

Abstract
During the 1950s, sheep ranchers in the western United States experienced episodic outbreaks of cyclopic lambs. In this highlight I describe how these mysterious incidents were traced to the grazing of Veratrum californicum wildflowers by pregnant ewes, leading to the discovery of cyclopamine (1) as a plant-derived teratogen. The precise mechanism of cyclopamine action remained enigmatic for 30 years, until this steroid alkaloid was found to be the first specific inhibitor of Hedgehog (Hh) signalling and a direct antagonist of the transmembrane receptor Smoothened (SMO). In addition to being a valuable probe of Hh pathway function, cyclopamine has been used to demonstrate the therapeutic potential of Hh pathway inhibitors. I discuss the development of SMO antagonists as anticancer therapies and emerging challenges.
1 Introduction
In Homer’s Odyssey, Odysseus and his shipmates escape the one-eyed giant Polyphemus by hiding under a flock of sheep. An equally remarkable account of cyclops, sheep, and men has emerged from the American West—a true story that begins like science fiction and culminates as medical drama. This fascinating chapter in the history of science commenced with the observation of severely malformed lambs in Idaho during the 1950s. Cyclopia was the most striking congenital defect, and the United States Department of Agriculture (USDA) surveyed the region’s soil, water, and flora in search of the cause. These studies led to the discovery of cyclopamine (1), a plant-derived steroid alkaloid with teratogenic activity.
In this highlight article, I recount how USDA scientists identified cyclopamine as the causative agent for these developmental defects. I also review the embryological, genetic, and biochemical investigations by the Beachy laboratory at the Johns Hopkins University School of Medicine that subsequently determined the mechanism of cyclopamine action. These studies revealed cyclopamine as the first small-molecule inhibitor of Hedgehog (Hh) signalling, a key regulator of embryonic patterning, and identified the transmembrane receptor Smoothened (SMO) as the direct target. Based on these findings, cyclopamine has been widely used to interrogate Hh pathway function in cells and animal models.
Cyclopamine also enabled the first preclinical studies of Hh pathway inhibition as a therapeutic strategy. The re-activation of developmental signalling pathways in children and adults can promote the onset and/or progression of several human cancers. In the case of Hh signalling, uncontrolled pathway activation can cause basal cell carcinoma (the most common human cancer), medulloblastoma (the most common pediatric brain tumor), and other malignancies. I discuss synthetic SMO antagonists have been developed for clinical use, their efficacy, and limitations.
2 The mysterious case of “monkey-faced” lambs
The appearance of lambs with craniofacial deformities must have been jarring to the sheepherders in south central and southwestern Idaho. The incidence rate varied between years and herds, with up to 25% of lambs affected in some cases.1,2 Lambs with the most severe deformities exhibited cyclopia, a domed cranium, cleft palate, shortening of the upper jaw, and malformation of the nose into a proboscis positioned above the eye (Figure 1A). The animals died shortly after birth due to compromised breathing or feeding, and post-mortem examinations revealed fused cerebral hemispheres, hydrocephalus, and absence of the olfactory bulbs and pituitary gland. The gestation period was also prolonged, allowing the foetus to grow up to four times the normal size.
Fig. 1. Discovery of cyclopamine as a plant-derived teratogen.

(A) Lamb with cyclopia and other craniofacial defects resulting from maternal ingestion of V. californicum. (B) Alpine meadow containing patches of V. californicum. Reproduced with permission (Ref. 5, copyright 2006, John Wiley and Sons). (C) Structures of teratogenic steroid alkaloids isolated from V. californicum (atomic numbering system shown).
The Basque herders tending these flocks referred to the craniofacial deformities as “chattos” disease, which translates into English as “monkey-face.” The occurrence of “monkey-faced” lambs was a significant economic hardship for the ranchers, with each afflicted animal representing a loss of about US$20 at the time (approximately US$150–US$300 per head today).3 Since the malformations were feared to be caused by genetic defects, the herders further worried that public knowledge of the cyclopic lambs would compromise the commercial value of their other livestock.
When the USDA began investigating these mysterious congenital defects in 1955, they first sought to rule in or out genetic causality. Binns and co-workers bred 48 “carrier” ewes that had given birth to malformed lambs with 12 developmentally normal rams birthed by these ewes, taking care to avoid inbreeding.1 Assuming a recessive disorder, 100% of the ewes and 50% of the rams would carry the genetic determinant. However, none of the resulting 88 lambs were malformed, and the USDA researchers concluded that the “monkey-faced” lambs did not arise from a hereditary disorder. They therefore turned their attention to possible environmental factors. The episodic nature of the lamb malformations provided some clues. First, the affected herds had grazed on ranges between 6,000 and 10,000 feet in elevation after breeding.1,2 Second, the congenital malformations typically arose within the first two to three weeks of the lambing season.4 These observations suggested that the causative agent was present briefly in alpine meadows at the start of the sheep-breeding season, typically August or early September, and/or that the foetuses were susceptible for a short period of time.
