

Abstract Abstract
To determine whether low ventricular filling pressures are a clinically relevant etiology of unexplained dyspnea on exertion, a database of 619 consecutive, clinically indicated invasive cardiopulmonary exercise tests (iCPETs) was reviewed to identify patients with low maximum aerobic capacity (V̇o2max) due to inadequate peak cardiac output (Qtmax) with normal biventricular ejection fractions and without pulmonary hypertension (impaired: n = 49, V̇o2max = 53% predicted [interquartile range (IQR): 47%–64%], Qtmax = 72% predicted [62%–76%]). These were compared to patients with a normal exercise response (normal: n = 28, V̇o2max = 86% predicted [84%–97%], Qtmax = 108% predicted [97%–115%]). Before exercise, all patients received up to 2 L of intravenous normal saline to target an upright pulmonary capillary wedge pressure (PCWP) of ≥5 mmHg. Despite this treatment, biventricular filling pressures at peak exercise were lower in the impaired group than in the normal group (right atrial pressure [RAP]: 6 [IQR: 5–8] vs. 9 [7–10] mmHg, P = 0.004; PCWP: 12 [10–16] vs. 17 [14–19] mmHg, P < 0.001), associated with decreased stroke volume (SV) augmentation with exercise (+13 ± 10 [standard deviation (SD)] vs. +18 ± 10 mL/m2, P = 0.014). A review of hemodynamic data from 23 patients with low RAP on an initial iCPET who underwent a second iCPET after saline infusion (2.0 ± 0.5 L) demonstrated that 16 of 23 patients responded with increases in Qtmax ([+24% predicted [IQR: 14%–34%]), V̇o2max (+10% predicted [7%–12%]), and maximum SV (+26% ± 17% [SD]). These data suggest that inadequate ventricular filling related to low venous pressure is a clinically relevant cause of exercise intolerance.
Keywords: exercise, cardiac output, hemodynamics, preload, postural orthostatic tachycardia syndrome
Dyspnea on exertion is a common presenting symptom with a broad differential diagnosis. Often the etiology remains unclear despite a thorough clinical and laboratory investigation.1,2 Cardiopulmonary exercise testing (CPET) may assist in the diagnostic evaluation by defining the degree of impairment in maximum aerobic capacity (V̇o2max), identifying the limiting organ system (e.g., heart vs. lung), and providing clues as to more specific pathophysiology. When CPET is coupled with invasive hemodynamic monitoring using radial and pulmonary arterial catheters (i.e., invasive CPET [iCPET]), the presence of peripheral and central cardiovascular abnormalities can be better characterized through direct measurements of systemic and pulmonary vascular pressures and systemic and mixed venous oxygen content as well as precise estimation of cardiac output (Qt).3 For example, these measurements have been used to characterize the exercise-induced increases in ventricular filling pressure and pulmonary arterial pressure in patients with heart failure4-8 and pulmonary arterial hypertension,9 respectively. Despite a detailed hemodynamic and metabolic evaluation, nearly 10% of symptomatic patients studied with iCPET in our laboratory had low V̇o2max and low maximum Qt (Qtmax) without a clearly identified cause.
During the normal exercise response, sympathetic stimulation and vagal withdrawal increase heart rate (HR), contractility, and mean systemic pressure. Increased respiratory efforts and vigorous limb skeletal muscle contractions also enhance venous return to the heart. Together, these responses increase stroke volume (SV) through the Frank-Starling mechanism. In this study, we tested the hypothesis that failure of these mechanisms to increase cardiac preload during exercise, as evidenced by persistently low ventricular filling pressures, may be the primary limitation of Qtmax in an undiagnosed population of patients with unexplained exercise intolerance.
Methods
Patients
For the single-test cohort, results from 619 consecutive, clinically indicated iCPET performed over a 9-year period at Massachusetts General Hospital were analyzed. For the sequential-testing cohort, results of 23 consecutive clinically indicated invasive cardiopulmonary exercise tests (iCPETs) performed between August 2012 and May 2013 at Brigham and Women’s Hospital were reviewed. The Partners Human Research Committee (2010P002423, 2008P000687) approved this retrospective chart review and waived the requirement for informed consent.
iCPET protocol
Before iCPET, radial and pulmonary artery catheters were placed by standard techniques, the latter by the internal jugular approach in the cardiac catheterization laboratory under fluoroscopic guidance. All patients completed a single bout of incremental exercise to exhaustion on an upright cycle ergometer. Two minutes of rest were followed by 3 minutes of unloaded cycling. Work was then continuously increased by 5–25 W/min on the basis of the patient’s history of exercise tolerance. Ventilation, O2, and CO2 were measured breath-by-breath with a commercially available metabolic cart (MGC Diagnostics, St. Paul, MN). Systemic mean arterial, right atrial, and mean pulmonary arterial pressures (MAP, RAP, and mPAP, respectively) were measured continuously. End-expiratory pulmonary capillary wedge pressure (PCWP) was obtained at rest and during each minute of exercise. Blood samples were simultaneously drawn from the radial line and the distal port of the pulmonary arterial catheter at rest and during the last 15 seconds of each minute of exercise and analyzed at 37°C for partial pressures of O2 and CO2, pH, hemoglobin concentration ([Hb]), and O2 saturation by co-oximetry.
