor-derived cytokines such as TGF-β and VEGF are also involved in MDSC expansion.61 Thrombin is a potent inducer of TGF-β, both by activating platelets to cause the release of TGF-β from the α-granules of platelets and via the release of latent TGF-β from extracellular matrix stores (Fig. 5). MDSCs are actively recruited to platelet-tumor microthrombi by secretion of chemokines from activated platelets, while the in vivo depletion of platelets prevents the accumulation of MDSCs in the tumor microenvironment.64 Interestingly, in some cases low dose CP has been reported to enhance chronic inflammation and increase the accumulation of MDSCs in tumor-bearing animals.65 We show that the anti-tumor effect of co-treatment with both dabigatran etexilate and CP correlates with the reduced accumulation of immunosuppressive MDSCs and splenic TGF-β levels.
Since thrombin appears to act as a tumor modifier, thrombin inhibition with dabigatran etexilate potentially impacts tumor growth and metastasis via multiple pathways. We found that the numbers of circulating TF+ microparticles were dramatically increased in tumor bearing mice, likely contributing to increased thrombin generation. Although thrombin inhibition with dabigatran etexilate significantly reduced circulating TF+ microparticles and reduced levels of thrombin-antithrombin complexes in 4T1 tumor bearing mice, dabigatran treatment alone was insufficient to inhibit tumor growth and metastasis. Treatment with both dabigatran etexilate and low dose CP was needed to inhibit tumor growth and metastasis.
Because thrombin promotes tumor growth and metastasis, administration of a thrombin inhibitor may not only alleviate the thrombotic events often associated with cancer, but also impacts tumor progression and metastasis. These results suggest that inhibition of thrombin contributes to the synergistic inhibition of tumor growth and metastasis with co-treatment with low dose CP and dabigatran etexilate. The use of thrombin inhibitors may be warranted not only to treat thrombotic events associated with cancer but also as adjunct therapy to directly treat malignant cancers.
Materials and Methods
Animals
Female Balb/c mice were obtained from Charles Rivers/NCI. Protocols for the use of animals in these studies were reviewed and approved by the Institutional Animal Care and Use Committee of the Lankenau Institute for Medical Research in accordance with the current US Department of Agriculture, Department of Health and Human Service regulations and standards.
Cell Culture
4T1 mouse mammary carcinoma cell line was obtained from American Type Culture Collection (Rockville, MD). Cells were cultured in DMEM supplemented with 10% FBS and 1X Penicillin/Streptomycin (Cellgro). 4T1 cells were treated with 1µg/mL of doxorubicin in 24 well plates with 750 µl of media to generate microparticles. After 24 hr culture, conditioned medium was spun at 2,300 rpm (500 × g) for 5 minutes to remove cells. Supernatants were collected and spun at 4,400 rpm (1,500 × g) as an additional clearance spin.
In vivo 4T1 tumor model
Female Balb/c mice were orthotopically injected in the mammary fat pad with 2.0 × 104 4T1 cells. Tumor growth was assessed morphometrically using calipers and tumor volumes were calculated using the formula V(mm3) = π/6 × A × B2 where A is the larger diameter and B is the smaller diameter of the tumor.66 Once tumors were between 25–50 mm3 in size (approximately 2–3 weeks after tumor cell injection), treatment was initiated. Cyclophosphamide (at doses of 15–50 mg/kg) was intraperitoneally (i.p.) injected once weekly with or without dabigatran etexilate administration. Mice were dosed with dabigatran etexilate by oral gavage twice daily (80 mg/kg) Monday through Friday and either placed on dabigatran chow (10 mg/g chow) or orally gavaged daily (120 mg/kg) over the weekends. Mice were sacrificed 3–7 weeks after tumor cell injection. Tumors and spleens were excised and either fixed in 4% paraformaldehyde, snap frozen in liquid nitrogen or processed for flow cytometry analysis. Lungs were perfused with India ink to count metastases. Blood was collected from the vena cava using a syringe pre-loaded with 100 ul of citrate-dextrose solution. 400 ul of blood was collected for a total volume of 500 ul. To count platelets, 50 ul of whole blood was diluted 1:20 with 1% ammonium oxalate monohydrate and set to shake at room temperature for 10 min to lyse red blood cells. After the red blood cells were lysed, platelets were manually counted using a hemocytometer and assessed for activation by flow cytometry. The remaining blood was sequentially centrifuged to separate platelet poor plasma (PPP) by spinning at 1400 rpm (200 × g) for 20 min and 4400 rpm (1500 × g) for 15 min at 4˚C. The PPP was frozen at −80˚C for later analysis.