Binns, James, and their co-workers conducted a seven-year survey of mineral elements and plants in the implicated grazing ranges.4 No unusual mineral composition could be found, and preliminary feeding trials with local grasses and broadleaf plants, Corydalis caseana (Sierra fumewort), and Allium validium (wild onion) did not reproduce the developmental defects. However, concurrent studies with pregnant rats and alpine flora found that Veratrum californicum (false hellebore; Figure 1B) caused fetal resorption, and embryonic lethality were also observed when pregnant sheep were fed this plant for up to two months after breeding. Shorter periods of maternal V. californicum ingestion resulted in lambs with congenital defects, providing the first evidence that a plant-derived teratogen could be responsible for the cyclopic lambs.
Large-scale range grazing and artificial feeding experiments were then conducted to follow up this lead.4,5 The USDA transported 48 pregnant sheep to Muldoon Canyon in the Challis National Forest, a region known to have abundant V. californicum. The ewes were grazed in this range for varied durations and then transferred to alfalfa and grass pastures. Eight of the ewes gave birth to lambs with craniofacial abnormalities, and the timing of breeding and range exposure indicated that these defects required feeding in V. californicum-containing areas between days 8 and 17 of gestation. Artificial feeding of dried or green V. californicum to 148 pregnant ewes confirmed that maternal ingestion of this wildflower was sufficient to disrupt fetal development. Moreover, when the ewes were rebred and not fed V. californicum, they all gave birth to normal lambs.
Subsequent experiments determined that the teratogenic agent(s) in V. californicum are most concentrated in its roots, with lower levels in the leaves and stems favoured by grazing sheep.4 As the plants mature or when they are subjected to drought or freezing conditions, the leaves and stems lose their teratogenic activity, perhaps explaining the episodic nature of the congenital malformations. A chronologic evaluation of V. californicum feeding also revealed gestation day 14 as the critical time of exposure for cyclopian-type defects.6 This point marks the onset of neural tube formation and patterning in sheep embryos, implicating this developmental process in teratogen action.
3 Teratogenic alkaloids of Veratrum plants
While the discovery of V. californicum teratogenicity solved the mystery of the “monkey-faced” lambs, the USDA continued to search for the causative natural products. Keeler and Binns sequentially extracted dried plant material with benzene/ammonium hydroxide and ethanol, and the ethanol-extractable compounds were fractionated further by alumina chromatography.7,8 The resulting alkaloid-rich extracts were then administered to pregnant ewes on gestation day 14. Successive rounds of crystallization also yielded individual alkaloids in purified form for further biological testing.
Through this animal-based screen, the USDA identified three structurally related alkaloids with teratogenic activities: cyclopamine (1), jervine (2), and cycloposine (3) (Figure 1C).8 Jervine had been previously identified as a steroid metabolite in Veratrum genus plants;9 cyclopamine and cycloposine were originally named alkaloids V and X, respectively, as their structures were unknown at the time of their isolation. It was later determined the cyclopamine is identical to 11-deoxojervine,10 which had been independently isolated from V. album by Masamune and co-workers,11 and cycloposine was found to be the glycoside derivative of cyclopamine.12
Cyclopamine was the primary teratogenic agent in V. californicum, and elucidating the structural elements required its activity was the next challenge. Since gram quantities of each compound were required for each tested sheep, Keeler explored the alkaloids’ effects on smaller vertebrates. He first investigated whether the teratogenic activity of cyclopamine would diverge between ruminant and non-ruminant animals, due to differences in gut microbes and acidity. In non-ruminant species, orally delivered cyclopamine was converted into non-teratogenic but toxic derivatives via acid-catalyzed spirofuran opening, a reaction that could be minimized by co-administration of calcium carbonate buffer.13,14 Jervine was comparatively resistant to acid due its 11-keto group. Interestingly, differences were observed between rodent species: hamster foetuses readily developed craniofacial defects upon oral administration of cyclopamine and jervine to their mothers, rats were sensitive only to the former, and mice were generally resistant to both teratogens.15 Cyclopamine and jervine even caused head malformations in chicken embryos,16 demonstrating their teratogenic activity in non-mammalian vertebrates.