For the single-test cohort, if the resting upright PCWP immediately before exercise was <5 mmHg, patients received 0.5-L intravenous normal saline boluses (repeated up to 4 times, to a maximum of 2.0 L) to target a resting PCWP of ≥5 mmHg. Right and left ventricular ejection fractions (RVEF and LVEF, respectively) and left ventricular end-diastolic volume (LVEDV) were measured upright at rest immediately before exercise and near peak exercise by first-pass radionuclide ventriculographic scanning (FPRVS; SIM-400, Philips Medical Systems, Andover, MA), as previously described and validated against simultaneous Fick SV measurements.10
For the sequential-testing cohort, an initial iCPET was performed as described above, but without saline administration before exercise. If, in the judgment of the supervising physician, hemodynamic measurements from the initial iCPET demonstrated low ventricular filling pressures (e.g., resting or peak RAP was <5 mmHg or RAP increased by <3 mmHg with exercise), intravenous normal saline was administered to target a resting upright RAP of >5 mmHg, and a second iCPET was performed. FPRVS measurements were not performed on this cohort.
Data analysis
Predicted values for V̇o2max incorporated age, sex, and height.11 The Fick principle was used to calculate Qt, where Qt = V̇o2/(Ca-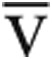 O2) and Ca-
O2) and Ca-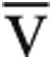 O2 is the difference between measured arterial and mixed venous oxygen content. Predicted Qtmax was calculated from the predicted V̇o2max, assuming a normal peak exercise Ca-
O2 is the difference between measured arterial and mixed venous oxygen content. Predicted Qtmax was calculated from the predicted V̇o2max, assuming a normal peak exercise Ca-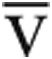 O2 of 140 mL/L.11 Right ventricular end-diastolic volume (RVEDV) was calculated from the Fick SV divided by the RVEF.
O2 of 140 mL/L.11 Right ventricular end-diastolic volume (RVEDV) was calculated from the Fick SV divided by the RVEF.
For the single-test cohort, the results of 619 iCPETs were reviewed to identify a population with an unexplained abnormal cardiac limit to exercise and a control population with a normal hemodynamic exercise response. This was accomplished by excluding tests in which data were incomplete because of technical difficulties with either hemodynamic or FPRVS measurements and tests that met any of the following criteria: (1) submaximum effort (maximum HR < 80% predicted [pred.] and peak respiratory exchange ratio [RER] < 1.05), (2) primary pulmonary mechanical limit (minute ventilation/maximum voluntary ventilation > 0.7 at the ventilatory threshold),12 (3) left or right ventricular systolic dysfunction (LVEF < 0.55 or RVEF < 0.45 at rest or maximum exercise, as measured by FPRVS), (4) pulmonary venous hypertension (PCWPmax > 20 mmHg), (5) pulmonary arterial hypertension (mPAPmax > 30 mmHg and pulmonary vascular resistance at maximum exercise > 80 dyne s/cm5),9 and (6) peripheral extraction limitation (V̇o2max < 80% pred. and Qtmax ≥ 80% pred.). For the remaining tests, patients were divided into the groups “normal” (V̇o2max ≥ 80% pred.) and “impaired” (V̇o2max < 80% pred.; i.e., patients with low V̇o2max due to low Qtmax with normal systolic function and without pulmonary hypertension; Fig. 1).
Figure 1.
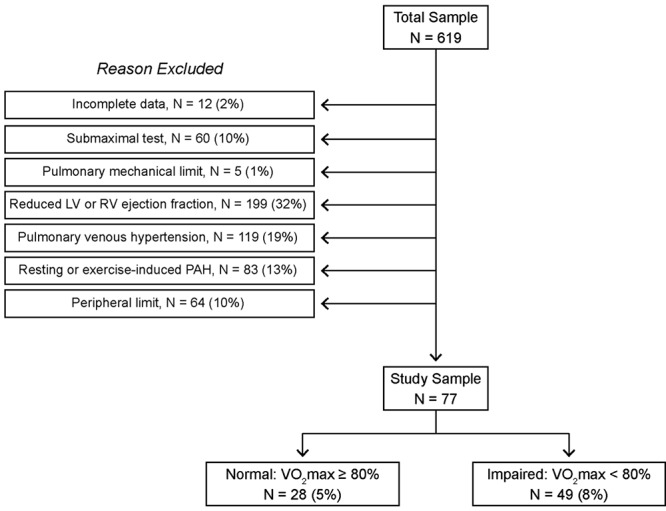
Identifying patients with an unexplained cardiac limitation to exercise and symptomatic normal controls from a historical database of invasive cardiopulmonary exercise testing parameters based on the exclusion criteria defined in “Methods.” LV: left ventricular; RV: right ventricular; PAH: pulmonary arterial hypertension; V̇o2 max: maximum aerobic capacity.
All statistical analyses were performed with Prism 6 (GraphPad Software, La Jolla, CA). The D’Agostino and Pearson omnibus normality test was used. All continuous, normally distributed data are reported as mean ± standard deviation. Nonnormally distributed data are reported as median (interquartile range). Comparisons between groups in the single-test cohort were made using the Student t test for normally distributed data, while the Mann-Whitney test was used for nonnormally distributed data. Comparisons between hemodynamic variables in the sequential-testing cohort were made with the Student t test for paired observations, as these data were normally distributed. The Fisher exact test was used for categorical variables. Correlations are presented as the Spearman ρ. A 2-tailed probability (P) value of <0.05 was considered significant.