Flow cytometry analysis of tissue samples and platelets
Spleens were placed in 5 mL red cell lysis buffer (0.17 M Tris-HCL, 0.16 M NH4Cl) for 15 m, pressed through a 70 µm filter and then incubated with an additional 5 mL of red cell lysis buffer for 15 m. Cells were spun down and resuspended in FACS buffer (1.5% heat inactivated FBS, 0.2% NaN3 in PBS). Equal numbers of viable cells were stained with goat polyclonal anti-arginase, PE-conjugated anti-CD11b, PE-Cy7-conjugated anti-Gr-1, and APC Cy7-conjugated anti-CD45 (all antibodies from eBioscience), followed by incubation with an FITC conjugated anti-goat IgG (Novus Biologicals) to detect arginase positive cells. Equal numbers of viable platelets were stained with PE-Cy7 conjugated anti-CD41 and PE conjugated anti-CD62P (eBioscience). Flow-cytometric data were acquired on a BD FACSCanto II and analyzed using FACSDiva software (BD Biosciences). Viable cells were gated based on forward and side scatter profiles.
Flow cytometry analysis of tissue factor positive microparticles
For flow-cytometric analysis, 30 µl of PPP or tissue culture media were diluted with 70 µl of Annexin V binding buffer (eBioscience) and stained with APC conjugated Annexin V (eBioscience) and goat polyclonal anti-mouse TF antibody (RD systems) followed by incubation with a FITC conjugated anti-goat IgG secondary antibody (Novus Biologicals).17 After incubation, samples were diluted with 1.5 ml of Annexin V binding buffer. Analysis of samples was conducted using a BD FACSCanto II and analyzed using FACSDiva software. Microparticles were identified by size (forward scatter) and annexin V binding. All annexin V positive events smaller than 1 µm were counted as microparticles. The size of the gate was adjusted using fluorescent microbeads measuring 0.5, 1.0 and 3.0 µm (Molecular Probes). Thresholds for TF-positive microparticles were set from annexin V binding positive microparticles. To identify the cell origin of microparticles, PPP was stained with PE-Cy7-conjugated anti-CD41 (eBioscience) as a platelet marker, APC-Cy7 conjugated anti-CD45 (BD PharMingen) as a leukocyte marker, PerCP-Cy5.5 conjugated anti-CD31 (BD PharMingen) as an endothelial marker, PE-conjugated anti-CD326 (BD PharMingen) as a marker for 4T1 tumor epithelial cells as well as staining for Annexin V and TF as described above.
Mouse thrombin-antithrombin (TAT) complex ELISA
PPP samples were diluted 1:4 and then assayed with the mouse TAT complex ELISA kit (AssayPro) as per the manufacturer's instructions. The absorbance was read on a microplate reader at a wavelength of 450 nm, and mouse TAT complex concentrations calculated by comparing with a standard curve.
Cytokine bead array analysis
Spleens were homogenized in PBS with protease inhibitors and DTT. The debris was cleared by centrifugation and the supernatants were analyzed using TGF-β Cytometric Bead Array reagents (BD Biosciences, San Jose, CA) and flow cytometry as per the manufacturer's protocol.
