

Abstract
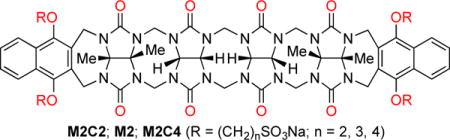
Two acyclic CB[n]-type molecular containers that differ in the length of the (CH2)n (M2C2, n=2, M2C4, n=4) linker between their aromatic sidewalls and SO3− solubilizing groups were prepared and studied. The inherent solubilities of M2C2 (68 mM) and M2C4 (196 mM) are higher than the analogue with a (CH2)3 linker (M2, 14 mM) studied previously. 1H NMR dilution experiments show that M2C2 and M2C4 do not self-associate in water, which enables their use as solubilizing excipients. We used phase solubility diagrams (PSD) to compare the solubilizing ability of M2, M2C2, M2C4, hydroxypropyl-β-cyclodextrin (HP-β-CD), and sulfobutylether-β-cyclodextrin (SBE-β-CD) toward 15 insoluble drugs. We find that M2C2 and M2C4 – as gauged by the slope of their PSDs – are less potent solubilizing agents than M2. However, the higher inherent solubility of M2C2 allows higher concentrations of drug to be formulated using M2C2 than with M2 in several cases. The solubilizing ability of M2C2 and SBE-β-CD were comparable in many cases with Krel values averaging 23 and 12, respectively, relative to HP-β-CD. In vitro cytotoxicity and in vivo maximum tolerated dose studies document the biocompatibility of M2C2.
Keywords: Cucurbituril, cyclodextrin, solubilizing excipient, molecular recognition, host-guest chemistry
TOC image
Acyclic cucurbit[n]uril-type molecular containers function as solubilizing excipients for insoluble drugs. The influence of the length of the linker between the aromatic wall and the sulfonate solubilizing group (e.g. M2C2, M2, M2C4) on their inherent aqueous solubility and recognition properties toward insoluble drugs are presented. The M2C2 does not display significant in vitro cytotoxicity and a maximal tolerated dose study indicates M2C2 is well tolerated in mice.
Introduction
A central topic in the field of supramolecular chemistry is the synthesis of new molecular container compounds, studies of their molecular recognition properties and incorporation into functional systems. The most popular classes of macrocyclic containers include the crown ethers, cyclodextrins, calixarenes, cyclophanes, cucurbiturils, pillararenes, and a variety of receptors created by self-assembly processes.1 The great interest in host•guest complexes stems from the fact that the properties of free guest and complexed guest (e.g. chemical stability, electrochemistry, conformational preferences, photophysical, pKa) often differ dramatically. Accordingly, host•guest complexes have been used to stabilize otherwise unstable compounds like white phosphorous (P4),2 to catalyze the bowl-to-bowl interconversion of corannulene,3 to shift the pKa of included guests (e.g. acetals) and thereby promote hydrolysis reactions under basic conditions,4 to create chemical sensing ensembles,5 and to contort alkanes into helical conformations.6 The supramolecular chemistry of the cucurbit[n]uril (CB[n]) family of molecular containers (Figure 1) is particularly rich, in large part due to their high affinity, high selectivity, and stimuli responsive host•guest complexes.7 For example, CB[n] compounds function as catalysts for cycloadditions reactions,8 components of biomembrane and chemical sensing systems,9 promotors of dimerization assembly for materials and biological applications,10 and sorption materials for gas purification.11
Figure 1.
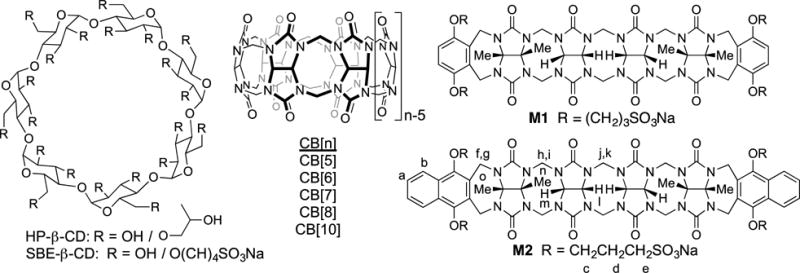
Chemical structures of HP-β-CD (average degree of substitution = 4.9), SBE-β-CD (average degree of substitution = 6.5), CB[n], M1 and M2 that have previously been used as solubilizing excipients for insoluble drugs.