The hamster models enabled Brown and Keeler to investigate the contributions of specific structural elements to teratogen activity.17,18 For example, the 3-hydroxyl group in jervine could be acylated or oxidized (with concomitant migration of the C5–C6 olefin to make the enone) without diminishing biological activity. Hydrogenation of the C12–C13 double bond and alkylation of the amine were also tolerated. In contrast, reduction of the 11-keto group to either alcohol epimer or N-acylation markedly reduced teratogenicity.
4 Mechanism of teratogen action
While cyclopamine became canonized in textbooks as a classic teratogen, its mechanism of action remained enigmatic. Its chemical structure led to early speculation that cyclopamine and its teratogenic derivatives might dysregulate steroid hormone receptors required for vertebrate development.8 However, biochemical or genetic evidence for this model failed to materialize. A key breakthrough would come nearly 30 years later, when Chiang and Beachy at the Johns Hopkins University School of Medicine generated the Sonic Hedgehog (Shh) knockout mouse to study the roles of this secreted morphogen in vertebrate development.19 The Shh loss-of-function phenotype was remarkably similar to the “monkey-faced” lambs observed by the Basque ranchers. The mutant embryos exhibited extensive craniofacial deformities, including cyclopia and a proboscis-like, displaced nose. Ventral cell fates were absent in the brain and spinal cord, and the axial skeleton, limbs, and digits were malformed (Figure 2A–B).
Fig. 2. Cyclopamine inhibits Hh signal transduction.
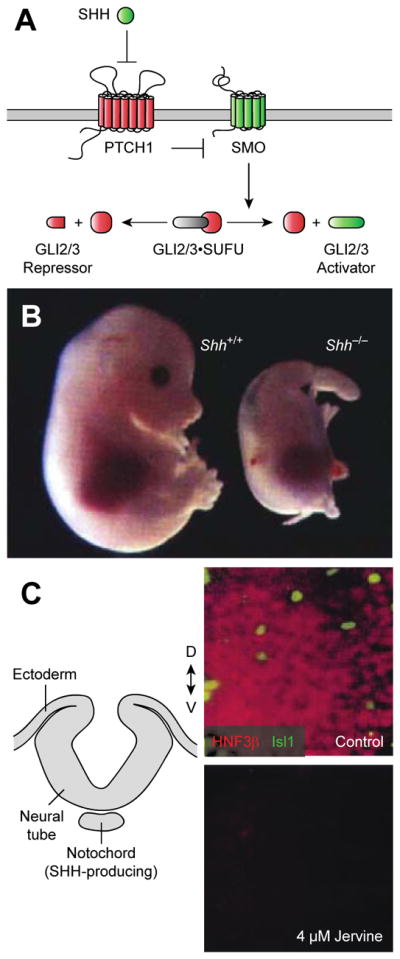
(A) The Hh signalling pathway with positive and negative regulators shown in green and red, respectively. (B) Comparison of wild type and Shh knockout mouse embryos (E11.5 stage). Adapted with permission (Ref. 19, copyright 1996, Nature Publishing Group). (C) Diagram of the chick neural tube prior to dorsal closure (transverse cross section) and micrographs of midline explants treated with either a vehicle control or jervine and then immunostained for SHH-dependent neuronal fates (floor plate cells, HNF3β; motor neurons, Isl1). Adapted with permission (Ref. 20, copyright 1998, The American Association for the Advancement of Science).
Hypothesizing that cyclopamine and the other alkaloids might act through SHH signalling inhibition, Cooper and Beachy exploited the teratogen sensitivity of chicken embryos and their amenability to tissue explant studies.20 They first demonstrated that neural plate explants containing SHH-producing cells fail to differentiate into ventral cell types (e.g., HNF3β-positive floor plate neurons) upon jervine treatment, recapitulating the Shh knockout mouse phenotype (Figure 2C). One possibility was that the steroid alkaloid interfered with SHH biogenesis, as this morphogen is converted from an inactive precursor into a cholesterol-modified N-terminal signalling domain (SHH-N).21 However, complementary studies with neural explants devoid of SHH-expressing cells revealed that jervine could also suppress ventral cell fates induced by recombinant SHH-N protein.20 Thus, the alkaloid targeted cellular responses to SHH-N rather than SHH-H production. Similar studies by Incardona and Roelink corroborated these findings.22
Determining the direct target of cyclopamine would involve the integration of genetic, synthetic organic, and biochemical approaches. SHH and other Hh ligands initiate cellular responses by binding to and inhibiting the 12-transmembrane receptor Patched1 (PTCH1), thereby alleviating its repression of the G protein-coupled receptor-like protein Smoothened (SMO) (Figure 2A).23 SMO in turn regulates the transcription factors GLI2 and GLI3, which bind to the scaffolding protein Suppressor of Fused (SUFU) and are proteolytically processed into N-terminal repressors in quiescent cells. SMO activation leads to the dissociation of SUFU/GLI complexes, enabling the full-length proteins to be converted into transcriptional activators. GLI2 and GLI3 activators then drive the expression of Hh target genes, which include the constitutively active transcription factor GLI1.