Statistics
All in vivo experiments were carried out using multiple animals (n = 4–8 per experimental group). All in vitro experiments were performed in at least triplicate, and data compiled from 2–3 separate experiments. All analyses, except for tumor growth curves, were done using a 1-way ANOVA with a tukey test for statistical significance. Tumor growth curves were analyzed using a linear mixed effects model that accounted for repeated measurements per animal, with fixed effects of treatment group, time and the interaction of time and treatment group.
Disclosure of Potential Conflicts of Interest
Susan Gilmour received funding support from Boehringer Ingelheim International GmbH. Joanne Van Ryn and Ashley Goss are employed by but not shareholders of Boehringer Ingelheim International GMbH.
Funding
Susan Gilmour received funding support from Boehringer Ingelheim International GmbH. Joanne Van Ryn and Ashley Goss are employed by but not shareholders of Boehringer Ingelheim International GMbH.
References
- 1.Wun T, White RH. Epidemiology of cancer-related venous thromboembolism. Best Pract Res Clin Haematol 2009; 22(1):p. 9-23; PMID:19285269; http://dx.doi.org/ 10.1016/j.beha.2008.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Khorana AA, Francis CW, Culakova E, Kuderer NM, Lyman GH. Frequency, risk factors, and trends for venous thromboembolism among hospitalized cancer patients. Cancer 2007; 110(10):p. 2339-46; PMID:17918266; http://dx.doi.org/ 10.1002/cncr.23062 [DOI] [PubMed] [Google Scholar]
- 3.Lee AY, Levine MN. Venous thromboembolism and cancer: risks and outcomes. Circulation 2003; 107(23 Suppl 1):p. I17-21; PMID:12814981 [DOI] [PubMed] [Google Scholar]
- 4.Sorensen HT, Johnsen SP, Nørgård B, Zacharski LR, Baron JA. Cancer and venous thromboembolism: a multidisciplinary approach. Clin Lab 2003; 49(11-12):p. 615-623; PMID:14651332 [PubMed] [Google Scholar]
- 5.Falanga A, Russo L. Epidemiology, risk and outcomes of venous thromboembolism in cancer. Hamostaseologie 2012; 32(2):p. 115-25; PMID:21971578; http://dx.doi.org/ 10.5482/ha-1170 [DOI] [PubMed] [Google Scholar]
- 6.Falanga A, Marchetti M, Vignoli A, Balducci D. Clotting mechanisms and cancer: implications in thrombus formation and tumor progression. Clin Adv Hematol Oncol 2003; 1(11):p. 673-8; PMID:16258469 [PubMed] [Google Scholar]
- 7.Barni S, Labianca R, Agnelli G, Bonizzoni E, Verso M, Mandalà M, Brighenti M, Petrelli F, Bianchini C, Perrone T, et al.. Chemotherapy-associated thromboembolic risk in cancer outpatients and effect of nadroparin thromboprophylaxis: results of a retrospective analysis of the PROTECHT study. J Transl Med 2011; 9:p. 179; PMID:22013950; http://dx.doi.org/ 10.1186/1479-5876-9-179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moore RA, Adel N, Riedel E, Bhutani M, Feldman DR, Tabbara NE, Soff G, Parameswaran R, Hassoun H. High incidence of thromboembolic events in patients treated with cisplatin-based chemotherapy: a large retrospective analysis. J Clin Oncol 2011; 29(25):p. 3466-73; PMID:21810688; http://dx.doi.org/ 10.1200/JCO.2011.35.5669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Otten HM, Mathijssen J, ten Cate H, Soesan M, Inghels M, Richel DJ, Prins MH. Symptomatic venous thromboembolism in cancer patients treated with chemotherapy: an underestimated phenomenon. Arch Intern Med 2004; 164(2):p. 190-4; PMID:14744843; http://dx.doi.org/ 10.1001/archinte.164.2.190 [DOI] [PubMed] [Google Scholar]