Over the past 20 years, the adoption of combinatorial chemistry tools by pharmaceutical industry has resulted in the rapid generation of new small molecules with good biological activity. Unfortunately, the solubility of an estimated 40–70% of these new chemical entities are so low that they cannot be formulated alone in water.12 The pharmaceutical industry has responded by developing tools to enable the formulation of these insoluble drugs including nanocrystalline solid forms, solid dispersions, co-solvent systems (e.g. Cremophore), prodrug approaches, co-crystals, and cyclodextrin derived solubilizing excipients (e.g. HP-β-CD and SBE-β-CD; Figure 1).13 Accordingly, researchers in the CB[n] field are actively investigating their potential in biomedical applications including the solubilization of insoluble drugs,14 their inherent cytotoxicity both in vitro and in vivo,15 their ability to delay or reverse the (side) effects of drugs,16 for controlled release,17 and for targeted drug delivery.18
Several years ago, the Isaacs group used our synthetic and mechanistic knowledge of the CB[n] forming reaction19 to design acyclic CB[n]-type receptors (M1 and M2, Figure 1) that retain many of the essential structural and molecular recognition features of the CB[n] family. Compounds M1 and M2 feature a central C-shaped glycoluril tetramer, two terminal aromatic walls, and four sulfonate solubilizing groups. Compound M1 has very good aqueous solubility (346 mM), dramatically increases the solubility of insoluble drugs, is not toxic when assayed in vitro and in vivo, and can be used to formulate and deliver albendazole to animals.20 Previously, we have studied the influence of the solubilizing group (e.g. SO3Na versus NH3+), the identity of the aromatic sidewall (e.g. (substituted) benzene versus (substituted) naphthalene, and the length of the central glycoluril oligomer on their ability as solubilizing agents for insoluble drugs.21 In brief, we found that M2 – with its SO3Na solubilizing groups, a central glycoluril tetramer, and naphthalene walls is the most potent general purpose solubilizing agent. Unfortunately, M2 is also the least soluble (14 mM) acyclic CB[n]-container in this series which limits the amount of insoluble drug that can be formulated by internal complexation. In this paper, we investigate two new containers (M2C2 and M2C4) that differ from M2 only in the length of the alkyl chain between the aromatic walls and the SO3Na groups with the goal of improving the inherent solubility of the host while retaining the excellent recognition properties of M2.
Results and Discussion
This results and discussion section is organized as follows. First, we describe the synthesis of the two new containers (M2C2 and M2C4) followed by the determination of their inherent solubility and self-association properties in aqueous solution. Next, we describe the creation of phase solubility diagrams (PSD) with 15 drugs and analyze the trends in the solubilization data. Then, we compare the data obtained with the more promising new host (M2C2) with that of SBE-β-CD which is an approved formulation method for several drugs administered to humans. Finally, we present the results of in vitro and in vivo toxicity studies conducted using M2C2.
