

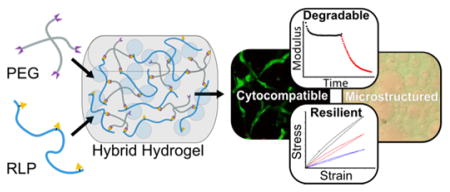
Abstract
Hydrogels derived from resilin-like polypeptides (RLPs) have shown outstanding mechanical resilience and cytocompatibility, and expanding the versatility of RLP-based materials via conjugation with other polypeptides and polymers would offer great promise in the design of a range of materials. Here, we present an investigation of the biochemical and mechanical properties of hybrid hydrogels composed of a recombinant RLP and a multi-arm PEG macromer. These hybrid hydrogels can be rapidly cross-linked through a Michael-type addition reaction between the thiols of cysteine residues on the RLP and vinyl sulfone groups on the multi-arm PEG. Oscillatory rheology and tensile testing confirmed the formation of elastomeric hydrogels with mechanical resilience comparable to aortic elastin; hydrogel stiffness was easily modulated through the cross-linking ratio. Macromolecular phase separation of the RLP-PEG hydrogels offers the unique advantage of imparting a heterogeneous microstructure, which can be used to localize cells, through simple mixing and crosslinking. Assessment of degradation of the RLP by matrix metalloproteinases (MMPs) illustrated the specific proteolysis of the polypeptide in both its soluble form and when cross-linked into hydrogels. Finally, the successful encapsulation and viable three-dimensional culture of human mesenchymal stem cells (hMSCs) demonstrated the cytocompatibility of the RLP-PEG gels. Overall, the cytocompatibility, elastomeric mechanical properties, micro-heterogeneity, and degradability of the RLP-PEG hybrid hydrogels offer a suite of promising properties for the development of cell-instructive, structured tissue engineering scaffolds.
Keywords: biomaterials, protein, hydrogels, degradation, phase separation
Graphical Abstract
INTRODUCTION
The elastomeric properties and supra-cellular architecture of tissues are critical determinants of function, and there is a significant need for biomaterials that can capture these properties and direct the formation of neo-tissue. Recombinant protein polymers have been widely investigated as biomaterials for tissue engineering applications1, 2 and elastomeric polypeptides represent a particularly exciting subset of materials designed for soft tissues that require flexible, reversible elasticity.3–17 In these applications, hydrogel scaffolds serve as temporary substitutes while healthy tissue regenerates18 and an ideal scaffold would be durable enough to withstand the repetitive mechanical forces experienced by some of these soft tissues.19–21 For example, collagen and elastin serve as critical structural components of cardiovascular tissues, which experience cyclic hemodynamic forces over extended time scales, and desirable biomaterials for cardiovascular applications would replicate the elasticity conferred by those proteins.21–24 Intrinsically disordered proteins (IDP) that serve as structural components of tissue such as elastin, resilin, abductin, and flagelliform spider silk are attractive candidates as these proteins display remarkable elasticity and fatigue resistance;11, 25–27 protein polymers based upon the characteristic sequences of IDPs have demonstrated mechanical properties comparable to those of the natural proteins.25 In particular, recently developed resilin-like polypeptide (RLPs) hydrogels have been shown to form elastic materials that are stable, resilient, and highly extensible.13, 14, 28 More recently, hybrid hydrogels, composed of recombinant elastomeric protein and PEG, have yielded materials in which encapsulated cells spread and adopt a spindle-like morphology.17, 29 In addition, protein-PEG hydrogels comprising other proteins such as collagen30–32 or fibrinogen,30, 33–36 have been shown to improve biocompatibility and cell-matrix interaction.35
Importantly, IDPs may be engineered to contain chimeric domains which impart specific bioactivity which facilitates cell-matrix interactions.10, 15, 29, 37–42 The development of hybrid materials that can also control the spatial organization of these interactions (e.g. spreading, migration, and adhesion) would offer critical advantages for controlling tissue morphogenesis.43 Accordingly, multiple strategies have been employed to develop materials with patterned or hierarchal structure and include: the 3D-patterning of bioactive molecules in cross-linked PEG gels,44, 45 the formation of macroporous scaffolds via solvent-induced phase separation46, 47 and the embedding of cell-adhesive microparticles within bulk gels.48 While not widely explored for generation of hydrogel microstructure, liquid-liquid partitioning of protein and PEG has been extensively employed in protein separation/purification and protein crystallization49–55 and has more recently been utilized in the preparation of microporous hydrogels through polymerization-induced phase separation.56–58 Similarly, PEG hydrogels incorporating Pluronic®F127 micelles have been shown to form nanostructured materials that facilitate cell spreading.59, 60 Opportunities to purposefully manipulate such liquid-liquid phase separation to create defined hydrogel microstructures and spatially control biochemical properties may be of significant value in scaffold design.
Another important design criteria for materials aimed at applications in tissue regeneration is scaffold degradation, either via chemical61–63 or biological means.64–67 Cell-mediated degradation through enzyme-sensitive cross-links has been widely employed to achieve this type of control64, 68 and recombinant protein polymers are easily equipped with degradable sequences.15, 37, 65, 69, 70 A number of elastin-like, silk-like and resilin-like polypeptides have been engineered with proteolytic domains15, 28, 37, 71 including matrix metalloproteinase (MMP)-sensitive domains,14, 15, 17 as MMPs are a key enzyme in the remodeling of ECM72, 73 as well as in tissue morphogenesis and repair.74
Here, we report microstructured degradable hybrid hydrogels composed of RLPs cross-linked with PEG macromers via a Michael-type addition reaction between the thiols of cysteine residues on the RLP and end-functionalized vinyl sulfone moieties on the four-arm star PEG (see Figure 1A). The mechanical properties of RLP-PEG hydrogels were investigated via oscillatory rheology and tensile testing. Domains conferring MMP-specific proteolysis were analyzed for degradation both when the RLP was in solution and when it was cross-linked into hydrogels. Encapsulation of hMSCs within RLP24-PEG gels was performed to investigate the cytocompatibilty of the materials using cell lines that are highly relevant to tissue engineering. Finally, phase contrast and confocal microscopy were employed to investigate the presence of a heterogeneous microstructure resulting from protein-PEG partitioning. Taken together, our results suggest the potential of these approaches for the production of cell-instructive materials that provide multiple cell-supportive domains and desirable elastomeric properties.
Figure 1.

Schematics illustrating the formation of hybrid hydrogels and the design of the RLP24 polypeptide. (A) Vinyl sulfone terminated four-arm star PEG macromers cross-link with the thiols of cysteine residues through a Michael-type addition reaction to form hydrogels. (B) The overall design of the RLPs and respective location of different bioactive domains. Additionally, the amino acid sequence of the protein is provided with important sequences colored. Parentheses indicate repeated sequences.
EXPERIMENTAL SECTION
MATERIALS
All chemicals or reagents were purchased from Sigma-Aldrich (St. Louis, MO) or Fisher Scientific (Waltham, MA) and used as received unless otherwise noted. Ni-NTA agarose used for protein purification was purchased from Qiagen (Valcencia, CA) or Thermo Scientific (Rockford, IL). Water for buffers or media was deionized and filtered using either a ThermoFisher Barnstead NANOpure Diamond water purifier or a Purelab Classic (Siemens, Munich, Germany). Hydroxyl terminated four-arm star PEG (10 kDa) was purchased from JenKem Technology USA (Plano, TX) and was used as received. The synthesis of the 4-arm PEG vinyl sulfone cross-linker was previously described17 and used established protocols;68 1H NMR analysis confirmed quantitative functionalization as has also been previously reported.17
EXPRESSION AND PURIFICATION OF RLP24
Detailed descriptions of the construction of the RLP24 gene, its expression and the purification of the RLP24 polypeptide have been previously reported.17 Briefly, the polypeptide was expressed using an E. coli host and Studier auto-inducing media (ZYP-5052 media) in a shaker-incubator;75 a 4-hour growth period at 37°C was followed with a 24-hour 24°C expression period. Cells were pelleted via centrifugation and frozen at −20°C. The cell pellet was later resuspended in native lysis buffer17, 76 containing 20 mM β-mercaptoethanol, sonicated to disrupt the cell walls and then centrifuged to clear the lysate of insoluble debris. The cleared lysate was then heated to 90°C for ten minutes and centrifuged again to remove precipitated protein. The heat stability and solubility of resilin-like polypeptides allows for the selective removal of contaminating bacterial proteins via a heating/precipitation procedure12 and it improved the final purity of the RLP.17 The cleared lysate was then purified under native conditions (10 mM β-mercaptoethanol) using Ni- NTA affinity chromatography and the purified protein was dialyzed against DI water to remove salts prior to lyophilization. The purity of the protein was confirmed using sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and visualized with Coomassie blue staining.