- 10.Agnelli G, M Verso. Thromboprophylaxis during chemotherapy in patients with advanced cancer. Thromb Res 2010; 125 Suppl 2:p. S17-20; PMID:20433999; http://dx.doi.org/ 10.1016/S0049-3848(10)70007-4 [DOI] [PubMed] [Google Scholar]
- 11.Noble S. A step in the right direction, but one size might not fit all. Lancet Oncol 2009; 10(10):p. 930-1; PMID:19796745; http://dx.doi.org/ 10.1016/S1470-2045(09)70293-1 [DOI] [PubMed] [Google Scholar]
- 12.Falanga A, Marchetti M. Anticancer treatment and thrombosis. Thromb Res 2012; 129(3):p. 353-9; PMID:22119391; http://dx.doi.org/ 10.1016/j.thromres.2011.10.025 [DOI] [PubMed] [Google Scholar]
- 13.Lechner D, Weltermann A. Chemotherapy-induced thrombosis: a role for microparticles and tissue factor? Semin Thromb Hemost 2008; 34(2):p. 199-203; PMID:18645926; http://dx.doi.org/ 10.1055/s-2008-1079261 [DOI] [PubMed] [Google Scholar]
- 14.Falanga A, Tartari CJ, Marchetti M. Microparticles in tumor progression. Thromb Res 2012; 129 Suppl 1:p. S132-6; PMID:22682124; http://dx.doi.org/ 10.1016/S0049-3848(12)70033-6 [DOI] [PubMed] [Google Scholar]
- 15.Boles JC, Williams JC, Hollingsworth RM, Wang JG, Glover SL, Owens AP 3rd, Barcel DA, Kasthuri RS, Key NS, Mackman N. Anthracycline treatment of the human monocytic leukemia cell line THP-1 increases phosphatidylserine exposure and tissue factor activity. Thromb Res 2012; 129(2):p. 197-203; PMID:21762960; http://dx.doi.org/ 10.1016/j.thromres.2011.06.022 [DOI] [PubMed] [Google Scholar]
- 16.Owens AP 3rd, Mackman N. Tissue factor and thrombosis: The clot starts here. Thromb Haemost 2010; 104(3):p. 432-9; PMID:20539911; http://dx.doi.org/ 10.1160/TH09-11-0771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hron G, Kollars M, Weber H, Sagaster V, Quehenberger P, Eichinger S, Kyrle PA, Weltermann A. Tissue factor-positive microparticles: cellular origin and association with coagulation activation in patients with colorectal cancer. Thromb Haemost 2007; 97(1):p. 119-23; PMID:17200778 [PubMed] [Google Scholar]
- 18.Tesselaar ME, Romijn FP, Van Der Linden IK, Prins FA, Bertina RM, Osanto S. Microparticle-associated tissue factor activity: a link between cancer and thrombosis? J Thromb Haemost 2007; 5(3):p. 520-7; PMID:17166244; http://dx.doi.org/ 10.1111/j.1538-7836.2007.02369.x [DOI] [PubMed] [Google Scholar]
- 19.Nierodzik ML, Karpatkin S. Thrombin induces tumor growth, metastasis, and angiogenesis: Evidence for a thrombin-regulated dormant tumor phenotype. Cancer Cell 2006; 10(5):p. 355-62; PMID:17097558; http://dx.doi.org/ 10.1016/j.ccr.2006.10.002 [DOI] [PubMed] [Google Scholar]
- 20.Falanga A, Panova-Noeva M, Russo L. Procoagulant mechanisms in tumour cells. Best Pract Res Clin Haematol 2009; 22(1):p. 49-60; PMID:19285272; http://dx.doi.org/ 10.1016/j.beha.2008.12.009 [DOI] [PubMed] [Google Scholar]