Taipale and Beachy used genetic mutations in murine Ptch1 and Smo to map the site of cyclopamine action within the Hh pathway.24 While cyclopamine and a more potent N-alkyl derivative, KAAD-cyclopamine (4) (Figure 3A), could inhibit the Hh ligand-independent pathway activity in Ptch1−/− embryonic fibroblasts, constitutively active SMO mutants exhibited varying degrees of chemoresistance. These findings suggsted that cyclopamine acts downstream of PTCH1 to regulate SMO activity state. Working with Beachy, I then demonstrated that cyclopamine inhibits Hh signalling by directly inhibiting SMO.25 This was achieved by synthesizing two N-alkyl-cyclopamine probes: a fluorescent derivative functionalized with a BODIPY FL dye (BODIPY-cyclopamine; 5) and a photoaffinity reagent containing an 125I-labeled aryl azide (PA-cyclopamine; 6) (Figure 3A). SMO-overexpressing cells could be stained with BODIPY-cyclopamine and visualized by microscopy or flow cytometry. Moreover, when live SMO-overexpressing cells were treated with PA-cyclopamine and irradiated with 254-nm light, probe-modified SMO protein could be detected by gel electrophoresis and autoradiography (Figure 3B). These receptor/ligand interactions could be competitively inhibited by KAAD-cyclopamine in both cases, confirming their specificity. Photocrosslinking studies with SMO truncation mutants also demonstrated that cyclopamine targets the heptahelical fold, rather than the extracellular cysteine-rich domain or cytoplasmic C-terminal tail. These findings have been recently corroborated by crystallographic analyses of the SMO/cyclopamine complex (Figure 3C).26
Fig. 3. Cyclopamine directly targets the transmembrane receptor SMO.

(A) Structures of chemical probes used to identify the cyclopamine target. (B) Autoradiograph of electrophoretically resolved lysates isolated from PA-cyclopamine-treated and UV-irradiated cells. Cells overexpressing wild type but not mutant, cyclopamine-resistant SMO exhibit PA-cyclopamine crosslinking that can be competitively inhibited by KAAD-cyclopamine. Adapted from Ref. 25 as allowed through the Creative Commons License. (C) Crystal structure of the SMO/cyclopamine complex (PDB ID: 4O9R).
It is noteworthy that this combination of genetics-enabled hypothesis building and chemical synthesis-enabled hypothesis testing was required to discover the SMO/cyclopamine connection. The Shh knockout mouse phenotype provided the initial clue, and the chick neural plate explant studies validated Hh signalling as plausible teratogen target. The subsequent mapping of cyclopamine action relative to PTCH1 and SMO revealed key functional relationships but not specific biochemical interactions. Conversely, unbiased cell-based experiments with BODIPY-cyclopamine and PA-cyclopamine were unsuccessful; endogenous SMO levels are too low to be detected with these chemical probes, necessitating directed studies with SMO-overexpressing cells. Elucidating the mechanism of cyclopamine action therefore depended upon both biological intuition and chemical innovation.
5 Targeted anticancer therapies
As the first small molecule known to specifically inhibit the Hh pathway, cyclopamine has been widely used to study Hh signalling mechanisms in cell-based models and Hh ligand-dependent patterning in vertebrate organisms. Cyclopamine has also played an important role in the development of SMO antagonists as therapeutic agents, as Hh pathway activation not only controls tissue patterning but also contributes to oncogenesis.
The connection between Hh pathway dysregulation and cancer was first uncovered in the late 1990s, when Gorlin syndrome, a rare genetic disorder, was linked to a microdeletion in the PTCH1 locus.27,28 Individuals with Gorlin syndrome are predisposed to basal cell carcinoma, medulloblastoma, rhabdomyosarcoma, and fibroma, and they often manifest facial dysmorphisms and skeletal abnormalities. Activating mutations in SMO and inactivating mutations in SUFU have subsequently been associated with basal cell carcinoma,29,30 and loss of SUFU function can cause medulloblastoma and meningioma (the most common brain cancer).31,32 SMO antagonists would therefore be expected to suppress Hh pathway-dependent tumor growth in some of these cases, and cyclopamine was used by Berman and Beachy to demonstrate the therapeutic potential of this approach.33 In this study, cyclopamine induced significant tumor regression in a murine allograft model of Ptch1+/−;p53−/− medulloblastoma.