Design and Synthesis of M2C2 and M2C4
As described previously, the design of acyclic CB[n]-type receptors (e.g. M2) is based on the use of a central glycoluril oligomer that imparts a preorganized C-shaped hydrophobic cation binding space with two naphthalene walls bearing four alkyl SO3Na groups. These compounds have the potential to bind to a wide variety of insoluble aromatic ring containing drugs by π–π interactions, thereby enhancing their aqueous solubility. The preparation of containers M2C2 and M2C4 were modeled on the synthesis of M2. The naphthalene alkyl-SO3Na groups were changed to ethyl or butyl to determine which variation would improve aqueous solubility. To prepare the sidewall with the ethyl or butyl-SO3Na groups, we initially reacted 1,4-dihydroxynaphthalene (1) with dibromoethane under basic conditions (K2CO3, 18-crown-6, CH3CN, 70 °C) to give 2 in 40% yield (Scheme 1). Transformation of dibromide 2 into the required (CH2)2SO3Na naphthalene sidewall 3 was accomplished by heating with 2.2 equiv. of Na2SO3 in H2O/DMF (1:1) at 100 °C to give 3 in 81% yield. Treatment of 1 with butanesultone (4) under basic conditions (NaOH, H2O, dioxane) at room temperature gave the required sidewall 5 with the (CH2)4SO3Na groups in 26% yield (Scheme 1). Reaction of 3 (5) with glycoluril tetramer bis(cyclic ether) 6 by a double electrophilic aromatic substitution reaction (CF3CO2H/Ac2O (1:1), 70 °C) delivered M2C2 (M2C4) in 58% (43%) yield. M2C2 and M2C4 were fully characterized by the standard methods (1H and 13C NMR, IR, high resolution mass spectrometry) which are in agreement with the depicted C2v symmetric structures.
Scheme 1.
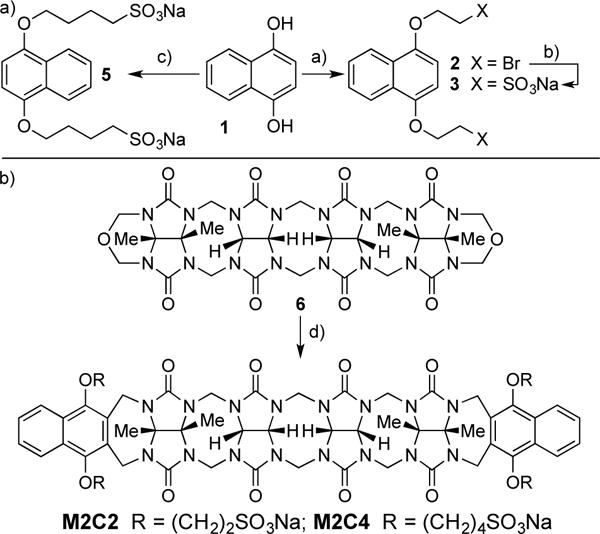
Synthesis of: a) aromatic walls 3 and 5, and b) acyclic CB[n]-type molecular containers M2C2 and M2C4. Conditions: a) K2CO3, 18-crown-6, BrCH2CH2Br, CH3CN, 70 °C; b) Na2SO3 in H2O/DMF (1:1),100 °C, 81%; c) butanesultone (4), NaOH, H2O, dioxane, RT, 26%; d) CF3CO2H/Ac2O (1:1), 70 °C, (M2C2, 58%; M2C4 43%).
X-ray Crystal Structure of M2C4
We were fortunate to obtain single crystals of M2C4 by slow evaporation of an aqueous solution that were suitable for determination of the x-ray crystal structure.22 Figure 2 shows a cross-eyed stereoview of one molecule of M2C4 contained within the unit cell. As expected, the methylene bridged glycoluril tetramer imparts an overall C-shape to the molecule and the aromatic naphthalene sidewalls further shape the hydrophobic cavity. As observed previously for M2, M2C4 exhibits a helical twist due to the out of plane splaying of its naphthalene walls; both senses of helicity are present in the crystal.20a Unlike M2, two of the O(CH2)4SO3Na arms of M2C4 appear to partially fill their own cavity. Specifically, one Ar-O-CH2 group on each sidewall is oriented such that the (CH2)4 group of one wall forms van der Waals interactions with the opposing sidewall and vice versa.20a These intramolecular non-covalent interactions would need to be broken in order for M2C4 to form M2C4•guest interactions and will therefore reduce the observed binding constants.
Figure 2.