- 21.Palumbo JS, Potter JM, Kaplan LS, Talmage K, Jackson DG, Degen JL. Spontaneous hematogenous and lymphatic metastasis, but not primary tumor growth or angiogenesis, is diminished in fibrinogen-deficient mice. Cancer Res 2002; 62(23):p. 6966-72; PMID:12460914 [PubMed] [Google Scholar]
- 22.Lal I, Dittus K, Holmes CE. Platelets, coagulation and fibrinolysis in breast cancer progression. Breast Cancer Res 2013; 15(4):p. 207; PMID:23905544; http://dx.doi.org/ 10.1186/bcr3425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goubran HA, Stakiw J, Radosevic M, Burnouf T. Platelet-cancer interactions. Semin Thromb Hemost 2014; 40(3):p. 296-305; PMID:24590421; http://dx.doi.org/ 10.1055/s-0034-1370767 [DOI] [PubMed] [Google Scholar]
- 24.Riedl J, Pabinger I, Ay C. Platelets in cancer and thrombosis. Hamostaseologie 2014; 34(1):p. 54-62; PMID:24305775; http://dx.doi.org/ 10.5482/HAMO-13-10-0054 [DOI] [PubMed] [Google Scholar]
- 25.Coughlin SR. Protease-activated receptors in hemostasis, thrombosis and vascular biology. J Thromb Haemost 2005; 3(8):p. 1800-14; PMID:16102047; http://dx.doi.org/ 10.1111/j.1538-7836.2005.01377.x [DOI] [PubMed] [Google Scholar]
- 26.Coughlin SR. Thrombin signalling and protease-activated receptors. Nature 2000; 407(6801):p. 258-64; PMID:11001069; http://dx.doi.org/ 10.1038/35025229 [DOI] [PubMed] [Google Scholar]
- 27.Rickles FR, Patierno S, Fernandez PM. Tissue factor, thrombin, and cancer. Chest 2003; 124(3 Suppl):p. 58S-68S; PMID:12970125; http://dx.doi.org/ 10.1378/chest.124.3_suppl.58S [DOI] [PubMed] [Google Scholar]
- 28.Kuderer NM, Ortel TL, Francis CW. Impact of venous thromboembolism and anticoagulation on cancer and cancer survival. J Clin Oncol 2009; 27(29):p. 4902-11; PMID:19738120; http://dx.doi.org/ 10.1200/JCO.2009.22.4584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Noble S. Low-molecular-weight heparin and survival in lung cancer. Thromb Res 2012; 129 Suppl 1:p. S114-8; PMID:22682120; http://dx.doi.org/ 10.1016/S0049-3848(12)70029-4 [DOI] [PubMed] [Google Scholar]
- 30.Lee AY, Rickles FR, Julian JA, Gent M, Baker RI, Bowden C, Kakkar AK, Prins M, Levine MN. Randomized comparison of low molecular weight heparin and coumarin derivatives on the survival of patients with cancer and venous thromboembolism. J Clin Oncol 2005; 23(10):p. 2123-9; PMID:15699480; http://dx.doi.org/ 10.1200/JCO.2005.03.133 [DOI] [PubMed] [Google Scholar]
- 31.Klerk CP, Smorenburg SM, Otten HM, Lensing AW, Prins MH, Piovella F, Prandoni P, Bos MM, Richel DJ, van Tienhoven G, et al.. The effect of low molecular weight heparin on survival in patients with advanced malignancy. J Clin Oncol 2005; 23(10):p. 2130-5; PMID:15699479; http://dx.doi.org/ 10.1200/JCO.2005.03.134 [DOI] [PubMed] [Google Scholar]
- 32.Eriksson BI, Dahl OE. Prevention of venous thromboembolism following orthopaedic surgery: clinical potential of direct thrombin inhibitors. Drugs 2004; 64(6):p. 577-95; PMID:15018589; http://dx.doi.org/ 10.2165/00003495-200464060-00002 [DOI] [PubMed] [Google Scholar]