While cyclopamine has been a valuable tool compound for basic research and preclinical models, its potential as a therapeutic agent is limited. In addition to its acid lability, cyclopamine can exert off-target effects on cell growth at concentrations that are only moderately higher than the doses required for Hh pathway blockade.34 Due to these constraints, academic and industrial scientists have sought new SMO inhibitors through high-throughput cell-based screens and medicinal chemistry. For example, Beachy and I identified four structurally distinct SMO antagonists (e.g., SANT-1 and SANT-2; 7 and 8),35 and several pharmaceutical companies have developed SMO-targeting drugs, including a cyclopamine derivative with improved stability and potency (saridegib; 9) (Figure 4A).36 Crystal structures of various SMO/ligand complexes have revealed a narrow hydrophobic cavity extending from the extracellular loops into the heptahelical bundle.26,37–39 These studies suggest that SMO ligands can be grouped into at least three general classes: compounds that primarily bind to the extracellular entrance of the cavity (e.g., cyclopamine; see Figure 3C), those that penetrate deeply into the transmembrane cavity (e.g., SANT-1), and others that engage both sites (e.g., MRT-92; 10, Figure 4A).
Fig. 4. Synthetic SMO antagonists.
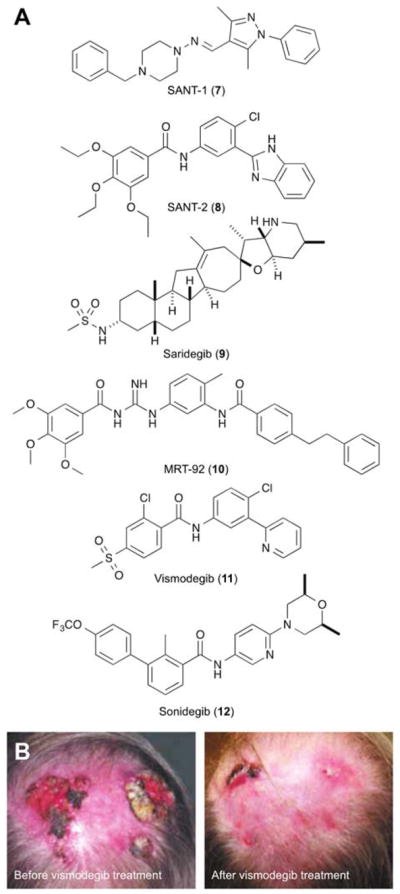
(A) Structures of selected SMO inhibitors. (B) Regression of locally advanced basal cell carcinoma after two months of vismodegib treatment. Adapted with permission (Ref. 40, copyright 2009, Massachusetts Medical Society).
To date, two SMO antagonists have been approved by the United States Food and Drug Administration for clinical use: vismodegib (Genentech; 11) and sonidegib (Novartis; 12) (Figure 4A). Both drugs can be highly effective treatments for advanced basal cell carcinomas that are not amenable to surgical removal (Figure 4B),40,41 and vismodegib was also reported to induce rapid but transient regression of metastatic medulloblastoma.42 However, these successes have been coupled with emerging challenges. As in the medulloblastoma case, chemoresistance occurs with a frequency that correlates with tumor grade.43,44 SMO mutations contribute to many of these relapses; however, genomic alterations involving downstream signalling components such as SUFU and GLI2 have been observed as well. In addition, systemic SMO blockade can cause on-target side effects, including hair loss, taste sensation deficits, and muscle cramps,45 and vismodegib (and cyclopamine) have been reported to activate non-canonical SMO functions.46 Overcoming these limitations through next-generation SMO inhibitors, alternative delivery methods, and/or new Hh pathway-targeting strategies will be necessary to complete the clinical vision initiated by cyclopamine.
6 Conclusions
The story of cyclopamine provides several take-home lessons. From a chemical perspective, it illustrates how challenging it can be to determine the mechanism(s) of biologically active small molecules. In this case, 30 years of speculation was finally resolved by advances in another research field, and both genetic and chemical approaches were required to establish SMO as the direct cyclopamine target. Neither method alone would have sufficed. As the search for new Hh pathway antagonists continues, the conceptual and technical insights gained through cyclopamine should help translate future molecular discoveries into mechanistic understanding.