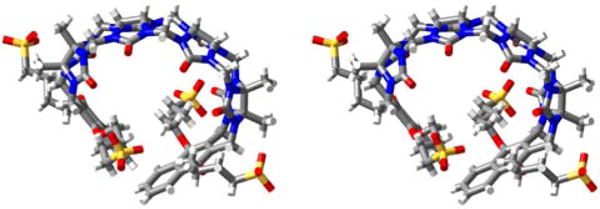
Cross-eyed stereoview of one molecule of M2C4 in the crystal. Color code: C, grey; H, white; N, blue; O, red; S, yellow; H-bonds, red-yellow stripped.
Determination of Inherent Solubility of M2C2 and M2C4
After having completed the synthesis of M2C2 and M2C4 we decided to measure the inherent solubility of these new containers in our standard 20 mM sodium phosphate buffered D2O at pD 7.4. A saturated solution of container was prepared by adding solid M2C2 or M2C4 to water until solid container remained undissolved. The insoluble material was removed by filtration and the concentration of M2C2 or M2C4 in solution was measured by a 1H NMR assay employing 1,3,5-benzenetricarboxylic acid as an internal standard of known concentration as described previously.20a,21a In this manner we determined the inherent solubilities of M2C2 and M2C4 as 68 and 196 mM, respectively. It is worth noting, however, that solutions of M2C4 higher than 100 mM become increasingly viscous. The high solubility of both M2C2 and M2C4 compared to M2 (14 mM) makes them potentially useful alternatives to M2 when high concentrations of formulated drug are required.
Containers M2C2 and M2C4 do not undergo strong self-association in water
Previously, we have measured the self-association constant (Ks) of M2 in 20 mM phosphate buffered D2O at pD 7.4 as 624 M−1 by monitoring the change in chemical shift of the aromatic wall protons as a function of concentration by 1H NMR spectroscopy and fitting the data to a standard 2-fold self-association model.21a We performed related 1H NMR dilution experiments for M2C2 (67 mM to 66 μM) and M2C4 (66 mM to 65 μM) and determined the Ks values to be 6 M−1 and 66 M−1, respectively (Supporting Information). More highly concentrated solutions of M2C4 flow somewhat slowly which suggests they are more viscous than less concentrated solutions. The observed lower intermolecular self-association constants establish that M2C2 and M2C4 will exist more fully in their monomeric forms in solution which could enhance their abilities as solubilizing excipients relative to M2. However, one must also consider the possibility that the O(CH2)nSO3Na arms can fold into their own cavity in solution as observed in the x-ray crystal structure of M2C4 (Figure 2). Such a self-folding event would tend to decrease the ability of the container to undergo intermolecular self-association and also decrease the ability to bind to insoluble drugs. Accordingly, we examined the 1H NMR chemical shifts of the O(CH2)nSO3Na arms of containers M2, M2C2, and M2C4 relative to the sidewalls 3 and 5 as model compounds for evidence of self-folding. For all three containers the CH2 groups become diastereotopic so two resonances are seen (Supporting Information). We find that the resonances for the arms of M2C2 and M2C4 are shifted slightly farther upfield relative to the sidewall model compounds as compared to M2. This suggests that containers M2C2 and M2C4 may undergo more significant self-folding phenomena than M2.