- 33.Hauel NH, Nar H, Priepke H, Ries U, Stassen JM, Wienen W. Structure-based design of novel potent nonpeptide thrombin inhibitors. J Med Chem 2002; 45(9):p. 1757-66; PMID:11960487; http://dx.doi.org/ 10.1021/jm0109513 [DOI] [PubMed] [Google Scholar]
- 34.Eriksson BI, Dahl OE, Ahnfelt L, Kälebo P, Stangier J, Nehmiz G, Hermansson K, Kohlbrenner V. Dose escalating safety study of a new oral direct thrombin inhibitor, dabigatran etexilate, in patients undergoing total hip replacement: BISTRO I. J Thromb Haemost 2004; 2(9):p. 1573-80; PMID:15333033; http://dx.doi.org/ 10.1111/j.1538-7836.2004.00890.x [DOI] [PubMed] [Google Scholar]
- 35.DeFeo K, Hayes C, Chernick M, Ryn JV, Gilmour SK. Use of dabigatran etexilate to reduce breast cancer progression. Cancer Biol Ther 2010; 10(10):p. 1001-8; PMID:20798593; http://dx.doi.org/ 10.4161/cbt.10.10.13236 [DOI] [PubMed] [Google Scholar]
- 36.Heppner GH, Miller FR, Shekhar PM. Nontransgenic models of breast cancer. Breast Cancer Res 2000; 2(5):p. 331-4; PMID:11250725; http://dx.doi.org/ 10.1186/bcr77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Diquelou A, Lemozy S, Dupouy D, Boneu B, Sakariassen K, Cadroy Y. Effect of blood flow on thrombin generation is dependent on the nature of the thrombogenic surface. Blood 1994; 84(7):p. 2206-13; PMID:7919337 [PubMed] [Google Scholar]
- 38.Wolberg AS, Kon RH, Monroe DM, Ezban M, Roberts HR, Hoffman M. Deencryption of cellular tissue factor is independent of its cytoplasmic domain. Biochem Biophys Res Commun 2000; 272(2):p. 332-6; PMID:10833414; http://dx.doi.org/ 10.1006/bbrc.2000.2783 [DOI] [PubMed] [Google Scholar]
- 39.Gil-Bernabe AM, Lucotti S, Muschel RJ. Coagulation and metastasis: what does the experimental literature tell us? Br J Haematol 2013; 162(4):p. 433-441; PMID:23691951; http://dx.doi.org/ 10.1111/bjh.12381 [DOI] [PubMed] [Google Scholar]
- 40.Thomas GM, Panicot-Dubois L, Lacroix R, Dignat-George F, Lombardo D, Dubois C. Cancer cell-derived microparticles bearing P-selectin glycoprotein ligand 1 accelerate thrombus formation in vivo. J Exp Med 2009; 206(9):p. 1913-27; PMID:19667060; http://dx.doi.org/ 10.1084/jem.20082297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.DuPre SA, Hunter KW Jr. Murine mammary carcinoma 4T1 induces a leukemoid reaction with splenomegaly: association with tumor-derived growth factors. Exp Mol Pathol 2007; 82(1):p. 12-24; PMID:16919266; http://dx.doi.org/ 10.1016/j.yexmp.2006.06.007 [DOI] [PubMed] [Google Scholar]
- 42.Blom JW, Vanderschoot JP, Oostindiër MJ, Osanto S, van der Meer FJ, Rosendaal FR. Incidence of venous thrombosis in a large cohort of 66,329 cancer patients: results of a record linkage study. J Thromb Haemost 2006; 4(3):p. 529-35; PMID:16460435; http://dx.doi.org/ 10.1111/j.1538-7836.2006.01804.x [DOI] [PubMed] [Google Scholar]
- 43.Horstman LL, Jy W, Jimenez JJ, Bidot C, Ahn YS. New horizons in the analysis of circulating cell-derived microparticles. Keio J Med 2004; 53(4):p. 210-30; PMID:15647627; http://dx.doi.org/ 10.2302/kjm.53.210 [DOI] [PubMed] [Google Scholar]
- 44.Key NS, Mackman N. Tissue factor and its measurement in whole blood, plasma, and microparticles. Semin Thromb Hemost 2010; 36(8):p. 865-75; PMID:21049387; http://dx.doi.org/ 10.1055/s-0030-1267040 [DOI] [PubMed] [Google Scholar]