Results
Single-test cohort patient characteristics
The single-test cohort was composed of 28 normal and 49 impaired patients and consisted primarily of women aged 35–60 years. There were more women in the impaired group (61% vs. 84%, P = 0.03), but the normal and impaired groups did not differ in terms of age, race, medical comorbidities, or laboratory test results. Likely as a result of the sex difference, body mass index (Table 1) and body surface area (BSA) were lower in the impaired group (BSA: 1.8 ± 0.2 vs. 1.9 ± 0.2 m2, P = 0.003); to address this, results are indexed to BSA. Cardiovascular medication use did not differ between the two groups (Table 1).
Table 1.
Demographic variables and baseline characteristics
| Single-test cohort | |||
|---|---|---|---|
| Characteristic | Normal (N = 28) | Impaired (N = 49) | Sequential-testing cohort (N = 21) |
| Age, median (IQR), years | 45 (40–60) | 46 (31–63) | 44 (25–58) |
| Male sex, no. (%) of patients | 11 (39.3) | 8 (16.3) | 6 (28.6) |
| BMI, median (IQR), kg/m2 | 27.3 (25.2–29.9) | 24.9 (21.1–28.3) | 23.0 (20.9–24.6) |
| Medications, no. (%) of patients | |||
| Diuretic | 3 (10.7) | 6 (12.2) | 2 (0.7) |
| BB | 4 (14.3) | 8 (16.3) | 2 (0.7) |
| CCB | 5 (17.9) | 12 (24.5) | 3 (14.3) |
| ACE inhibitor/ARB | 4 (14.3) | 8 (16.3) | 2 (0.7) |
| [Hb], median (IQR), g/dL | 12.8 (12.1–13.7) | 11.9 (11.1–13.5) | 13.9 (13.6–14.5) |
| V̇o2max, mean ± SD, mL/kg/min | 24.4 ± 9.4 | 16.6 ± 5.0 | 23.2 ± 7.2a |
| V̇o2max, median (IQR), % pred. | 86 (84–97) | 53 (47–64) | 80 (70–102)a |
| Preexercise fluid, median (IQR), L | 0 (0–0.75) | 0.1 (0–0.9) | 2.0 (1.9–2.3)b |
BMI: body mass index; BB: β-adrenergic receptor antagonist; CCB: calcium channel blocker; ACE: angiotensin-converting enzyme; ARB: angiotensin receptor blocker; [Hb]: hemoglobin concentration; IQR: interquartile range; V̇o2max: maximum aerobic capacity; SD: standard deviation; % pred.: percent of predicted value.
V̇o2max from the first invasive cardiopulmonary exercise test (iCPET).
Volume of normal saline infused between iCPETs.
iCPET results: normal versus impaired
In the upright position at rest, impaired and normal patients had similar cardiac indices (CIs), although impaired patients had a decreased resting SV index that was compensated for by a slightly increased resting HR (Table 2). Resting MAP was decreased in impaired compared to normal patients (Table 2); however, RAP, mPAP, and PCWP were similar between the two groups after receipt of a similar amount of intravenous normal saline before the iCPET (Tables 1, 2; Fig. 2).
Table 2.
Single-test cohort iCPET hemodynamic measurements
| Normal | Impaired | P | ||||
|---|---|---|---|---|---|---|
| Variable | Rest | Peak | Rest | Peak | Rest | Peak |
| Cardiac index, L/min/m2 | 2.7 (2.2–3.5) | 8.6 (6.9–10.2) | 2.6 (1.9–3.2) | 6.3 (5.2–7.1) | 0.33 | <0.01 |
| Cardiac output, % predicted | 108 (97–115) | 72 (62–76) | <0.01 | |||
| Heart rate, bpm | 78 ± 13 | 153 ± 20 | 85 ± 17 | 141 ± 28 | 0.07 | 0.03 |
| Heart rate, % predicted | 92 ± 10 | 84 ± 13 | <0.01 | |||
| SV index, mL/m2 | 37 ± 12 | 56 ± 10 | 32 ± 9 | 44 ± 9 | 0.03 | <0.01 |
| MAP, mmHg | 101 ± 14 | 124 ± 16 | 93 ± 13 | 112 ± 16 | 0.02 | <0.01 |
| SVR index, dyn·s/cm5/m2 | 2,666 (2,220–3,691) | 1,067 (868–1,322) | 2,832 (2,190–3,801) | 1,369 (1,148–1,698) | 0.90 | <0.01 |
| Mean PA pressure, mmHg | 15 (12–17) | 29 (26–31) | 14 (12–16) | 24 (22–27) | 0.76 | <0.01 |
| PVR index, dyn·s/cm5/m2 | 257 (198–350) | 130 (99–155) | 285 (182–422) | 148 (105–198) | 0.42 | 0.07 |
Ca- O2, mL O2/dL O2, mL O2/dL |
5.5 (5.0–6.4) | 12.5 (11.1–13.0) | 5.7 (4.7–6.5) | 10.1 (9.0–11.4) | 0.91 | <0.01 |
| RVEDV index, mL/m2 | 70 (61–85) | 79 (71–97) | 70 (62–82) | 81 (69–95) | 0.48 | 0.63 |
| LVEDV index, mL/m2 | 58 (51–69) | 64 (59–81) | 56 (50–66) | 64 (53–80) | 0.43 | 0.55 |
“Peak” indicates the value at maximum exercise, while “rest” indicates the upright, resting value before exercise. Nonnormally distributed data are reported as median (interquartile range); continuous, normally distributed data are reported as mean ± standard deviation. iCPET: invasive cardiopulmonary exercise testing; SV: stroke volume; MAP: mean arterial pressure; SVR: systemic vascular resistance; PA: pulmonary artery; PVR: pulmonary vascular resistance; Ca- O2: difference between arterial and mixed venous oxygen content; RVEDV: right ventricular end diastolic volume; LVEDV: left ventricular end diastolic volume.