PREPARATION OF THIOL-FUNCTIONALIZED RLP24
Pure lyophilized protein was reduced using tris(2-carboxyethyl)phosphine hydrochloride (TCEP-HCl) in a 10 mM MES, 500 mM NaCl buffer (pH 5.5). Depending upon the initial free thiol content of the RLP, the quantity of TCEP-HCl and the incubation time were modulated. However, conditions usually consisted of a 1–3x TCEP-HCl to thiol ratio and incubation at room temperature, with stirring, for a period of approximately one hour. The protein was reduced at a 20 mg/mL concentration, but was diluted to 10 mg/mL with additional MES buffer in preparation for desalting.
Two different methods were employed for the removal of salts and TCEP-HCl prior to freezing and lyophilization of the polypeptides reported in this study. The initial method utilized disposable Zeba™ Spin desalting columns (7 kDa MWCO, ThermoFisher, Rockford, IL); the protein solution was eluted from the column via centrifugation, collected, immediately frozen using liquid N2 and then lyophilized.17 For polypeptide samples employed later, fast protein liquid chromatography (FPLC) was employed to improve the desalting process and to track the differential elution of protein and salt. A GE Life Sciences AKTA™ Explorer100 and Purifier 10 high performance FPLC (Delaware Biotechnology Institute, Newark, DE) equipped with a HiPrep™ 26/10 Desalting column was utilized. The reduced RLP24 protein solution was eluted at a rate of 8–10 mL/min using a mobile phase of chilled DI water. With UV-Vis detection at 280 nm. The eluted RLP24 protein was collected, immediately frozen in liquid N2, and then lyophilized. The free thiol content of the lyophilized protein was determined through an Ellman’s colorimetric assay and UV-Vis spectroscopy as previously reported17 and by using established protocols.77, 78
PRECURSOR PREPARATION AND HYDROGEL COMPOSITION
The RLP24-PEG hydrogels investigated in this study were formed from a 20 wt% precursor solution at three different molar ratios (3:2, 1:1 and 1:2) of vinyl sulfone to thiol (precursor conditions are summarized in Table S1). To prepare cross-linked RLP-PEG hydrogels, the precursors were first dissolved separately in PBS buffer (pH 7.4) and then dispensed into individual aliquots prior to mixing for gel formation. The pH sensitive dye, phenolphthalein (1 mM), was included in the RLP solution and 0.2 uL drops of concentrated NaOH were used to adjust the final pH to ~8.2. This was indicated by a color change in the solution to a slightly pinkish hue which corresponds to the color change (colorless → pink) phenolphthalein undergoes at pH 8.2. For all investigations involving hydrogels, samples were prepared using different preparations (expression, purification, reduction) of RLP24 to provide multiple experimental repeats used to generate the reported averages.
OSCILLATORY RHEOLOGY
The oscillatory rheology experiments were conducted on a stress-controlled AR-G2 rheometer (TA Instruments, New Castle, DE) using a 20 mm diameter stainless steel cone-on-plate geometry with a 1° cone angle and a 25 μm gap distance. The precursor solutions were prepared as described above. To slow the rate of cross-linking, the precursors were chilled on ice prior to being briefly mixed using a vortex mixer. The RLP-PEG solution was deposited quickly onto a prechilled (4°C) rheometer stage while the cone-on-plate geometry was brought to the appropriate gap (25 μm). Mineral oil was used to seal the geometry and prevented dehydration; the temperature was then quickly raised to 37°C where it was maintained for the remainder of the experiment. The formation of elastic hydrogels was monitored using a time sweep conducted in the linear viscoelastic regime (see Figure S1) at 1% strain and an angular frequency of 6 rad/s. This experiment was followed with a frequency sweep from 0.1 rad/s to 100 rad/s conducted at 1% strain. Experiments were repeated on three to four samples for each cross-linking ratio and the shear modulus was reported as the simple mean. The error is reported as the standard deviation of the samples tested.
HYDROGEL SWELLING
For the swelling experiments, the precursors were briefly mixed, vortexed, and cross-linked overnight on slips of Parafilm™ at 37°C in a humidified chamber. The gels were swelled for five days at 37°C in a PBS buffer containing a solution of 1mM ethylenediaminetetraacetic acid (EDTA), 0.002% sodium azide and antibiotic/antimycotic (Life Technologies, Carlsbad, CA) to prevent enzymatic degradation or microorganism contamination. The swollen hydrogels were removed from the buffer, carefully blotted dry, and measured gravimetrically on a microbalance. To measure the dry weight of the RLP-PEG hydrogel, the hydrogels were frozen in liquid N2 and lyophilized. The swelling ratio and water content were calculated using the following equations:14, 15, 37
where q is the swelling ratio at equilibrium, WC is the water content at equilibrium, ms is the mass of the swollen hydrogel, and md is the mass of the dried hydrogel. The data reported are the simple mean of five samples with the error reported as the standard deviation.
TENSILE TESTING
The tensile testing of the RLP24-PEG hydrogels was carried out on an RSA-G2 dynamic mechanical analyzer (DMA) (TA Instruments) using the axial tensile geometry. Rectangular RLP-PEG hydrogels were cast in polytetrafluoroethylene (PTFE) molds sealed with a glass slide (see Figure S2). Precursor solutions, prepared as described above, were mixed briefly using a vortex mixer and injected into the mold via a channel on the reverse side of the PTFE/glass plate mold (see Figure S3). The precursors were cross-linked overnight in a humidified chamber at 37°C before being swelled for a minimum of 48 hours at 37°C in a PBS buffer used in the swelling studies. For long-term storage, gels were kept at 4°C in sealed containers with a small quantity of the PBS to prevent dehydration. Large gels were utilized for uniaxial cyclic strain testing and smaller gels for strain-to-break measurements (see Table S2); dimensions of swollen gels were measured using calipers. An immersion chamber filled with room temperature PBS prevented dehydration during tensile experiments. For the cyclic strain experiments, the hydrogels were strained to 20%, 40% and 60% strain at a constant rate of 0.1 mm/s for five cycles per sample; since RLP24-PEG hydrogels were prone to slippage in the tensile geometry, the total number of cycle per sample was limited. Strain-to-break measurements were similarly strained at a 0.1 mm/s strain rate. The raw data were processed using Origin™ 8.5.1 (OriginLab Corporation, Northampton, MA). An adjacent averaging function (see Figure S4) was applied to the raw data to generate extension and contraction curves for cyclic testing and for the strain-to-break curves. Resilience, or the percent of energy recovered following deformation, was determined by integrating the extension and contraction curves and then dividing the area of the contraction curve by the area of the extension curve.13, 14 The resilience for a particular sample was calculated as the simple mean of five cycles at one of the strains (20, 40, or 60% strain). The final resilience for a given cross-linking ratio and strain was calculated as the average of at least three samples. The Young’s modulus for a particular sample was determined from the slope of the extension curve (up to 10% strain) for each cycle of a 60% strain experiment.79 The final Young’s modulus reported for a given sample was determined as the simple mean of at least three samples. The strain-to-break was determined as the percent strain at which the stress dropped by 20% or more (tearing was considered breaking). The final strain-to-break values were reported as the mean of at least three samples for each cross-linking ratio. The errors reported for all of tensile data are the standard deviation of at least three separate samples, prepared from separate batches of bacterial cell culture.
HYDROGEL MICROSTRUCTURE
Microscale heterogeneity in RLP24-PEG hydrogels (20 wt%, 3:2 VS:thiol) was investigated via phase contrast imaging with an Eclipse Ti-E fluorescence microscope (Nikon Instruments, Melville, NY). RLP24-PEG hydrogels were compared to pure RLP hydrogels cross-linked with an amine-reactive small molecule, tris(hydroxymethyl)phosphine, at a 3:1 ratio of hydroxyl group to lysine in 20wt% solutions of RLP24. Both sets of hydrogels were cross-linked at 37°C in a humidified environment for three hours. Phase contrast images of the hydrogels (5x and 10x magnification) were collected immediately following cross-linking, as this provided the best contrast to visualize heterogeneity in the hydrogels.
Confocal microscopy was also used to investigate the heterogeneity of the RLP24-PEG hydrogels. For this analysis, RLP24 and PEGVS were labeled with tetramethylrhodamine isothiocyanate (TRITC) (Thermo Fisher) and 5-((2-(and-3)-S-(acetylmercapto) succinoyl) amino) fluorescein, (SAMSA fluorescein, Life Technologies), respectively. RLP24 was dissolved in a sodium carbonate buffer (100 mM pH 9.0) at a 5 mg/mL concentration and was reacted overnight at 4°C with the TRITC fluorophore at a 1:250 ratio of isothiocyanate to lysine. The protein was then desalted using a Zeba™ Spin desalting column (MWCO 7kDa) to remove the unreacted fluorophore; it was then reduced and lyophilized using the protocols described above. The PEGVS was labeled via the reaction of the vinyl sulfone group with thiolated SAMSA fluorescein (1:250 thiol:vinyl sulfone); the SAMSA was prepared according the manufacturer-provided protocol and was reacted with the PEGVS (150 mg) for 1 hour at room temperature in 10.5 mL of buffer (20 mM sodium phosphate, pH 7.5). The PEGVS was similarly desalted to remove residual fluorophore before being lyophilized to a dry powder. The fluorescently-tagged precursors were diluted into solutions of unlabeled precursors at a 100:1 weight ratio (unlabeled:labeled) and were then mixed to form the RLP24-PEGVS hydrogels. Hydrogels were cross-linked at 37°C within CoverWell™ perfusion chambers (Grace Bio-Labs, Bend, OR) on glass slides and were also imaged directly.