- 45.Callander NS, Varki N, Rao LV. Immunohistochemical identification of tissue factor in solid tumors. Cancer 1992; 70(5):p. 1194-201; PMID:1381270; http://dx.doi.org/ 10.1002/1097-0142(19920901)70:5%3c1194::AID-CNCR2820700528%3e3.0.CO;2-E [DOI] [PubMed] [Google Scholar]
- 46.Yu JL, May L, Lhotak V, Shahrzad S, Shirasawa S, Weitz JI, Coomber BL, Mackman N, Rak JW. Oncogenic events regulate tissue factor expression in colorectal cancer cells: implications for tumor progression and angiogenesis. Blood 2005; 105(4):p. 1734-41; PMID:15494427; http://dx.doi.org/ 10.1182/blood-2004-05-2042 [DOI] [PubMed] [Google Scholar]
- 47.Magnus N, D'Asti E, Meehan B, Garnier D, Rak J. Oncogenes and the coagulation system–forces that modulate dormant and aggressive states in cancer. Thromb Res 2014; 133 Suppl 2:p. S1-9; PMID:24862126; http://dx.doi.org/ 10.1016/S0049-3848(14)50001-1 [DOI] [PubMed] [Google Scholar]
- 48.Piccioli A, Falanga A, Baccaglini U, Marchetti M, Prandoni P. Cancer and venous thromboembolism. Semin Thromb Hemost 2006; 32(7):p. 694-9; PMID:17024596; http://dx.doi.org/ 10.1055/s-2006-951297 [DOI] [PubMed] [Google Scholar]
- 49.Magnus N, Garnier D, Rak J. Oncogenic epidermal growth factor receptor up-regulates multiple elements of the tissue factor signaling pathway in human glioma cells. Blood 2010; 116(5):p. 815-8; PMID:20462964; http://dx.doi.org/ 10.1182/blood-2009-10-250639 [DOI] [PubMed] [Google Scholar]
- 50.Mukherjee SD, Swystun LL, Mackman N, Wang JG, Pond G, Levine MN, Liaw PC. Impact of chemotherapy on thrombin generation and on the protein C pathway in breast cancer patients. Pathophysiol Haemost Thromb 2010; 37(2-4):p. 88-97; PMID:21430357; http://dx.doi.org/ 10.1159/000324166 [DOI] [PubMed] [Google Scholar]
- 51.Swystun LL, Mukherjee S, Liaw PC. Breast cancer chemotherapy induces the release of cell-free DNA, a novel procoagulant stimulus. J Thromb Haemost 2011; 9(11):p. 2313-21; PMID:21838758; http://dx.doi.org/ 10.1111/j.1538-7836.2011.04465.x [DOI] [PubMed] [Google Scholar]
- 52.Swystun LL, Mukherjee S, Levine M, Liaw PC. The chemotherapy metabolite acrolein upregulates thrombin generation and impairs the protein C anticoagulant pathway in animal-based and cell-based models. J Thromb Haemost 2011; 9(4):p. 767-75; PMID:21320281; http://dx.doi.org/ 10.1111/j.1538-7836.2011.04232.x [DOI] [PubMed] [Google Scholar]
- 53.Nierodzik ML, Chen K, Takeshita K, Li JJ, Huang YQ, Feng XS, D'Andrea MR, Andrade-Gordon P, Karpatkin S. Protease-activated receptor 1 (PAR-1) is required and rate-limiting for thrombin-enhanced experimental pulmonary metastasis. Blood 1998; 92(10):p. 3694-700; PMID:9808563 [PubMed] [Google Scholar]
- 54.Ossovskaya VS, Bunnett NW. Protease-activated receptors: contribution to physiology and disease. Physiol Rev 2004; 84(2):p. 579-621; PMID:15044683; http://dx.doi.org/ 10.1152/physrev.00028.2003 [DOI] [PubMed] [Google Scholar]
- 55.Huang YQ, Li JJ, Hu L, Lee M, Karpatkin S. Thrombin induces increased expression and secretion of angiopoietin-2 from human umbilical vein endothelial cells. Blood 2002; 99(5):p. 1646-50; PMID:11861279; http://dx.doi.org/ 10.1182/blood.V99.5.1646 [DOI] [PubMed] [Google Scholar]