O2: difference between arterial and mixed venous oxygen content; RVEDV: right ventricular end diastolic volume; LVEDV: left ventricular end diastolic volume.
Figure 2.
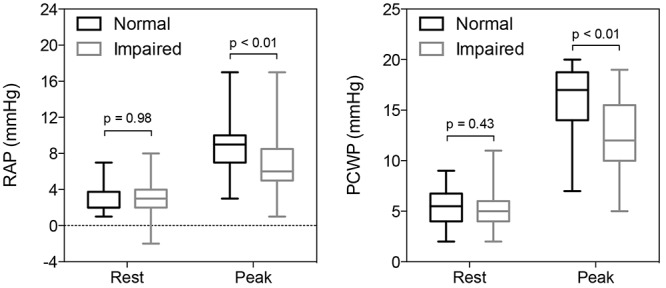
Right and left ventricular filling pressures at rest and maximum exercise in normal and impaired patients. Box and whiskers indicate interquartile range and the minimum/maximum values, respectively. PCWP: pulmonary capillary wedge pressure; RAP: right atrial pressure.
With exercise, impaired patients had blunted increases in RAP and PCWP (Fig. 2). This was associated with a reduced V̇o2max due to low peak CI (Tables 1, 2). The latter was primarily a consequence of decreased SV augmentation from rest to peak exercise (ΔSV index: +13 ± 10 vs. +18 ± 10 mL/m2, impaired vs. normal, respectively, P = 0.01). A decreased HR response in impaired patients made a small contribution to the reduction in maximum CI, as impaired patients achieved 92% of the peak HR of normal patients but only 79% of their peak SV index.
Of the determinants of SV, impaired patients were found to have decreased absolute biventricular filling pressures at maximum exercise (Fig. 2) as well as less augmentation of RAP and PCWP with exercise (ΔRAP: 4 [2–5] vs. 6.0 [4–7] mmHg, P = 0.007; ΔPCWP: 7 ± 3 vs. 10 ± 4 mmHg, P < 0.001, impaired vs. normal, respectively). By FPRVS, normal and impaired patients had similar RVEDV and LVEDV indices at rest and peak exercise (Table 2).
A pooled analysis of all patients in the single-test cohort demonstrated that peak RAP and PCWP were significantly correlated with V̇o2max. Similarly, peak PCWP correlated with Qtmax (% pred.) and SV index (Fig. 3). Together, these data suggest that the deficiency in peak Qt observed in impaired patients was due to low ventricular filling pressures, a marker of inadequate preload.
Figure 3.
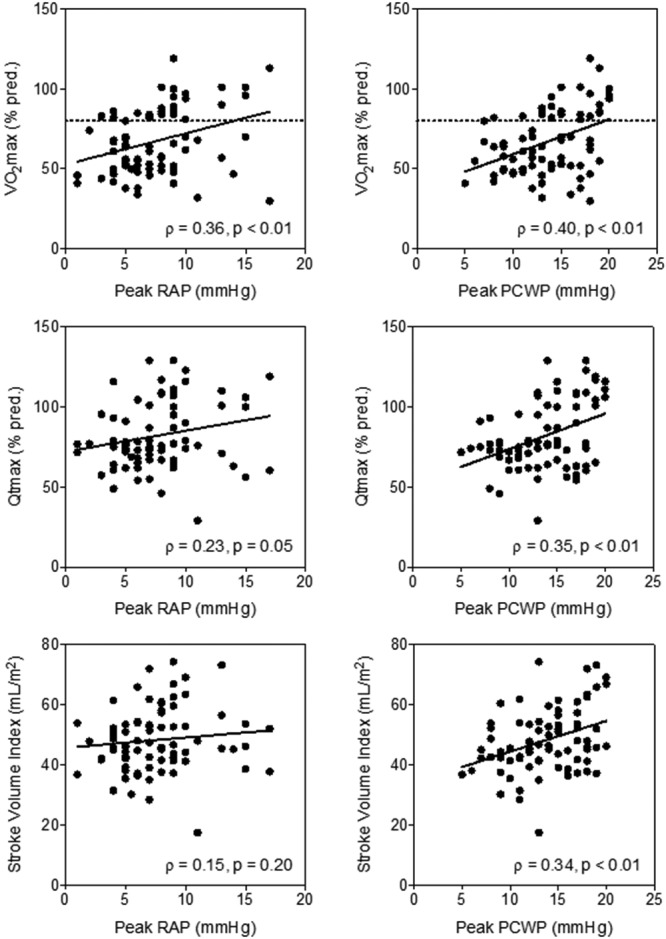
Scatter plots with linear regression of peak right atrial and pulmonary capillary wedge pressures (RAP and PCWP, respectively) versus percent of predicted maximum aerobic capacity (V̇o2max), percent of predicted peak cardiac output (Qtmax), and stroke volume index in patients in the single-test cohort. Dashed line at 80% reflects the cut-off between normal and impaired patients.
Sequential-testing cohort
A small number of patients in our exercise lab underwent clinically indicated sequential exercise tests to assess the impact, if any, of volume loading on their exercise capacity and exertional symptoms in consideration of therapy with fludrocortisone, salt supplementation, or other intravascular volume–expanding therapies. On the basis of the analysis of the single-test cohort above, we reviewed the impact of saline infusion on hemodynamic and metabolic variables in this group of patients. Of the 23 patients who underwent sequential iCPETs, 2 were excluded from analysis because of submaximum effort (RER < 1.05). The remaining patients were thinner and less limited, compared to impaired patients in the single-test cohort, while medication use was similar (Table 1). Subjects in the sequential-testing group received 2.0 ± 0.5 L of normal saline between tests.