HYDROGEL DEGRADATION
Hydrogel degradation by rhMMP1 was monitored in situ via compression testing utilizing the RSA-G2 DMA and a machined PTFE cup holding a small volume (1.5 mL) of enzyme-containing buffer (see Figure S5). Cylinder-shaped RLP24-PEG hydrogels (50 μL, 20wt%, 3:2 ratio of vinyl sulfone to thiol) were cross-linked in modified syringes utilizing the protocols described above. These hydrogels were swelled for 3 days in bacteriostatic/antimicrobial PBS buffer at 37°C and stored at 4°C. The dimensions of the swollen hydrogel were approximately 5 mm in diameter and 3 mm in height as measured by calipers. Hydrogels were prestrained to 10% strain to prevent detachment from the plates during the experiment. The experiment, conducted at room temperature, was divided into a 12-hour control period over which the gel was tested in enzyme buffer lacking MMP and a 10-hour experimental period during which the gel was incubated with the rhMMP1 enzyme at a 171 nM (3.33 mg/L) concentration. An oscillatory time sweep experiment was performed at 5% strain and at a frequency of 1 Hz; data points were collected in 15-minute intervals. The raw data were normalized to the initial modulus and a simple mean of the normalized data points of three separate samples were used to generate curves; the error given is the standard deviation of the modulus of the three samples.
ENCAPSULATION, VIABILITY, AND PROLIFERATION OF hMSCs
Human mesenchymal stem cells from bone marrow (Lonza, Basel, Switzerland) were cultured according to the supplier’s recommendations at 37°C, 5% CO2 and using the BulletKit™ hMSC growth medium; cell culture confluency was kept below 80% and passage numbers varied between four and nine. The encapsulation of the hMSC-derived cells was performed using the same protocol as reported previously for the encapsulation of human aortic adventitial fibroblasts in RLP24-PEG hydrogels.17 Hydrogels were produced at a 20wt% total polymer concentration and a 3:2 cross-linking ratio (vinyl sulfone:thiol). For the imaging experiments to determine cell viability and proliferation, hydrogels with a volume of 50 μL were seeded at a density of 200,000 per mL (10,000 cells per hydrogel).
Over a two week period, cell viability within the RLP24-PEG hydrogels was analyzed via laser scanning confocal microscopy and Live/Dead® staining (Life Technologies). Hydrogels were placed in PBS buffer containing 2 μM Calcein-AM and 4 μM ethidium homodimer-1 for 30 minutes prior to imaging with a Zeiss LSM 510 NLO multiphoton confocal microscope (Carl Zeiss, Inc., Thornwood, NY). Experiments were run in triplicate with several z-stacks acquired from every sample for each time point. Representative maximum intensity projections are presented.
Proliferation of encapsulated cells was investigated using a Click-iT EdU cell proliferation assay (Life Technologies). Cell-gels were incubated for 48 hours with a 20 mM concentration of 5-ethynyl-2′-deoxyuridine (EdU) starting on day 3 of culture. The gels were then fixed with paraformaldehyde on day 5 and reacted, according the manufacturer’s instructions, with Alexa Fluor® 555. Following a destaining procedure to remove the Alexa dye, the cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) and the gels were imaged using an Eclipse TiE fluorescence microscope (Nikon, Tokyo, Japan). Experiments were run in triplicate and z-stacks were acquired.
RESULTS AND DISCUSSION
DESIGN AND PREPARATION OF RLP24
All of the studies presented in this work were performed using a single resilin-like polypeptide, RLP24 (see Figure 1B), derived from a previously reported family of RLPs.17 The polypeptide was chosen due to its high expression yield and its ability to be effectively cross-linked into elastic hydrogels using a vinyl-sulfone functionalized, multi-arm PEG macromer; its gene was constructed through recursive ligation of an RLP gene as reported previously.10, 14, 17 The RLP24 protein was designed to contain 24 repeats of a modified resilin-like sequence (GGRPSDSFGAPGGGN) derived from the Drosophila melanogaster CG15920 gene.10, 17, 80 To impart RLP24 with cell adhesion properties, four integrin binding domains (RGDSP) were incorporated at regular intervals along the polypeptide chain.17, 81 Two matrix metalloproteinase (MMP)-sensitive sequences (GPQGIWGQ) were included to confer enzyme-specific degradation. Additionally, the polypeptide design incorporated two heparin-binding domains (KAAKRPKAAKDKQTK) that could permit sequestration of heparin and growth factors.17, 82 Finally, three cysteine residues provided the polypeptide with its cross-linking functionality.17
As previously reported, RLP24 was expressed using Studier auto-induction and purified under native conditions using affinity chromatography17 with yields routinely as high as ~100 mg/L of expression culture volume and ~5.6 mg/g of wet cell pellet mass. As with other RLP proteins, heating the cell-free lysate to 80–90°C selectively precipitated contaminating bacterial proteins from solution12 and, when performed prior to Ni-affinity chromatography, helped enhance the purity of the final product. The inclusion of β-mercaptoethanol (10–20 mM) in the lysis and wash buffers helped prevent disulfide formation with contaminating bacterial proteins, but it left an unreactive adduct that had to be removed following purification. Use of a TCEP-HCl reduction step and dialysis at low temperatures70 was not a successful method for RLP24 reduction; residual TCEP could not be removed completely, perhaps due to electrostatic interactions between basic residues of the RLP and TCEP acid groups. Therefore, the RLP24 was reduced in a high-salt buffer (10 mM MES, 500 mM NaCl buffer pH 5.5) and initial polypeptide samples were purified via the use of disposable desalting columns. Polypeptides produced subsequently were purified via fast protein liquid chromatography (FPLC) equipped with a desalting column, with chilled ddH2O as the mobile phase (see Figure S6); this improved the desalting process as larger quantities of protein could be prepared using FPLC and the separation of eluates could be easily monitored. The purified and reduced RLP24 exhibited thiol functionality ranging between 85% and 95% of total thiol content as determined via an Ellman’s assay.17, 77
OSCILLATORY RHEOLOGY
In situ oscillatory rheology was performed to investigate the gelation and mechanical properties of RLP24-PEG hydrogels that were produced at three different cross-linking ratios (3:2, 1:1 and 1:2 vinyl sulfone:thiol). Figure 2A depicts representative oscillatory time sweeps of RLP24-PEG hydrogels cross-linked at the different ratios. Regardless of reactive group stoichiometry, the hydrogels rapidly cross-linked (within 2–4 minutes) with gelation times comparable to other recombinant protein-PEG hydrogels that utilized the same chemistry and similar conditions;70 the rapid gelation of the RLP24-PEG hydrogels supports their handling via injection. Additionally, the gels exhibited the frequency-independent behavior consistent with elastic, solid-like materials with permanently cross-linked networks (see Figure S7).
Figure 2.

Oscillatory rheology of RLP24-PEG hydrogels. (A) Representative time sweeps of RLP24-PEG hydrogels cross-linked at different ratios of vinyl sulfone to thiol. (1% strain, 6 rad/s) (B) The summary of the effect the cross-linking ratio had on the storage modulus (G′)(Pa) for RLP24-PEG hydrogels as analyzed via oscillatory rheology.
The average storage moduli for the RLP24-PEG hydrogels are presented in Figure 2B. RLP24-PEG hydrogels cross-linked at a 3:2 ratio of vinyl sulfone to thiol formed hydrogels with storage modulus of approximately 12–13 kPa while the hydrogels cross-linked at 1:1 and 1:2 exhibited moduli of approximately 8–9 kPa and 3–4 kPa, respectively. Consistent with observations found in the literature, a slight stoichiometric imbalance was necessary to overcome non-ideal cross-linking behavior common in polymer networks.70, 83, 84 The expected increase in storage modulus with increasing cross-linking stoichiometry has been observed in other protein-PEG hydrogels70 and was also observed in RLP hydrogels cross-linked using small molecules.14 The characteristic solubility of most RLPs and RLP24, which is soluble at concentrations as high as 40 wt% during precursor preparation, offers opportunities to produce concentrated materials with increased mechanical robustness. In contrast, other structural proteins with limited solubility may require additional modification; for example, some silk-like polypeptides require disruption of β-sheet formation through the incorporation of elastin-like domains which impart greater solubility.85, 86
TENSILE TESTING
Tensile testing of RLP24-PEG hydrogels was used to investigate the mechanical properties of the swollen hydrogels under uniaxial strain; multiple samples of each cross-linking ratio were measured under repeated cycles of extension and contraction at different percent strain in order to determine the elasticity and resilience of the hybrid hydrogel networks. Figure 3A depicts representative loading and unloading cycles at 60% strain for all three cross-linking ratios. In agreement with the rheological data, the more highly cross-linked RLP24-PEG gels exhibited steeper stress/strain curves which reflect their greater stiffness. Table 1 reports the Young’s modulus for each sample as determined from the slope of the first 10% strain of the extension curve. The 3:2 RLP24-PEG gel had a Young’s modulus of approximately 11.5 kPa while the 1:1 and 1:2 RLP24-PEG gels had Young’s moduli of approximately 7.0 kPa and 4.3 kPa, respectively. Overall, the tensile moduli were lower than expected based upon the relationship between Young’s modulus and shear modulus for isotropic, rubber-like materials (see Supporting Information).87, 88 However, the disparity is likely due to the lack of swelling in the RLP24-PEG gels tested via rheometry. As indicated in Figure S8A/B, the RLP24-PEG hydrogels absorb a significant amount of water which reduces the polymer volume fraction of the gels prepared for tensile testing. Conversely, the gels formed between the platens of the rheometer remain at a higher precursor concentration, which, when combined with a greater number of chain entanglements and noncovalent interactions increases gel stiffness.