From a biological perspective, cyclopamine highlights the intimate link between ontogeny and oncogenesis. Many other signalling pathways that regulate embryonic patterning also contribute to cancer later on in life. For example, nearly all colorectal cancers are caused by uncontrolled activation of the Wnt pathway,47 and mutations that activate Notch signalling are a major cause of T-cell acute lymphoblastic leukemia.48 Finally, cyclopamine exemplifies the unpredictable yet intrinsic role of basic science in medicine. If not for the V. californicum extracts characterized by the USDA in the 1950s, vismodegib and sonidegib might not have been developed fifty years later. Few could have envisioned that a molecule associated with such monstrosities in the mountains would inspire new anticancer therapies in the clinic.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (R01 GM113100).
References
- 1.Binns W, Thacker EJ, James LF, Huffman WT. J Am Vet Med Assoc. 1959;134:180–183. [PubMed] [Google Scholar]
- 2.Binns W, James LF, Shupe JL, Thacker EJ. Arch Environ Health. 1962;5:106–108. doi: 10.1080/00039896.1962.10663251. [DOI] [PubMed] [Google Scholar]
- 3.Babbott FL, Jr, Binns W, Ingalls TH. Arch Environ Health. 1962;5:109–113. doi: 10.1080/00039896.1962.10663252. [DOI] [PubMed] [Google Scholar]
- 4.Binns W, James LF, Shupe JL, Everett G. Am J Vet Res. 1963;24:1164–1175. [PubMed] [Google Scholar]
- 5.Binns W, James LF, Shupe JL. Ann N Y Acad Sci. 1964;111:571–576. doi: 10.1111/j.1749-6632.1964.tb53124.x. [DOI] [PubMed] [Google Scholar]
- 6.Binns W, Shupe JL, Keeler RF, James LF. J Am Vet Med Assoc. 1965;147:839–842. [PubMed] [Google Scholar]
- 7.Keeler RF, Binns W. Can J Biochem. 1966;44:819–828. doi: 10.1139/o66-100. [DOI] [PubMed] [Google Scholar]
- 8.Keeler RF, Binns W. Can J Biochem. 1966;44:829–838. doi: 10.1139/o66-101. [DOI] [PubMed] [Google Scholar]
- 9.Jacobs WA, Craig LC. J Biol Chem. 1943;148:51–55. [Google Scholar]
- 10.Keeler RF. Phytochemistry. 1969;8:223–225. [Google Scholar]
- 11.Masamune T, Mori Y, Takasugi M, Murai A, Ohuchi S, Sato N, Katsui N. Bull Chem Soc Jpn. 1965;38:1374–1378. doi: 10.1246/bcsj.38.1374. [DOI] [PubMed] [Google Scholar]
- 12.Keeler RF. Steroids. 1969;13:579–588. doi: 10.1016/s0039-128x(69)80012-7. [DOI] [PubMed] [Google Scholar]
- 13.Keeler RF. Teratology. 1970;3:175–180. doi: 10.1002/tera.1420030210. [DOI] [PubMed] [Google Scholar]
- 14.Wilson SR, Strand MF, Krapp A, Rise F, Petersen D, Krauss S. J Pharm Biomed Anal. 2010;52:707–713. doi: 10.1016/j.jpba.2010.02.017. [DOI] [PubMed] [Google Scholar]
- 15.Keeler RF. Proc Soc Exp Biol Med. 1975;149:302–306. doi: 10.3181/00379727-149-38794. [DOI] [PubMed] [Google Scholar]
- 16.Bryden MM, Perry C, Keeler RF. Teratology. 1973;8:19–25. doi: 10.1002/tera.1420080104. [DOI] [PubMed] [Google Scholar]
- 17.Brown D, Keeler RF. J Agric Food Chem. 1978;26:561–563. doi: 10.1021/jf60217a063. [DOI] [PubMed] [Google Scholar]
- 18.Brown D, Keeler RF. J Agric Food Chem. 1978;26:564–566. doi: 10.1021/jf60217a069. [DOI] [PubMed] [Google Scholar]
- 19.Chiang C, Litingtung Y, Lee E, Young KE, Corden JL, Westphal H, Beachy PA. Nature. 1996;383:407–413. doi: 10.1038/383407a0. [DOI] [PubMed] [Google Scholar]
- 20.Cooper MK, Porter JA, Young KE, Beachy PA. Science. 1998;280:1603–1607. doi: 10.1126/science.280.5369.1603. [DOI] [PubMed] [Google Scholar]