- 56.Ollivier V, Chabbat J, Herbert JM, Hakim J, de Prost D. Vascular endothelial growth factor production by fibroblasts in response to factor VIIa binding to tissue factor involves thrombin and factor Xa. Arterioscler Thromb Vasc Biol 2000; 20(5):p. 1374-81; PMID:10807756; http://dx.doi.org/ 10.1161/01.ATV.20.5.1374 [DOI] [PubMed] [Google Scholar]
- 57.Tsopanoglou NE, Maragoudakis ME. On the mechanism of thrombin-induced angiogenesis. Potentiation of vascular endothelial growth factor activity on endothelial cells by up-regulation of its receptors. J Biol Chem 1999; 274(34):p. 23969-76; PMID:10446165; http://dx.doi.org/ 10.1074/jbc.274.34.23969 [DOI] [PubMed] [Google Scholar]
- 58.Nieswandt B, Hafner M, Echtenacher B, Männel DN. Lysis of tumor cells by natural killer cells in mice is impeded by platelets. Cancer Res 1999; 59(6):p. 1295-1300; PMID:10096562 [PubMed] [Google Scholar]
- 59.Kopp HG, Placke T, Salih HR. Platelet-derived transforming growth factor-beta down-regulates NKG2D thereby inhibiting natural killer cell antitumor reactivity. Cancer Res 2009; 69(19):p. 7775-83; PMID:19738039; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-2123 [DOI] [PubMed] [Google Scholar]
- 60.Labelle M, Begum S, Hynes RO. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 2011; 20(5):p. 576-90; PMID:22094253; http://dx.doi.org/ 10.1016/j.ccr.2011.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 2009; 9(3):p. 162-74; PMID:19197294; http://dx.doi.org/ 10.1038/nri2506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bunt SK, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S. Inflammation induces myeloid-derived suppressor cells that facilitate tumor progression. J Immunol 2006; 176(1):p. 284-90; PMID:16365420; http://dx.doi.org/ 10.4049/jimmunol.176.1.284 [DOI] [PubMed] [Google Scholar]
- 63.Turpin B, Miller W, Rosenfeldt L, Kombrinck K, Flick MJ, Steinbrecher KA, Harmel-Laws E, Mullins ES, Shaw M, Witte DP, et al.. Thrombin drives tumorigenesis in colitis-associated colon cancer. Cancer Res 2014; 74(11):p. 3020-30; PMID:24710407; http://dx.doi.org/ 10.1158/0008-5472.CAN-13-3276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Labelle M, Begum S, Hynes RO. Platelets guide the formation of early metastatic niches. Proc Natl Acad Sci U S A 2014; 111(30):p. E3053-61; PMID:25024172; http://dx.doi.org/ 10.1073/pnas.1411082111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sevko A, Sade-Feldman M, Kanterman J, Michels T, Falk CS, Umansky L, Ramacher M, Kato M, Schadendorf D, Baniyash M, et al.. Cyclophosphamide promotes chronic inflammation-dependent immunosuppression and prevents antitumor response in melanoma. J Invest Dermatol 2013; 133(6):p. 1610-9; PMID:23223128; http://dx.doi.org/ 10.1038/jid.2012.444 [DOI] [PubMed] [Google Scholar]
- 66.Buzzai M, Jones RG, Amaravadi RK, Lum JJ, DeBerardinis RJ, Zhao F, Viollet B, Thompson CB. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res 2007; 67(14):p. 6745-52; PMID:17638885; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-4447 [DOI] [PubMed] [Google Scholar]