Comparison of the Qtmax responses after saline administration identified a group of 16 responders (ΔQtmax: 25% ± 15% pred.) associated with increases in peak SV, RAP, and PCWP (Fig. 4). The average increase in V̇o2max (12% ± 8% pred.) was less because of a fall in Ca-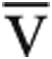 O2max after saline infusion (Fig. 4). Of the 5 nonresponders (i.e., no increase in Qt), 2 developed pulmonary venous hypertension with PCWP > 20 mmHg after fluid administration, and 3 had evidence of impaired peripheral oxygen extraction with Ca-
O2max after saline infusion (Fig. 4). Of the 5 nonresponders (i.e., no increase in Qt), 2 developed pulmonary venous hypertension with PCWP > 20 mmHg after fluid administration, and 3 had evidence of impaired peripheral oxygen extraction with Ca-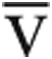 O2max/[Hb] < 0.8 on the initial test. A comparison of the demographic and exercise parameters from the two tests revealed no other differences between responders and nonresponders.
O2max/[Hb] < 0.8 on the initial test. A comparison of the demographic and exercise parameters from the two tests revealed no other differences between responders and nonresponders.
Figure 4.

A, Change in peak cardiac output (Qtmax) and maximum aerobic capacity (V̇o2max) in a subset of patients with evidence of low ventricular filling pressures after administration of intravenous fluid. B, This was associated with increases in V̇o2max and in peak stroke volume (SVmax), right atrial pressure (RAPmax), and pulmonary capillary wedge pressure (PCWPmax) in responders (filled symbols) compared to nonresponders (open symbols). Statistical significance was determined using the Student paired t test, and P values are noted above the brackets. Ca-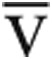 O2: difference between measured arterial and mixed venous oxygen content.
O2: difference between measured arterial and mixed venous oxygen content.
Neuroendocrine evaluation of patients in the single-test cohort
In order to address the etiology of preload insufficiency during exercise, medical records for patients in the single-test cohort were reviewed for the results of any neuroendocrine evaluation. Data were available for 16 impaired and 2 normal patients. Of the impaired patients, 7 underwent tilt-table testing (TTT), and 5 of these had tests consistent with the postural orthostatic tachycardia syndrome (POTS). Of the 4 patients who underwent nerve conduction studies, 2 had abnormal results: 1 with dysautonomia and 1 with diffuse axonal sensorimotor polyneuropathy. Of the 8 patients tested for adrenal function, 4 had abnormal results: 1 with an abnormal cortisol stimulation test, 2 with low random cortisol measurements, and 1 with a noon adrenocorticotropic hormone (ACTH) of <10 pg/mL. The patient with low ACTH also had abnormal TTT results. The patient with dysautonomia by nerve conduction study had normal TTT results. Thus, of 16 patients, 10 had abnormal results. Of the normal patients, 1 had a normal nerve conduction study and 1 had a remote history of positive TTT results. The latter patient achieved a V̇o2max of 86% pred. after 1 L of saline before iCPET.
The impaired patients with a known diagnosis of POTS (n = 5) were all young (28.4 ± 8.9 years) women. Compared to those in normal patients, the resting upright and maximum HRs were higher (106 ± 16 bpm, P < 0.01 and 173 ± 13 bpm, P = 0.04, respectively), although maximum HR was not different (96% pred. vs. 92% pred.). The POTS patients had lower V̇o2max and Qtmax (53% pred. [49%–70%] and 77% pred. [75%–77%], respectively, P < 0.001 for both) with low RAPmax (5 [3–5] mmHg, P = 0.002) and a trend toward a lower peak SV index (50 ± 5 mL/m2, P = 0.06). In this group, resting RAP was also low (1.0 [0–2.5] mmHg, P = 0.03) despite administration of 1 ± 0.7 L of intravenous normal saline before exercise.
Discussion
In this retrospective analysis of iCPET data, we have identified a group of patients with exercise intolerance as a consequence of impaired Qt augmentation associated with low biventricular filling pressures. This contrasts with the more common hemodynamic profiles of patients with cardiac limits related to heart failure or pulmonary vascular disease, which are characterized by exercise-induced elevations of PCWP or mPAP, respectively.4-9 These low filling pressures were associated with blunted SV augmentation, suggesting inadequate preload as an etiology for low Qt. When such patients were challenged with intravenous saline, the majority demonstrated improvements in Qtmax on subsequent testing, supporting the hypothesis that inadequate ventricular filling accounts for the pathologic exercise response.
Preload is strictly defined as the precontractile stretch of ventricular cardiomyocytes. Ventricular-wall stress is the clinical surrogate for preload and can be estimated with measurements of ventricular end-diastolic pressure and volume. Preload normally increases during exercise, as a consequence of increased venous blood return caused by venoconstriction of capacitance vessels and increased actions of the respiratory and skeletal muscle pumps. The observations that impaired patients failed to increase ventricular filling pressures and SV during exercise seem to suggest failure of these mechanisms to support circulatory demands during exercise, thereby limiting V̇o2max.