Figure 3.

Tensile testing of RLP24-PEG hydrogels. (A) Representative loading/unloading curves of the RLP24-PEG hydrogels strained to 60% at a rate 0.1 mm/s. (B) Deformation responses at 20, 40 and 60% strain for RLP24-PEG hydrogels cross-linked at 3:2 vinyl sulfone to thiol ratio. (C) Representative strain-to-break curves for each of the RLP24-PEG cross-linking ratios. Strain rate 0.1mm/s.
Table 1.
Summary of the mechanical and physical properties of RLP24-PEG hydrogels at three cross-linking ratios.
| Cross-linking Ratio (VS:CYS) | Water Content (%) | Shear Modulus (Pa) | Young’s Modulus (Pa) | Strain to Break (%) |
|---|---|---|---|---|
| 3:2 | 92.1 ± 0.5 | 12300 ± 1601a | 11530 ± 2340 | 68.1 ± 7.9 |
| 1:1 | 93.8 ± 0.4 | 8040 ±1385a | 7070 ± 1350 | 85.6 ± 9.0 |
| 1:2 | 95.5 ± 0.3 | 4205 ± 415 | 4250 ± 1250 | 173.4 ± 34.9 |
previously reported data.17
Figure 3B depicts a representative deformation response of the fourth loading/unloading cycle at 20, 40 and 60% strain for the 3:2 hydrogel composition. The 3:2 gel exhibited a slight increase in hysteresis with increasing deformation, but overall remained remarkably resilient. Table 2 reports the resilience values, which varied between 87 and 98%, for the different hydrogel compositions and corresponding strains; generally, the RLP24-PEG hydrogels exhibited little hysteresis (see Figure 3A, Figure S9A–C) or plastic deformation (see Figure S9D) over the number of cycles investigated. The 3:2 RLP24-PEG hydrogels exhibited resilience between 88 and 93%, which is slightly lower than that observed for the hydroxymethyl phosphine cross-linked RLPs (90–96% resilience under similar conditions),13, 14 as might be expected with the introduction of the PEG. The RLP24-PEG hydrogels show greater resilience, however, when compared to polybutadiene rubber (~80%)11 and available data of crosslinked ELP films,89–91 which suggest mechanical resilience on the order of 50–60%.89–91 More recently reported silk/elastin-like materials have demonstrated resilience up to approximately 80–90%.92, 93 The resilience values for these RLP-based hydrogels are comparable to that of aortic elastin (approximately 80%), suggesting the potential suitability for developing the RLP24-PEG gels for cardiovascular applications.
Table 2.
Resilience of RLP24-PEG hydrogels. Each value represents the mean of three separate sample measurements.
| Cross-linking Ratio (VS:CYS) | Resilience at percent strain (%)
|
||
|---|---|---|---|
| 20% | 40% | 60% | |
| 3:2 | 93 ± 4 | 90 ± 4 | 88 ± 4 |
| 1:1 | 90 ± 4 | 89 ± 3 | 87 ± 3 |
| 1:2 | 98 ± 2 | 92 ± 1 | 89 ± 1 |
Each value represents the simple mean of 3 samples; the error is reported as the standard deviation.
In addition to the cyclic strain measurements, the RLP24-PEG hydrogels were subjected to strain-to-break experiments in order to explore the extensibility of the gels. Figure 3C depicts representative strain-to-break measurements for the three RLP24-PEG cross-linking ratios; the 3:2 ratio RLP24-PEG gel had an average strain-to-break of approximately 68% while the 1:1 and 1:2 ratios had average strain-to-break values of approximately 85% and 173%, respectively (see Table 1). Pure RLP hydrogels exhibited maximum strain values that are far greater (up to 400% strain) than these values for the RLP24-PEG hybrid hydrogels.13, 14, 16 The use of PEG as a cross-linker in the hybrid hydrogels would not immediately suggest such a disparity in extensibility; in fact, wholly PEG hydrogels formed through step-polymerization cross-linking were extensible to 300% strain.79 The disparities here may result, at least in part, from the fact that the RLP24-PEG hydrogels were more swollen and softer than the RLP hydrogels, and thus may have been more susceptible to damage when compressed by the metal clamps of the axial tensile geometry during sample loading. The water content for each RLP24-PEG hydrogel composition was greater than 90% (see Figure S8A/B), which while consistent with values reported for other protein-PEG hydrogels,70, 94 is higher than those observed for the RLP-only hydrogels (approximately 80–90%).14,15 As elastomers absorb large quantities of solvent, the molecular interactions that lead to their toughness diminish: first, fewer network chains occupy the cross-sectional area of the hydrogel, which implies fewer chains need to be fractured to propagate a crack; second, separation between segments of the network would provide less opportunity for crystallization and viscoelastic energy dissipation.95 In addition, and perhaps a main contribution, the RLP24-PEG hydrogels were found to partition into different protein-rich and PEG-rich phases (see below). As a result, less than expected cross-linking may occur within these domains; a factor which could be contrasted with microgel-reinforced hydrogel films where the continuous phase is hardened using highly cross-linked double-network microparticles,96 Heterogeneous microstructure in RLP24-PEG hydrogels may dampen the toughness of hydrogels as fracture can propagate through more loosely cross-linked matrix.95 While the heterogeneity and the highly swollen nature of the RLP24-PEG hydrogels both likely contribute to the lower extensibility the materials retain resilience that is characteristic of high-performance resilin-based materials.
HYDROGEL MICROSTRUCTURE
Upon mixing, transparent precursor solutions of RLP24 and PEG instantly turn opaque before gradually regaining optically clarity as the hydrogels were cross-linked; phase contrast microscopy revealed a unique spherical heterogeneity within the hydrogels. To determine the qualitative composition of this microstructure, the RLP24 and PEGVS precursors were labeled with rhodamine and fluorescein, respectively, prior to being cross-linked into hydrogels and imaged via confocal microscopy. Figure 4 depicts all three channels and a composite acquired from imaging a fluorescent RLP24-PEG hydrogel; the PEG phase is highlighted in green (Figure 4A) while the RLP24 phase is in red (Figure 4B). Figure 4C displays the transmitted channel and Figure 4D is a composite image of all three channels. The figure clearly indicates that the RLP24-PEG hydrogel (20 wt%, 3:2 ratio of vinyl sulfone to thiol) comprises a heterogeneous structure with RLP-rich (red spheres) and PEG-rich (green interstitial region) phases. Importantly, the two precursor macromolecules did not precipitate and have only partially partitioned into separate phases; this avoids the complete exclusion of one macromolecule from a particular phase and thus permits cross-linking of the hybrid gels.49, 97, 98 As indicated in Figure S10, the hydrogel microstructure persisted even after twelve hours of swelling in PBS buffer. The phase separation observed in RLP24-PEG hydrogels was distinct from the behavior of the RLP-only hydrogels (cross-linked with THP) and was also exclusive to those cross-linked with a higher molecular weight (~10 kDa) PEG precursor (see Figure S11). RLP24 hydrogels cross-linked with a smaller PEG (~2 kDa, Figure S11) lacked phase-separated heterogeneity, but also lacked the mechanical integrity necessary for further testing.
Figure 4.

Confocal microscopy of the fluorescently-labeled RLP24-PEGVS hydrogels. Panels depict the (A) fluorescein channel and the PEG component of the hydrogel, (B) the rhodamine channel and the RLP24 component of the hydrogel, and (C) the transmitted channel. Panel (D) provides a composite image of all three channels. Scale bar is 200 μm.