- 21.Porter JA, Young KE, Beachy PA. Science. 1996;274:255–259. doi: 10.1126/science.274.5285.255. [DOI] [PubMed] [Google Scholar]
- 22.Incardona JP, Gaffield W, Kapur RP, Roelink H. Development. 1998;125:3553–3562. doi: 10.1242/dev.125.18.3553. [DOI] [PubMed] [Google Scholar]
- 23.Briscoe J, Therond PP. Nat Rev Mol Cell Biol. 2013;14:416–429. doi: 10.1038/nrm3598. [DOI] [PubMed] [Google Scholar]
- 24.Taipale J, Chen JK, Cooper MK, Wang B, Mann RK, Milenkovic L, Scott MP, Beachy PA. Nature. 2000;406:1005–1009. doi: 10.1038/35023008. [DOI] [PubMed] [Google Scholar]
- 25.Chen JK, Taipale J, Cooper MK, Beachy PA. Genes Dev. 2002;16:2743–2748. doi: 10.1101/gad.1025302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weierstall U, James D, Wang C, White TA, Wang D, Liu W, Spence JC, Bruce Doak R, Nelson G, Fromme P, Fromme R, Grotjohann I, Kupitz C, Zatsepin NA, Liu H, Basu S, Wacker D, Han GW, Katritch V, Boutet S, Messerschmidt M, Williams GJ, Koglin JE, Marvin Seibert M, Klinker M, Gati C, Shoeman RL, Barty A, Chapman HN, Kirian RA, Beyerlein KR, Stevens RC, Li D, Shah ST, Howe N, Caffrey M, Cherezov V. Nat Commun. 2014;5:3309. doi: 10.1038/ncomms4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Unden AB, Holmberg E, Lundh-Rozell B, Stahle-Backdahl M, Zaphiropoulos PG, Toftgard R, Vorechovsky I. Cancer Res. 1996;56:4562–4565. [PubMed] [Google Scholar]
- 28.Chidambaram A, Goldstein AM, Gailani MR, Gerrard B, Bale SJ, DiGiovanna JJ, Bale AE, Dean M. Cancer Res. 1996;56:4599–4601. [PubMed] [Google Scholar]
- 29.Xie J, Murone M, Luoh SM, Ryan A, Gu Q, Zhang C, Bonifas JM, Lam CW, Hynes M, Goddard A, Rosenthal A, Epstein EH, Jr, de Sauvage FJ. Nature. 1998;391:90–92. doi: 10.1038/34201. [DOI] [PubMed] [Google Scholar]
- 30.Pastorino L, Ghiorzo P, Nasti S, Battistuzzi L, Cusano R, Marzocchi C, Garre ML, Clementi M, Scarra GB. Am J Med Genet A. 2009;149A:1539–1543. doi: 10.1002/ajmg.a.32944. [DOI] [PubMed] [Google Scholar]
- 31.Taylor MD, Liu L, Raffel C, Hui CC, Mainprize TG, Zhang X, Agatep R, Chiappa S, Gao L, Lowrance A, Hao A, Goldstein AM, Stavrou T, Scherer SW, Dura WT, Wainwright B, Squire JA, Rutka JT, Hogg D. Nat Genet. 2002;31:306–310. doi: 10.1038/ng916. [DOI] [PubMed] [Google Scholar]
- 32.Aavikko M, Li SP, Saarinen S, Alhopuro P, Kaasinen E, Morgunova E, Li Y, Vesanen K, Smith MJ, Evans DG, Poyhonen M, Kiuru A, Auvinen A, Aaltonen LA, Taipale J, Vahteristo P. Am J Hum Genet. 2012;91:520–526. doi: 10.1016/j.ajhg.2012.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berman DM, Karhadkar SS, Hallahan AR, Pritchard JI, Eberhart CG, Watkins DN, Chen JK, Cooper MK, Taipale J, Olson JM, Beachy PA. Science. 2002;297:1559–1561. doi: 10.1126/science.1073733. [DOI] [PubMed] [Google Scholar]
- 34.Yauch RL, Gould SE, Scales SJ, Tang T, Tian H, Ahn CP, Marshall D, Fu L, Januario T, Kallop D, Nannini-Pepe M, Kotkow K, Marsters JC, Rubin LL, de Sauvage FJ. Nature. 2008;455:406–410. doi: 10.1038/nature07275. [DOI] [PubMed] [Google Scholar]