We doubt that these findings are simply a consequence of intravascular volume depletion in the impaired group. In the single-test cohort, all patients received intravenous saline to target an upright PCWP of ≥5 mmHg before exercise. Indeed, the upright resting RAP and PCWP were identical in the normal and impaired groups. With exercise, however, these pressures failed to augment appropriately in impaired patients. Given the results in the sequential-testing cohort, it seems possible that the routine practice of volume loading in the single-test cohort likely moved some patients from the impaired to the normal group by supporting Qt and V̇o2max above their at-home values. A limitation of our study, however, is the absence of a quantitative assessment of circulating blood volume, which should be incorporated in prospective studies of these patients.
Similarly, these results cannot be explained by either submaximal effort or deconditioning. All patients included in the analysis demonstrated maximal effort by achieving a maximum HR of ≥80% pred. or an RER of ≥1.05, suggesting that their metabolic demand should have been sufficient to stimulate a normal Qt response to exercise. Moreover, a comparison of trained versus deconditioned male patients using iCPET demonstrated that less fit subjects had higher PCWP and RAP at the same workload of 150 W associated with decreased SV and end-diastolic volume (determined by echocardiography). This study indicates that deconditioning is associated with higher ventricular filling pressures and suggests improved lusitropy with training.13 In contrast, our impaired group had decreased SV and low filling pressures.
Presuming adequate intravascular volume and similarly functioning respiratory and muscle pumps, the etiology of inadequate venous return may be a consequence of impaired venoconstriction of capacitance vessels in the impaired population. Indeed, the observation that 10 patients in the impaired group had abnormal neuroendocrine testing with evidence of POTS (5 cases), adrenal insufficiency (3 cases), or autonomic neuropathy (2 cases) supports this hypothesis. POTS is characterized by orthostatic tachycardia without significant hypotension.14 Yet these patients often experience presyncope, palpitations, and exercise intolerance that resolve with lying supine. The pathophysiologic basis of POTS is unclear, and the diagnosis itself likely reflects a conglomeration of different mechanisms. A report demonstrated that POTS patients have reduced left ventricular mass and decreased blood volume, leading to low peak SV and Qt with compensatory increases in HR.15 The invasive hemodynamic profile of the 5 POTS patients in this study confirms a significantly reduced peak SV compared to normal patients, consistent with a previous study.16 We further show that POTS patients had persistently low RAP despite receiving an average of 1 L of normal saline before the test, suggesting that venous capacitance is the issue rather than total intravascular volume. Other studies have also suggested that inadequate peripheral vasoconstriction,17 cardiac sympathetic dysautonomia,18 and autoimmune autonomic neuropathy19 may contribute to symptoms of POTS. All POTS patients in this study had improvement in subjective exercise tolerance with medical therapy, 4 with β-adrenergic receptor antagonists and 1 with midodrine, in addition to increased fluid intake, compression stockings, and monitored exercise training. These observations can likely serve as a starting point for additional investigations into the mechanistic underpinnings of the exercise limitation in patients with preload insufficiency.
Interestingly, impaired patients had decreased systemic oxygen extraction normalized to [Hb], as compared to normal ones (0.81 ± 0.12 vs. 0.87 ± 0.09, P = 0.04), which is consistent with abnormal blood flow distribution to metabolically inactive vascular beds (e.g., impaired splanchnic vasoconstriction with exercise), shunting past oxidative muscle fiber capillary beds, or intrinsic mitochondrial dysfunction. Regardless of the etiology, this finding is suggestive of generalized circulatory dysregulation as a component of the pathogenesis of exercise intolerance in impaired patients. Anecdotally, several of these patients report a severe illness before symptom onset, in many cases occurring 1 year or more before their evaluation, suggesting that an infectious or inflammatory etiology may contribute. In addition, structural limitations to venous return, such as inferior vena cava thrombosis, should be considered in the differential diagnosis.
Notably, the improvement in V̇o2max after volume administration in the sequential-testing cohort was less striking than the improvement in Qtmax. This is similar to the recent noninvasive study of POTS patients undergoing an intravenous fluid challenge.20 Our data indicate that this is entirely due to the effects of dilutional anemia on oxygen extraction (Fig. 4), as the Ca-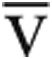 O2/[Hb] ratio did not change after volume administration in the sequential-testing cohort (+1 ± 9%, P = 0.5). Human studies demonstrate a decrease in the V̇o2max of exercising leg muscle after isovolemic reduction in [Hb], two-thirds of which is attributable to reduced diffusion of oxygen;21 while the precise mechanism is unknown, oxygen delivery may depend on intracapillary red blood cell spacing, changes in oxygen dissociation rates, or increased red blood cell flow heterogeneity.21,22 This suggests that therapeutic interventions to increase intravascular volume (e.g., oral hydration, fludrocortisone, salt tabs) may be less effective than therapies directed at vascular tone (e.g., midodrine, pyridostigmine). Indeed, pyridostigmine has shown promise in patients with POTS, particularly when associated with antecedent viral infection or secondary to an autoimmune disorder.23,24 The role of these medications in treating patients with exercise intolerance due to abnormal venous return, as described here, is a promising area for future investigation.
O2/[Hb] ratio did not change after volume administration in the sequential-testing cohort (+1 ± 9%, P = 0.5). Human studies demonstrate a decrease in the V̇o2max of exercising leg muscle after isovolemic reduction in [Hb], two-thirds of which is attributable to reduced diffusion of oxygen;21 while the precise mechanism is unknown, oxygen delivery may depend on intracapillary red blood cell spacing, changes in oxygen dissociation rates, or increased red blood cell flow heterogeneity.21,22 This suggests that therapeutic interventions to increase intravascular volume (e.g., oral hydration, fludrocortisone, salt tabs) may be less effective than therapies directed at vascular tone (e.g., midodrine, pyridostigmine). Indeed, pyridostigmine has shown promise in patients with POTS, particularly when associated with antecedent viral infection or secondary to an autoimmune disorder.23,24 The role of these medications in treating patients with exercise intolerance due to abnormal venous return, as described here, is a promising area for future investigation.