The partitioning of two different polymers in aqueous solutions has been a well-known phenomenon for more than a century.99 The behavior is most simply described using the statistical mechanical treatment of ternary polymer solutions in Flory-Huggins theory,100, 101 in which the free energy of mixing is dominated by polymer-polymer interactions owing to the high molecular weight of the polymer chains. Two dissimilar polymers that interact unfavorably phase separate even at low concentrations of polymer solute.49, 50, 100, 101 Aqueous mixtures of PEG and protein have been widely studied owing to their relevance to protein crystallization,51, 52 separation/purification strategies,53–55 and the analysis of disease progression.97 However, most protein-PEG solutions eventually form macroscopic phases;49, 97 in the RLP-PEG hydrogels, the cross-linking reaction competes with the process of macroscale phase separation and effectively captures heterogeneous micron-size domains of different compositions. Other work within our lab further supports that PEG-induced phase separation, rather than the cross-linking chemistry, is a driving factor behind the generation of microstructure in these hydrogels. RLP-PEG hydrogels employing amine-functionalized precursors cross-linked with THP, exhibit similar phase separation (unpublished observations), and recently reported photocross-linkable RLP-PEG hybrid hydrogels also share the observed heterogeneity. The domains in the photocross-linked gels were smaller (1 to 10 μm) than those observed here for the RLP24-PEG gels (10–100 μm), owing to the more rapid gelation (30–45 seconds) imparted by the photo-initiated thiol-ene click chemistry; the separated phases have less time to coalesce into larger domains as compared to the RLP24-PEG hydrogels reported here.38 Simple strategies, such as controlling gelation times, thus provide powerful alternatives for the generation of microstructured materials that may direct cell location and signaling.
HYDROGEL DEGRADATION
The RLP24 polypeptide was designed with two MMP cleavage domains (GPQGIWGQ) derived from human α(I) collagen72 to impart cell-directed degradability to the material and because of the role MMPs play in tissue remodeling,72, 73 cell migration, and cell proliferation.65, 67, 68, 102 Therefore, it is important that these domains remain sensitive to degradation even when the polypeptide is cross-linked into a hydrogel network; to confirm this sensitivity to enzyme-mediated degradation, proteolytic degradation of RLP24-PEG hydrogels by rhMMP1 was investigated through in situ monitoring of hydrogel mechanical properties via compression testing. The RLP24-PEG hydrogels (20wt%, 3:2 vinyl sulfone to thiol ratio) were tested without enzyme for 12 hours before buffer containing rhMMP1 (171 nM, 1.5 mL total volume) was substituted for an additional 10 hours of testing. Figure 5 depicts the simple mean and error (standard deviation) of the normalized compressive modulus for three different RLP24-PEG hydrogels. The black squares, indicating the control period, demonstrated an approximate 20% decrease in the normalized storage modulus following the start of the experiment; this initial decrease likely resulted from the swelling of the RLP24-PEG hydrogel as it stabilized after only three hours of testing. Importantly, after introduction of the enzyme, the normalized storage modulus decreased from approximately 80% to 20% of its original value over the remaining course of the experiment, indicating the cleavage of the MMP domains in the hydrogel (consistent with observed degradation of the soluble RLP, see Supporting Information, Figure S13) and suggesting opportunities for engineering cell-directed degradability to these biomaterials. The decrease in RLP24-PEG hydrogel storage moduli was more rapid than that reported for either ELP hydrogels with uPA-degradable domains37 or pure RLP hydrogels with MMP-sensitive domains (unpublished data). In both of these other reports, the networks were formed using small-molecule cross-linkers that likely decrease the network mesh size and may consequently restrict access of the enzyme; both approaches offer select advantages in tuning degradation rates of these materials.
Figure 5.
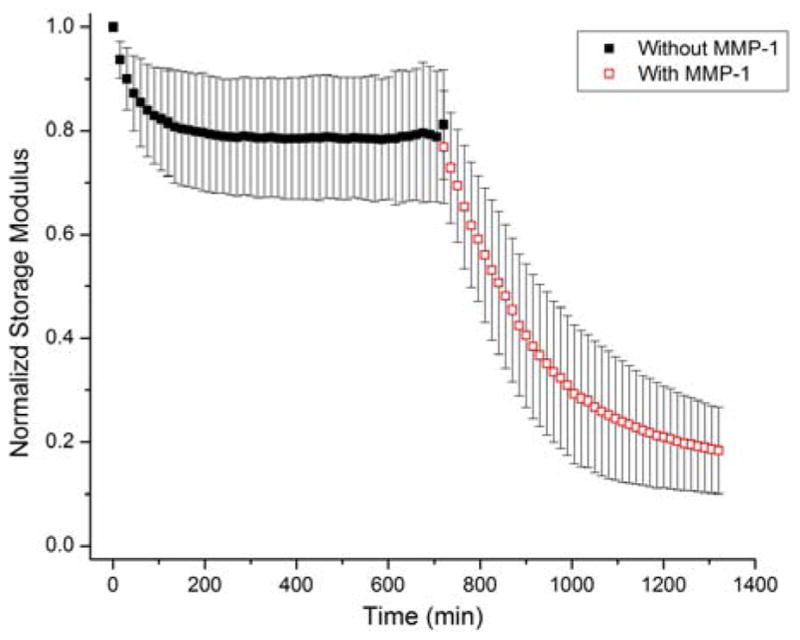
Normalized compressive storage modulus of RLP24-PEG hydrogels (20wt%, 3:2 vinyl sulfone to thiol) incubated in MMP1 enzyme buffer without (closed, black) and with (open, red) MMP1 enzyme. Data points were acquired every 15 minutes using 5% strain at a frequency of 1Hz.
ENCAPSULATION, VIABILITY AND PROLIFERATION OF hMSCS
As potential injectable scaffolds for tissue engineering, the RLP24-PEG hydrogels should have the capacity to deliver viable cells to target tissues. Therefore, the cytocompatiblity of RLP24-PEG hydrogels was investigated by testing the viability of hMSCs encapsulated within the gels and cultured in 3D over a two-week period. Adult human mesenchymal stem cells (hMSCs) are particularly useful as they are easily expanded, relatively non-immunogenic, and capable of multilineage differentiation.103 It has been suggested that hMSCs can function as vascular progenitors104–106 and support the development of healthy vascular tissue following coronary bypass surgery. hMSCs therefore may have significant utility in cardiovascular tissue engineering applications. Bone-marrow-derived hMSCs were chosen for this study owing to their capacity for self-renewal and differentiation107 as well as their vascular differentiation potential.108 Encouraging results from MSC-seeded vascular grafts suggest their potential as a source of an allogenic cell line for vascular therapies.108 For example, MSC-seeded nanofibrous grafts demonstrated greater anti-thrombogenic properties and developed a more highly organized elastic lamina layer as compared to acellular grafts.109
Figure 6 presents representative results of the LIVE/DEAD confocal microscopy assay used to assess cell viability. The day 0 image, depicted in Figure 6A, illustrates the high viability of the hMSCs immediately following encapsulation; panels B, C, and D depict results for days 5, 10, and 15, respectively. Consistent with our previous studies encapsulating AoAFs within RLP24-PEG hydrogels,17 the hMSC-laden hydrogels formed rapidly and within 5–7 minutes displayed elastic solid-like behavior, similar to that of cell-lacking hydrogels as assessed qualitatively via tilt-test and observations of physical properties during handling. Following encapsulation, the hMSCs demonstrated good viability; a majority of the cells were stained green, indicating intact, metabolically active cells. Importantly, as depicted in the subsequent time points (see Figure 6B–D), the hMSCs remained viable throughout the entire culture period, though in later time points the cell density was reduced due to gel degradation and swelling. The high and extended viability of the bone-derived hMSCs in this study suggests the suitability of these matrices for the delivery and use of hMSCs in the types of applications described above.
Figure 6.

Analysis of viability of hMSCs encapsulated in RLP24-PEG hydrogels (20wt% 3:2 vinyl sulfone:thiol) at day 0 (A), day 5 (B), day 10 (C) and day 15 (D). Maximum intensity projections of z-stacks (200–250 μm thick) are presented. The objective was a 10x water lens and the scale bars represent 200 μm.
As also illustrated in Figures 6B–D, the hMSCs were able to spread out and adopt 3D spindle-like morphologies when encapsulated in the RLP24-PEG gels, similar to our previous observations of the spreading of adventitial fibroblasts in the RLP-PEG gels,17 but in contrast to the lack of spreading of cells in early examples of cell encapsulation within RLP and ELP hydrogels cross-linked via small molecules.15, 110 More recently, pure ELP hydrogels cross-linked via tetrakis(hydroxymethyl)phosphonium chloride (THPC) have exhibit improved spreading, but these gels are formed at lower precursor concentrations (≤5 wt%).111, 112 Therefore, the improved cell spreading in these matrices and in the RLP-PEG hydrogels most likely results, at least in part, from the greater mesh size of the hydrogels and improved accessibility of the MMP-sensitive domains when they exist. Photocross-linkable RLP-PEG hydrogels developed by our lab and reported previously cross-linked rapidly and demonstrated remarkable hMSC viability, but there was minimal cell spreading even in the presence of RGD ligands;38 the lack of MMP degradable domains in the photoreactive RLPs may have been detrimental to cell spreading. Anderson et al., demonstrated the necessity of MMP-sensitive cross-links in facilitating the spreading behavior of hMSCs encapsulated within PEG networks.102 The observed spreading of the hMSCs is also consistent with other reports of cell encapsulation and migration within recombinant protein-PEG hybrid hydrogels containing MMP sensitive domains.65
Interestingly, the hMSCs were also found to spread around and onto the phase-separated domains depicted in Figure 5; a sequence of composite confocal images, presented in Figure S14, illustrates this behavior. Recently, Lee et al. demonstrated the use of PEG as a porogenic agent in fibrinogen-based hydrogels; during the photocross-linking of fibrinogen-PEG diacrylate conjugates, phase separation was induced by the presence of free hydroxyl-terminated PEG. The free PEG was subsequently leached from the gels, but left porosity that enhanced the spreading of encapsulated smooth muscle cells.58 While the PEG component in the RLPL24-PEG hydrogels remains cross-linked within the hydrogels reported here, the heterogeneity (and corresponding interfaces) introduced by phase separation is suggested to enhance cell spreading behavior.