- 35.Chen JK, Taipale J, Young KE, Maiti T, Beachy PA. Proc Natl Acad Sci U S A. 2002;99:14071–14076. doi: 10.1073/pnas.182542899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tremblay MR, Lescarbeau A, Grogan MJ, Tan E, Lin G, Austad BC, Yu LC, Behnke ML, Nair SJ, Hagel M, White K, Conley J, Manna JD, Alvarez-Diez TM, Hoyt J, Woodward CN, Sydor JR, Pink M, MacDougall J, Campbell MJ, Cushing J, Ferguson J, Curtis MS, McGovern K, Read MA, Palombella VJ, Adams J, Castro AC. J Med Chem. 2009;52:4400–4418. doi: 10.1021/jm900305z. [DOI] [PubMed] [Google Scholar]
- 37.Wang C, Wu H, Katritch V, Han GW, Huang XP, Liu W, Siu FY, Roth BL, Cherezov V, Stevens RC. Nature. 2013;497:338–343. doi: 10.1038/nature12167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang C, Wu H, Evron T, Vardy E, Han GW, Huang XP, Hufeisen SJ, Mangano TJ, Urban DJ, Katritch V, Cherezov V, Caron MG, Roth BL, Stevens RC. Nat Commun. 2014;5:4355. doi: 10.1038/ncomms5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hoch L, Faure H, Roudaut H, Schoenfelder A, Mann A, Girard N, Bihannic L, Ayrault O, Petricci E, Taddei M, Rognan D, Ruat M. FASEB J. 2015;29:1817–1829. doi: 10.1096/fj.14-267849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Von Hoff DD, LoRusso PM, Rudin CM, Reddy JC, Yauch RL, Tibes R, Weiss GJ, Borad MJ, Hann CL, Brahmer JR, Mackey HM, Lum BL, Darbonne WC, Marsters JC, Jr, de Sauvage FJ, Low JA. N Engl J Med. 2009;361:1164–1172. doi: 10.1056/NEJMoa0905360. [DOI] [PubMed] [Google Scholar]
- 41.Skvara H, Kalthoff F, Meingassner JG, Wolff-Winiski B, Aschauer H, Kelleher JF, Wu X, Pan S, Mickel L, Schuster C, Stary G, Jalili A, David OJ, Emotte C, Antunes AM, Rose K, Decker J, Carlson I, Gardner H, Stuetz A, Bertolino AP, Stingl G, De Rie MA. J Invest Dermatol. 2011;131:1735–1744. doi: 10.1038/jid.2011.48. [DOI] [PubMed] [Google Scholar]
- 42.Rudin CM, Hann CL, Laterra J, Yauch RL, Callahan CA, Fu L, Holcomb T, Stinson J, Gould SE, Coleman B, LoRusso PM, Von Hoff DD, de Sauvage FJ, Low JA. N Engl J Med. 2009;361:1173–1178. doi: 10.1056/NEJMoa0902903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sharpe HJ, Pau G, Dijkgraaf GJ, Basset-Seguin N, Modrusan Z, Januario T, Tsui V, Durham AB, Dlugosz AA, Haverty PM, Bourgon R, Tang JY, Sarin KY, Dirix L, Fisher DC, Rudin CM, Sofen H, Migden MR, Yauch RL, de Sauvage FJ. Cancer Cell. 2015;27:327–341. doi: 10.1016/j.ccell.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Atwood SX, Sarin KY, Whitson RJ, Li JR, Kim G, Rezaee M, Ally MS, Kim J, Yao C, Chang AL, Oro AE, Tang JY. Cancer Cell. 2015;27:342–353. doi: 10.1016/j.ccell.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sekulic A, Migden MR, Oro AE, Dirix L, Lewis KD, Hainsworth JD, Solomon JA, Yoo S, Arron ST, Friedlander PA, Marmur E, Rudin CM, Chang AL, Low JA, Mackey HM, Yauch RL, Graham RA, Reddy JC, Hauschild A. N Engl J Med. 2012;366:2171–2179. doi: 10.1056/NEJMoa1113713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Teperino R, Amann S, Bayer M, McGee SL, Loipetzberger A, Connor T, Jaeger C, Kammerer B, Winter L, Wiche G, Dalgaard K, Selvaraj M, Gaster M, Lee-Young RS, Febbraio MA, Knauf C, Cani PD, Aberger F, Penninger JM, Pospisilik JA, Esterbauer H. Cell. 2012;151:414–426. doi: 10.1016/j.cell.2012.09.021. [DOI] [PubMed] [Google Scholar]
- 47.Najdi R, Holcombe RF, Waterman ML. J Carcinog. 2011;10:5. doi: 10.4103/1477-3163.78111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Van Vlierberghe P, Ferrando A. J Clin Invest. 2012;122:3398–3406. doi: 10.1172/JCI61269. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.