The primary clinical impact of this study is to highlight the concept of preload insufficiency in the differential diagnosis of unexplained dyspnea or other effort intolerance. The majority (64%) of patients studied in our laboratory with low V̇o2max due to low Qtmax are found to have elevated ventricular filling pressures (Fig. 1) characteristic of impaired lusitropy, contractility, or increased ventricular afterload, primarily as a result of heart failure with preserved or reduced ejection fraction or pulmonary arterial hypertension. The invasive hemodynamic profiles of these patients have been well described in the literature.4-6,8,9 In contrast, the impaired group accounted for 8% of iCPETs performed in our exercise laboratory, which represents a notable minority of patients who present for the clinical evaluation of unexplained dyspnea.
From these data, a diagnosis of preload insufficiency should be considered in patients with RAPmax < 6.5 mmHg, PCWPmax < 12.5 mmHg (Fig. 3), ΔRAP < 5.5 mmHg, or ΔPCWP < 6.75 mmHg based on receiver-operator characteristic analyses of the single-test cohort (data not shown), particularly in the absence of other etiologies of impaired Qtmax. The addition of volume loading and sequential iCPET may facilitate this diagnosis, particularly in clinical scenarios where absolute systemic venous pressure is not a direct measure of preload, as there are cases where inadequate preload limits cardiac output despite normal or elevated filling pressures.25
Given the retrospective and cross-sectional nature of this study, patients in the impaired group did not follow a uniform clinical evaluation of their exertional symptoms, and only limited follow-up data are available. None of the patients in the study has been referred for a subsequent noninvasive or invasive CPET.
Similarly, the normal group had to be defined on the basis of the invasive hemodynamic exercise profile of symptomatic patients, given a paucity of normative invasive hemodynamic data and the ethical considerations of performing such tests in normal patients. Moreover, the scarcity of “normal” exercise tests in this population limits the utility of using a higher cut-off for V̇o2max for the analysis.
Comparing the single-test and sequential-testing cohorts demonstrates a low number of relatively less impaired patients in the sequential-testing cohort. Since the decision to perform a second iCPET is based primarily on evidence of low ventricular filling pressures, only 10 of 21 had V̇o2max < 80% pred., with only 4 of those having Qtmax < 80% pred. (i.e., meeting our definition of impaired from the single-test cohort). These 4 patients responded with an average absolute increase in Qtmax of 16% pred.; 1 of them had a decrease in Qtmax after receipt of fluid. Although the remaining patients had “normal” V̇o2max and Qtmax % pred., all underwent iCPET in evaluation of new-onset exertional symptoms of dyspnea and/or fatigue. Historically, these patients reported fair aerobic fitness and regular exercise, suggesting that a relative decrease in their personal V̇o2max accounted for their symptoms rather than a low V̇o2max in population-based terms.
These data suggest that the normal SV response to exercise and aerobic capacity are governed in part by biventricular preload. We have directly demonstrated that a subset of unexplained-dyspnea patients have low ventricular filling pressures as their sole abnormality of cardiopulmonary function during exercise. This may be as limiting as high filling pressures or impaired contractility and should be considered in the evaluation of patients with unexplained exertional symptoms. This work provides a foundation for prospective, controlled studies on the role of saline loading in the diagnosis of preload insufficiency, the prevalence of abnormal neuroendocrine function in these patients, and their response to treatment.
Acknowledgments
We gratefully acknowledge Teresa Van Horn for assistance with the iCPET database; Paul Pappagianopoulos, Julie Tracy, and Arlene Schiro for expert supervision and analysis of exercise testing and results; and Dr. Joseph Loscalzo for critical review of the manuscript while in preparation.
Source of Support: This work was supported by the National Heart, Lung, and Blood Institute (NHLBI; grants K23HL091106 to GDL and T32HL007633 to WMO) and NHLBI Heart Failure Network grant U01HL084877 to GDL and DMS.
Conflict of Interest: None declared.