The proliferation of hMSCs encapsulated within the RLP24-PEG hydrogels was analyzed between days 3 and 5 of culture via fluorescence microscopy and a Clickit™ EdU assay. Figure 7 depicts the results from a representative cell-gel construct with panels A and B representing cell nuclei stained with DAPI and the Alexa Fluor™ 555 dye, respectively, and panel C depicting a merged image. DAPI non-specifically stains all nuclei present while the Alexa Fluor™ 555 dye stains only the small fraction of nuclei that recently divided. Although the cell density is reduced in these images owing to swelling (as noted above), the composite image clearly depicts that several nuclei, appearing in purple, recently divided and that the hMSCs are proliferating within the RLP24-PEG hydrogels. These results compare well with our previous encapsulation results with aortic adventitial fibroblasts17 as well as with hMSC proliferation within bioactive PEG hydrogels102 and various silk-composite gels.113–115 The Clickit™ EdU was the most successful method that provided evidence for hMSC proliferation within RLP24-PEG gels; other methods such as alamarBlue® and PicoGreen® assays were unsuccessful due to dye retention and poor DNA recovery, respectively. The clear cytocompatibility and mechanical properties of the RLP24-PEG hydrogels illustrate their utility as bioactive hydrogels for tissue engineering applications that require soft elasticity.
Figure 7.

Proliferation of hMSCs analyzed via Click-it™ Edu Assay and fluorescence microscopy between days 3 and 5 of cell culture. All cell nuclei are stained with DAPI (A) and proliferative cell nuclei stained with the Alexa Fluor™ 555 (B). The final panel (C) provides a merged image with the proliferative nuclei appearing purple. Maximum intensity projection of z-stacks are depicted.
Increasingly important to the design of tissue engineering scaffolds is nano-/microscale control over material properties like topography, matrix stiffness or biological signal distribution;43 morphogenesis of complex tissues, as well as the associated behaviors of differentiation, proliferation, adhesion, and migration, within a 3D scaffold relies on precise signaling to seeded or infiltrating cells.43 Currently, post-gelation methods, including photolithographic patterning44, 45, 116 and leaching inert elements,58, 117, 118 offer methods for creating hierarchal structures within gels, but these techniques suffer from either low-throughput or extensive post-processing. The phase separation action of concentrated protein-polymer solutions and its consequent generation of hydrogel microstructure may offer unique, one-step opportunities to localize biological signals, cells or physical material properties (e.g. matrix elasticity) within a single 3D material to better mimic the hierarchal structure of native ECM.
CONCLUSIONS
Hybrid hydrogels comprising a resilin-like polypeptide, RLP24, and vinyl sulfone functionalized PEG macromer are elastic, resilient materials capable of the successful encapsulation and 3D culture of hMSCs. Cross-linked via a Michael-type addition between vinyl sulfone and thiol, these hydrogels rapidly form and have rubber-like properties that would be useful for mechanically-demanding tissue engineering applications, especially those aiming to remedy cardiovascular pathologies. The resilin-like polypeptides are sensitive to degradation via rhMMP1 both as soluble polypeptide and as cross-linked hydrogels. Encapsulated hMSCs remain viable and proliferate within the hydrogels over a 15 day time period and adopt a spread, spindle-like morphology. Finally, the RLP24-PEG hydrogels demonstrate liquid-liquid partitioning behavior that gives rise to heterogeneous microstructure which might have implications for controlling the mechanical and biological properties of these hydrogels and directing cell behavior.
Supplementary Material
Acknowledgments
Funding Sources
This publication was supported by the National Institutes of Health; specifically the Delaware COBRE program, supported by a grant from the National Institute of General Medical Sciences (1 P30 GM110758-01), the National Institute on Deafness and Other Communication Disorders (RO1 DC011377A to K.L.K), and the National Heart, Lung, and Blood Institute (5 R01 HL108110-02; to R.E.A.).
This publication was made possible by the Delaware COBRE program, supported by a grant from the National Institute of General Medical Sciences (NIGMS, 1 P30 GM110758-01) from the National Institutes of Health, and also by the National Institute on Deafness and Other Communication Disorders (RO1 DC011377A) and the National Heart Lung and Blood Institute (RO1 HL108110). The authors would like to thank Dr. Xinqiao Jia for the generous access to the RSA-G2 mechanical analyzer and Derrick Allen (Department of Chemistry & Biochemistry, University of Delaware, Newark, DE) for the preparation of the Teflon molds.
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
References
- 1.Werkmeister JA, Ramshaw JAM. Biomedical Materials. 2012;7(1) doi: 10.1088/1748-6041/7/1/012002. [DOI] [PubMed] [Google Scholar]
- 2.Gomes S, Leonor IB, Mano JF, Reis RL, Kaplan DL. Prog Polym Sci. 2012;37(1):1–17. doi: 10.1016/j.progpolymsci.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gomes SC, Leonor IB, Mano JF, Reis RL, Kaplan DL. Biomaterials. 2011;32(18):4255–4266. doi: 10.1016/j.biomaterials.2011.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kundu B, Kurland NE, Bano S, Patra C, Engel FB, Yadavalli VK, Kundu SC. Prog Polym Sci. 2014;39(2):251–267. [Google Scholar]
- 5.Spiess K, Lammel A, Scheibel T. Macromol Biosci. 2010;10(9):998–1007. doi: 10.1002/mabi.201000071. [DOI] [PubMed] [Google Scholar]
- 6.van Eldijk M, McGann C, Kiick K, van Hest JM. Elastomeric polypeptides. In: Deming T, editor. Peptide-Based Materials. Vol. 310. Springer; Berlin Heidelberg: 2012. pp. 71–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Annabi N, Mithieux SM, Camci-Unal G, Dokmeci MR, Weiss AS, Khademhosseini A. Biochem Eng J. 2013;77(0):110–118. doi: 10.1016/j.bej.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nettles DL, Chilkoti A, Setton LA. Adv Drug Del Rev. 2010;62(15):1479–1485. doi: 10.1016/j.addr.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lim DW, Nettles DL, Setton LA, Chilkoti A. Biomacromolecules. 2007;8(5):1463–1470. doi: 10.1021/bm061059m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Charati MB, Ifkovits JL, Burdick JA, Linhardt JG, Kiick KL. Soft Matter. 2009;5(18):3412–3416. doi: 10.1039/b910980c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elvin CM, Carr AG, Huson MG, Maxwell JM, Pearson RD, Vuocolo T, Liyou NE, Wong DCC, Merritt DJ, Dixon NE. Nature. 2005;437(7051):999–1002. doi: 10.1038/nature04085. [DOI] [PubMed] [Google Scholar]
- 12.Kim M, Elvin C, Brownlee A, Lyons R. Protein Expression Purif. 2007;52(1):230–236. doi: 10.1016/j.pep.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 13.Li L, Kiick K. Frontiers in Chemistry. 2014;2(21) doi: 10.3389/fchem.2014.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li L, Teller S, Clifton RJ, Jia X, Kiick KL. Biomacromolecules. 2011;12(6):2302–2310. doi: 10.1021/bm200373p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li L, Tong Z, Jia X, Kiick KL. Soft Matter. 2013;9(3):665–673. doi: 10.1039/C2SM26812D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lyons RE, Nairn KM, Huson MG, Kim M, Dumsday G, Elvin CM. Biomacromolecules. 2009;10(11):3009–3014. doi: 10.1021/bm900601h. [DOI] [PubMed] [Google Scholar]
- 17.McGann CL, Levenson EA, Kiick KL. Macromol Chem Phys. 2013;214(2):203–213. doi: 10.1002/macp.201200412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu J. Biomaterials. 2010;31(17):4639–4656. doi: 10.1016/j.biomaterials.2010.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Daamen WF, Veerkamp JH, van Hest JCM, van Kuppevelt TH. Biomaterials. 2007;28(30):4378–4398. doi: 10.1016/j.biomaterials.2007.06.025. [DOI] [PubMed] [Google Scholar]
- 20.Konig G, McAllister TN, Dusserre N, Garrido SA, Iyican C, Marini A, Fiorillo A, Avila H, Wystrychowski W, Zagalski K, Maruszewski M, Jones AL, Cierpka L, de la Fuente LM, L’Heureux N. Biomaterials. 2009;30(8):1542–1550. doi: 10.1016/j.biomaterials.2008.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mitchell SL, Niklason LE. Cardiovasc Pathol. 2003;12(2):59–64. doi: 10.1016/s1054-8807(02)00183-7. [DOI] [PubMed] [Google Scholar]
- 22.Long JL, Tranquillo RT. Matrix Biol. 2003;22(4):339–350. doi: 10.1016/s0945-053x(03)00052-0. [DOI] [PubMed] [Google Scholar]
- 23.Patel A, Fine B, Sandig M, Mequanint K. Cardiovasc Res. 2006;71(1):40–49. doi: 10.1016/j.cardiores.2006.02.021. [DOI] [PubMed] [Google Scholar]
- 24.Silver FH, Snowhill PB, Foran DJ. Ann Biomed Eng. 2003;31(7):793–803. doi: 10.1114/1.1581287. [DOI] [PubMed] [Google Scholar]
- 25.Whittaker J, Balu R, Choudhury NR, Dutta NK. Polym Int. 2014;63(9):1545–1557. [Google Scholar]
- 26.Gosline J, Lillie M, Carrington E, Guerette P, Ortlepp C, Savage K. Philos Trans R Soc London, Ser B. 2002;357(1418):121–132. doi: 10.1098/rstb.2001.1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Su RSC, Renner JN, Liu JC. Biomacromolecules. 2013;14(12):4301–4308. doi: 10.1021/bm401162g. [DOI] [PubMed] [Google Scholar]
- 28.Li L, Charati MB, Kiick KL. Polymer Chemistry. 2010;1(8):1160–1170. doi: 10.1039/b9py00346k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang H, Cai L, Paul A, Enejder A, Heilshorn SC. Biomacromolecules. 2014;15(9):3421–3428. doi: 10.1021/bm500969d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gonen-Wadmany M, Oss-Ronen L, Seliktar D. Biomaterials. 2007;28(26):3876–3886. doi: 10.1016/j.biomaterials.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 31.Munoz-Pinto DJ, Jimenez-Vergara AC, Gharat TP, Hahn MS. Biomaterials. 2015;40:32–42. doi: 10.1016/j.biomaterials.2014.10.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sargeant TD, Desai AP, Banerjee S, Agawu A, Stopek JB. Acta Biomater. 2012;8(1):124–132. doi: 10.1016/j.actbio.2011.07.028. [DOI] [PubMed] [Google Scholar]
- 33.Shapira-Schweitzer K, Seliktar D. Acta Biomater. 2007;3(1):33–41. doi: 10.1016/j.actbio.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 34.Almany L, Seliktar D. Biomaterials. 2005;26(15):2467–2477. doi: 10.1016/j.biomaterials.2004.06.047. [DOI] [PubMed] [Google Scholar]
- 35.Seliktar D. Science. 2012;336(6085):1124–1128. doi: 10.1126/science.1214804. [DOI] [PubMed] [Google Scholar]
- 36.Dikovsky D, Bianco-Peled H, Seliktar D. Biomaterials. 2006;27(8):1496–1506. doi: 10.1016/j.biomaterials.2005.09.038. [DOI] [PubMed] [Google Scholar]
- 37.Straley KS, Heilshorn SC. Soft Matter. 2009;5(1):114–124. [Google Scholar]
- 38.McGann CL, Dumm RE, Jurusik AK, Sidhu I, Kiick KL. Macromol Biosci. 2015 doi: 10.1002/mabi.201500305. n/a-n/a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim Y, Renner JN, Liu JC. Biomaterials Science. 2014;2(8):1110–1119. doi: 10.1039/c3bm60333d. [DOI] [PubMed] [Google Scholar]
- 40.Renner JN, Kim Y, Cherry KM, Liu JC. Protein Expression Purif. 2012;82(1):90–96. doi: 10.1016/j.pep.2011.11.019. [DOI] [PubMed] [Google Scholar]
- 41.Sun F, Zhang WB, Mahdavi A, Arnold FH, Tirrell DA. Proceedings of the National Academy of Sciences. 2014;111(31):11269–11274. doi: 10.1073/pnas.1401291111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang WB, Sun F, Tirrell DA, Arnold FH. J Am Chem Soc. 2013;135(37):13988–13997. doi: 10.1021/ja4076452. [DOI] [PubMed] [Google Scholar]
- 43.Dvir T, Timko BP, Kohane DS, Langer R. Nature nanotechnology. 2011;6(1):13–22. doi: 10.1038/nnano.2010.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hahn MS, Miller JS, West JL. Adv Mater. 2006;18(20):2679–2684. [Google Scholar]
- 45.Lee SH, Moon JJ, West JL. Biomaterials. 2008;29(20):2962–2968. doi: 10.1016/j.biomaterials.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bailey BM, Hui V, Fei R, Grunlan MA. J Mater Chem. 2011;21(46):18776–18782. doi: 10.1039/C1JM13943F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bailey BM, Fei R, Munoz-Pinto D, Hahn MS, Grunlan MA. Acta Biomater. 2012;8(12):4324–4333. doi: 10.1016/j.actbio.2012.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Krishna OD, Jha AK, Jia X, Kiick KL. Biomaterials. 2011;32(27):6412–6424. doi: 10.1016/j.biomaterials.2011.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Albertsson PA. Partition of Cell Particles and Macromolecules. Wiley; 1986. [Google Scholar]
- 50.Baskir JN, Hatton TA, Suter UW. Biotechnol Bioeng. 1989;34(4):541–558. doi: 10.1002/bit.260340414. [DOI] [PubMed] [Google Scholar]
- 51.Hedrich HC, Spener F, Menge U, Hecht HJ, Schmid RD. Enzyme Microb Technol. 1991;13(10):840–847. [Google Scholar]
- 52.McPherson A. J Cryst Growth. 1991;110(1–2):1–10. [Google Scholar]
- 53.Diederich P, Amrhein S, Hämmerling F, Hubbuch J. Chem Eng Sci. 2013;104(0):945–956. [Google Scholar]
- 54.Hatti-Kaul R, Mattiasson B. Isolation and Purification of Proteins. Taylor & Francis; 2003. [Google Scholar]
- 55.Lima ÁS, Alegre RM, Meirelles AJA. Carbohydr Polym. 2002;50(1):63–68. [Google Scholar]
- 56.Lee AG, Arena CP, Beebe DJ, Palecek SP. Biomacromolecules. 2010;11(12):3316–3324. doi: 10.1021/bm100792y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Elbert DL. Acta Biomater. 2011;7(1):31–56. doi: 10.1016/j.actbio.2010.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee BH, Tin SPH, Chaw SY, Cao Y, Xia Y, Steele TWJ, Seliktar D, Bianco-Peled H, Venkatraman SS. J Biomater Sci, Polym Ed. 2014;25(4):394–409. doi: 10.1080/09205063.2013.862401. [DOI] [PubMed] [Google Scholar]
- 59.Frisman I, Seliktar D, Bianco-Peled H. Acta Biomater. 2012;8(1):51–60. doi: 10.1016/j.actbio.2011.07.030. [DOI] [PubMed] [Google Scholar]
- 60.Frisman I, Seliktar D, Bianco-Peled H. Biomaterials. 2011;32(31):7839–7846. doi: 10.1016/j.biomaterials.2011.06.078. [DOI] [PubMed] [Google Scholar]
- 61.Baldwin AD, Kiick KL. Polymer Chemistry. 2013;4(1):133–143. doi: 10.1039/C2PY20576A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kharkar PM, Kloxin AM, Kiick KL. Journal of Materials Chemistry B. 2014;2(34):5511–5521. doi: 10.1039/c4tb00496e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zustiak SP, Leach JB. Biomacromolecules. 2010;11(5):1348–1357. doi: 10.1021/bm100137q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lutolf MP, Raeber GP, Zisch AH, Tirelli N, Hubbell JA. Adv Mater. 2003;15(11):888–892. [Google Scholar]
- 65.Rizzi SC, Ehrbar M, Halstenberg S, Raeber GP, Schmoekel HG, Hagenmuller H, Muller R, Weber FE, Hubbell JA. Biomacromolecules. 2006;7(11):3019–3029. doi: 10.1021/bm060504a. [DOI] [PubMed] [Google Scholar]
- 66.Aimetti AA, Machen AJ, Anseth KS. Biomaterials. 2009;30(30):6048–6054. doi: 10.1016/j.biomaterials.2009.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Patterson J, Hubbell JA. Biomaterials. 2010;31(30):7836–7845. doi: 10.1016/j.biomaterials.2010.06.061. [DOI] [PubMed] [Google Scholar]
- 68.Lutolf MP, Lauer-Fields JL, Schmoekel HG, Metters AT, Weber FE, Fields GB, Hubbell JA. Proc Natl Acad Sci U S A. 2003;100(9):5413–5418. doi: 10.1073/pnas.0737381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kiick KL. Polymer Reviews. 2007;47(1):1–7. [Google Scholar]
- 70.Rizzi SC, Hubbell JA. Biomacromolecules. 2005;6(3):1226–1238. doi: 10.1021/bm049614c. [DOI] [PubMed] [Google Scholar]
- 71.Gustafson JA, Price RA, Frandsen J, Henak CR, Cappello J, Ghandehari H. Biomacromolecules. 2013;14(3):618–625. doi: 10.1021/bm3013692. [DOI] [PubMed] [Google Scholar]
- 72.Nagase H, Fields GB. Biopolymers. 1996;40(4):399–416. doi: 10.1002/(SICI)1097-0282(1996)40:4%3C399::AID-BIP5%3E3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 73.Turk BE, Huang LL, Piro ET, Cantley LC. Nat Biotech. 2001;19(7):661–667. doi: 10.1038/90273. [DOI] [PubMed] [Google Scholar]
- 74.Vu TH, Werb Z. Genes Dev. 2000;14(17):2123–2133. doi: 10.1101/gad.815400. [DOI] [PubMed] [Google Scholar]
- 75.Studier FW. Protein Expression Purif. 2005;41(1):207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 76.The QIAexpressionist. 5. QIAGEN, Inc; 2003. [Google Scholar]
- 77.Interchim Quantitation of sulfhydryls DTNB. Ellman’s reagent. 2012 Jul; http://www.interchim.fr/ft/0/01566H.pdf.