References
- 1.DePaso WJ, Winterbauer RH, Lusk JA, Dreis DF, Springmeyer SC. Chronic dyspnea unexplained by history, physical examination, chest roentgenogram, and spirometry: analysis of a seven-year experience. Chest 1991;100(5):1293–1299. [DOI] [PubMed]
- 2.Pratter MR, Curley FJ, Dubois J, Irwin RS. Cause and evaluation of chronic dyspnea in a pulmonary disease clinic. Arch Intern Med 1989;149(10):2277–2282. [PubMed]
- 3.Maron BA, Cockrill BA, Waxman AB, Systrom DM. The invasive cardiopulmonary exercise test. Circulation 2013;127(10):1157–1164. [DOI] [PubMed]
- 4.Mancini D, Katz S, Donchez L, Aaronson K. Coupling of hemodynamic measurements with oxygen consumption during exercise does not improve risk stratification in patients with heart failure. Circulation 1996;94(10):2492–2496. [DOI] [PubMed]
- 5.Metra M, Faggiano P, D’Aloia A, Nodari S, Gualeni A, Raccagni D, Dei Cas L. Use of cardiopulmonary exercise testing with hemodynamic monitoring in the prognostic assessment of ambulatory patients with chronic heart failure. J Am Coll Cardiol 1999;33(4):943–950. [DOI] [PubMed]
- 6.Kitzman DW, Higginbotham MB, Cobb FR, Sheikh KH, Sullivan MJ. Exercise intolerance in patients with heart failure and preserved left ventricular systolic function: failure of the Frank-Starling mechanism. J Am Coll Cardiol 1991;17(5):1065–1072. [DOI] [PubMed]
- 7.Borlaug BA, Nishimura RA, Sorajja P, Lam CS, Redfield MM. Exercise hemodynamics enhance diagnosis of early heart failure with preserved ejection fraction. Circ Heart Fail 2010;3(5):588–595. [DOI] [PMC free article] [PubMed]
- 8.Santos M, Opotowsky AR, Shah AM, Tracy J, Waxman AB, Systrom DM. Central cardiac limit to aerobic capacity in patients with exertional pulmonary venous hypertension: implications for heart failure with preserved ejection fraction. Circ Heart Fail 2015;8(2):278–285. [DOI] [PMC free article] [PubMed]
- 9.Tolle JJ, Waxman AB, Van Horn TL, Pappagianopoulos PP, Systrom DM. Exercise-induced pulmonary arterial hypertension. Circulation 2008;118(21):2183–2189. [DOI] [PMC free article] [PubMed]
- 10.Maroni JM, Oelberg DA, Pappagianopoulos P, Boucher CA, Systrom DM. Maximum cardiac output during incremental exercise by first-pass radionuclide ventriculography. Chest 1998;114(2):457–461. [DOI] [PubMed]
- 11.Wasserman K, Hansen JE, Sue DY, Stringer WW, Whipp BJ. Principles of exercise testing and interpretation. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins, 2005.
- 12.Medoff BD, Oelberg DA, Kanarek DJ, Systrom DM. Breathing reserve at the lactate threshold to differentiate a pulmonary mechanical from cardiovascular limit to exercise. Chest 1998;113(4):913–918. [DOI] [PubMed]
- 13.Stickland MK, Welsh RC, Petersen SR, Tyberg JV, Anderson WD, Jones RL, Taylor DA, Bouffard M, Haykowsky MJ. Does fitness level modulate the cardiovascular hemodynamic response to exercise? J Appl Physiol 2006;100(6):1895–1901. [DOI] [PubMed]
- 14.Low PA, Sandroni P, Joyner M, Shen WK. Postural tachycardia syndrome (POTS). J Cardiovasc Electrophysiol 2009;20(3):352–358. [DOI] [PMC free article] [PubMed]
- 15.Fu Q, VanGundy TB, Galbreath MM, Shibata S, Jain M, Hastings JL, Bhella PS, Levine BD. Cardiac origins of the postural orthostatic tachycardia syndrome. J Am Coll Cardiol 2010;55(25):2858–2868. [DOI] [PMC free article] [PubMed]
- 16.Masuki S, Eisenach JH, Schrage WG, Johnson CP, Dietz NM, Wilkins BW, Sandroni P, Low PA, Joyner MJ. Reduced stroke volume during exercise in postural tachycardia syndrome. J Appl Physiol 2007;103(4):1128–1135. [DOI] [PubMed]
- 17.Stewart JM, Ocon AJ, Clarke D, Taneja I, Medow MS. Defects in cutaneous angiotensin-converting enzyme 2 and angiotensin-(1–7) production in postural tachycardia syndrome. Hypertension 2009;53(5):767–774. [DOI] [PMC free article] [PubMed]
- 18.Goldstein DS, Holmes C, Frank SM, Dendi R, Cannon RO III, Sharabi Y, Esler MD, Eisenhofer G. Cardiac sympathetic dysautonomia in chronic orthostatic intolerance syndromes. Circulation 2002;106(18):2358–2365. [DOI] [PubMed]
- 19.Vernino S, Low PA, Fealey RD, Stewart JD, Farrugia G, Lennon VA. Autoantibodies to ganglionic acetylcholine receptors in autoimmune autonomic neuropathies. N Engl J Med 2000;343(12):847–855. [DOI] [PubMed]
- 20.Figueroa RA, Arnold AC, Nwazue VC, Okamoto LE, Paranjape SY, Black BK, Diedrich A, et al. Acute volume loading and exercise capacity in postural tachycardia syndrome. J Appl Physiol 2014;117(6):663–668. [DOI] [PMC free article] [PubMed]
- 21.Schaffartzik W, Barton ED, Poole DC, Tsukimoto K, Hogan MC, Bebout DE, Wagner PD. Effect of reduced hemoglobin concentration on leg oxygen uptake during maximal exercise in humans. J Appl Physiol 1993;75(2):491–498; discussion 489–490. [DOI] [PubMed]
- 22.Kurdak SS, Grassi B, Wagner PD, Hogan MC. Effect of [Hb] on blood flow distribution and O2 transport in maximally working skeletal muscle. J Appl Physiol 1995;79(5):1729–1735. [DOI] [PubMed]
- 23.Raj SR, Black BK, Biaggioni I, Harris PA, Robertson D. Acetylcholinesterase inhibition improves tachycardia in postural tachycardia syndrome. Circulation 2005;111(21):2734–2740. [DOI] [PubMed]
- 24.Grubb BP. Postural tachycardia syndrome. Circulation 2008;117(21):2814–2817. [DOI] [PubMed]
- 25.Opotowsky AR, Halpern D, Kulik TJ, Systrom DM, Wu F. Inadequate venous return as a primary cause for Fontan circulatory limitation. J Heart Lung Transplant 2014;33(11):1194–1196. [DOI] [PubMed]