- 78.Bulaj G, Kortemme T, Goldenberg DP. Biochemistry. 1998;37(25):8965–8972. doi: 10.1021/bi973101r. [DOI] [PubMed] [Google Scholar]
- 79.Cui J, Lackey MA, Madkour AE, Saffer EM, Griffin DM, Bhatia SR, Crosby AJ, Tew GN. Biomacromolecules. 2012;13(3):584–588. doi: 10.1021/bm300015s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ardell DH, Andersen SO. Insect Biochem Mol Biol. 2001;31(10):965–970. doi: 10.1016/s0965-1748(01)00044-3. [DOI] [PubMed] [Google Scholar]
- 81.Ruoslahti E, Pierschbacher MD. Science. 1987;238(4826):491–497. doi: 10.1126/science.2821619. [DOI] [PubMed] [Google Scholar]
- 82.Liu SC, Zhou FY, Hook M, Carson DD. Proc Natl Acad Sci U S A. 1997;94(5):1739–1744. doi: 10.1073/pnas.94.5.1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chambon F, Winter HH. J Rheol. 1987;31(8):683–697. [Google Scholar]
- 84.Winter HH, Morganelli P, Chambon F. Macromolecules. 1988;21(2):532–535. [Google Scholar]
- 85.Chow D, Nunalee ML, Lim DW, Simnick AJ, Chilkoti A. Mater Sci Eng R-Rep. 2008;62(4):125–155. doi: 10.1016/j.mser.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cappello J, Crissman J, Dorman M, Mikolajczak M, Textor G, Marquet M, Ferrari F. Biotechnol Prog. 1990;6(3):198–202. doi: 10.1021/bp00003a006. [DOI] [PubMed] [Google Scholar]
- 87.Mubeen A. Mechanics of Solids. Pearson Education; 2002. [Google Scholar]
- 88.Mott PH, Roland CM. Phys Rev B. 2009;80(13):132104. [Google Scholar]
- 89.Wu X, Sallach RE, Caves JM, Conticello VP, Chaikof EL. Biomacromolecules. 2008;9(7):1787–1794. doi: 10.1021/bm800012x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sallach RE, Cui W, Wen J, Martinez A, Conticello VP, Chaikof EL. Biomaterials. 2009;30(3):409–422. doi: 10.1016/j.biomaterials.2008.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Krishna UM, Martinez AW, Caves JM, Chaikof EL. Acta Biomater. 2012;8(3):988–997. doi: 10.1016/j.actbio.2011.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Teng W, Cappello J, Wu X. Biomacromolecules. 2009;10(11):3028–3036. doi: 10.1021/bm900651g. [DOI] [PubMed] [Google Scholar]
- 93.Qiu W, Huang Y, Teng W, Cohn CM, Cappello J, Wu X. Biomacromolecules. 2010;11(12):3219–3227. doi: 10.1021/bm100469w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Halstenberg S, Panitch A, Rizzi S, Hall H, Hubbell JA. Biomacromolecules. 2002;3(4):710–723. doi: 10.1021/bm015629o. [DOI] [PubMed] [Google Scholar]
- 95.Naficy S, Brown HR, Razal JM, Spinks GM, Whitten PG. Aust J Chem. 2011;64(8):1007–1025. [Google Scholar]
- 96.Hu J, Hiwatashi K, Kurokawa T, Liang SM, Wu ZL, Gong JP. Macromolecules. 2011;44(19):7775–7781. [Google Scholar]
- 97.Annunziata O, Asherie N, Lomakin A, Pande J, Ogun O, Benedek GB. Proc Natl Acad Sci U S A. 2002;99(22):14165–14170. doi: 10.1073/pnas.212507199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Atha DH, Ingham KC. J Biol Chem. 1981;256(23):12108–17. [PubMed] [Google Scholar]
- 99.Beijerinck MW. Kolloidz. 1910;7:16. [Google Scholar]
- 100.Walter H. Partitioning In Aqueous Two – Phase System: Theory, Methods, Uses, And Applications To Biotechnology. Elsevier Science; 2012. [Google Scholar]
- 101.Flory PJ. Principles of Polymer Chemistry. Cornell University Press; 1953. [Google Scholar]
- 102.Anderson SB, Lin CC, Kuntzler DV, Anseth KS. Biomaterials. 2011;32(14):3564–3574. doi: 10.1016/j.biomaterials.2011.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chamberlain G, Fox J, Ashton B, Middleton J. Stem Cells. 2007;25(11):2739–2749. doi: 10.1634/stemcells.2007-0197. [DOI] [PubMed] [Google Scholar]
- 104.Au P, Tam J, Fukumura D, Jain RK. Blood. 2008;111(9):4551–4558. doi: 10.1182/blood-2007-10-118273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Davani S, Marandin A, Mersin N, Royer B, Kantelip B, Hervé P, Etievent JP, Kantelip JP. Circulation. 2003;108(10S):253–258. doi: 10.1161/01.cir.0000089186.09692.fa. [DOI] [PubMed] [Google Scholar]
- 106.Nagaya N, Fujii T, Iwase T, Ohgushi H, Itoh T, Uematsu M, Yamagishi M, Mori H, Kangawa K, Kitamura S. American Journal of Physiology - Heart and circulatory physiology. 2004;287(6):H2670–H2676. doi: 10.1152/ajpheart.01071.2003. [DOI] [PubMed] [Google Scholar]
- 107.Bianco P, Robey PG. Nature. 2001;414(6859):118–121. doi: 10.1038/35102181. [DOI] [PubMed] [Google Scholar]
- 108.Huang NF, Li S. Regen Med. 2008;3(6):877–892. doi: 10.2217/17460751.3.6.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hashi CK, Zhu Y, Yang GY, Young WL, Hsiao BS, Wang K, Chu B, Li S. Proc Natl Acad Sci U S A. 2007;104(29):11915–11920. doi: 10.1073/pnas.0704581104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lim DW, Nettles DL, Setton LA, Chilkoti A. Biomacromolecules. 2008;9(1):222–230. doi: 10.1021/bm7007982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lampe KJ, Antaris AL, Heilshorn SC. Acta Biomater. 2013;9(3):5590–5599. doi: 10.1016/j.actbio.2012.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Chung C, Lampe KJ, Heilshorn SC. Biomacromolecules. 2012;13(12):3912–3916. doi: 10.1021/bm3015279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Haider M, Cappello J, Ghandehari H, Leong K. Pharm Res. 2008;25(3):692–699. doi: 10.1007/s11095-007-9282-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Jeon O, Alsberg E. Tissue Eng Part A. 2013;19(11–12):1424–1432. doi: 10.1089/ten.tea.2012.0581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wang X, Partlow B, Liu J, Zheng Z, Su B, Wang Y, Kaplan DL. Acta Biomater. 2015;12(0):51–61. doi: 10.1016/j.actbio.2014.10.027. [DOI] [PubMed] [Google Scholar]
- 116.Culver JC, Hoffmann JC, Poche RA, Slater JH, West JL, Dickinson ME. Adv Mater. 2012;24(17):2344–8. doi: 10.1002/adma.201200395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Chiu YC, Larson JC, Isom A, Jr, Brey EM. Tissue Eng Part C Methods. 2010;16(5):905–12. doi: 10.1089/ten.TEC.2009.0646. [DOI] [PubMed] [Google Scholar]
- 118.Annabi N, Nichol JW, Zhong X, Ji C, Koshy S, Khademhosseini A, Dehghani F. Tissue Eng Part B Rev. 2010;16(4):371–83. doi: 10.1089/ten.teb.2009.0639. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.