Theory of Phase Solubility Diagrams
In order to motivate the discussion of the experimentally determined PSDs created with M2C2, M2C4, and SBE-β-CD it is useful to first discuss PSDs from a theoretical viewpoint.21a,23 PSDs are plots of the concentration of insoluble drug in solution versus the concentration of container used (Figure 3). PSDs can display a variety of forms, but linear PSDs (known as AL-type) are common and are taken as evidence that container and drug form soluble well defined complexes of 1:1 stoichiometry.23 When AL-type PSDs are observed, the system behaves according to equation 1 which allows for a determination of the binding constant Ka for the container•drug complex based on the experimentally determined slope of the PSD and inherent solubility of the drug (S0). The slope of the PSD is a measure of the efficiency of solubilization of drug; we consider a container that solubilizes 50 mole percent (e.g. [container] = 100 mM gives [drug] = 50 mM) to be an efficient solubilizing agent. If a slope of 0.5 is inserted into equation 1, then it is easy to demonstrate that Ka × S0 = 1. Accordingly, if we wish to efficiently solubilize a drug (e.g. slope ≥ 0.5) which has an inherent solubility 10 μM (1 μM) then the container must bind to the drug with a Ka value of at least 105 M−1 (106 M−1). In this context, the generally higher Ka values observed for CB[n]•guest complexes relative to cyclodextrin•guest complexes promises to enable the formulation of drugs of lower inherent solubility. Figure 3 shows calculations of PSDs for a hypothetical container-drug system that obeys equation 1, with an inherent drug solubility of 10 μM and different values of Ka for the container•drug complex. As can readily be seen, increasing (decreasing) the Ka value 9-fold changes the slope of the PSD from 0.5 to 0.9 (0.1) respectively. Conversely, differences in experimentally determined slope can be translated into differences in Ka. In this paper we intend to compare the solubilizing abilities of different containers (e.g. C1 and C2) toward a given drug (D1) so it is also useful to consider equation 2 which relates the slopes of two PSDs to the relative binding constant Krel. Advantageously, a precise knowledge of So,D1 is not required to determine the relative binding constant (Krel) of two different containers toward a given drug since it cancels out on the top and bottom of equation 2.
| (1) |
| (2) |
Figure 3.
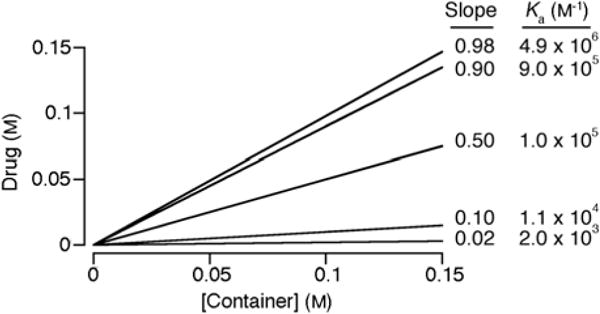
Simulated PSDs for container•drug systems that obey equation 1 with S0 = 10 μM, different values of slope and the corresponding values of Ka.
Use of M2C2, M2C4, and SBE-β-CD as Solubilizing Agents for Insoluble Drugs
To assess the abilities of the newly synthesized containers M2C2 and M2C4 as solubilizing agents for insoluble drugs relative to M2, HP-β-CD,20a,21a and SBE-β-CD we created PSDs for the containers with 15 insoluble drugs (Figures 4 and 5). Of these 15 drugs, 13 are used in practice whereas PBS-108624 and MEPBZ25 are not in clinical use. We created the PSDs as described previously.20a,21a In brief, we stir a known concentration of container with an excess of insoluble drug overnight to establish equilibrium, filter or centrifuge away the excess insoluble drug, and then measure the concentration of drug in solution by 1H NMR integration relative to added 1,3,5-benzenetricarboxylic acid as internal standard of known concentration. In this manner we constructed PSDs for the solubilization of 15 drugs with M2C2 and SBE-β-CD and a subset of these drugs with M2C4 due to limitations discussed below (Supporting Information). In most cases, the newly measured PSD are linear at low container concentration, which is consistent with well defined 1:1 complexes. Table 1 presents the results of these studies including the inherent solubility of the drug (S0), the slope of the PSD, the Krel value referenced to the weakest binding container (generally HP-β-CD), and Ka values. The values for M2 and HP-β-CD measured previously20a,21a under identical conditions are also presented in Table 1 for comparison. The uncertainty in Ka and Krel values are generally ≤ 24%, as derived from the error in the linear regression fitting of the slope of the PSD, although larger uncertainties are noted for PSDs with slope approaching unity which leads to large errors in the (1 – slope) terms of equations 1 and 2 (Supporting Information).
Figure 4.
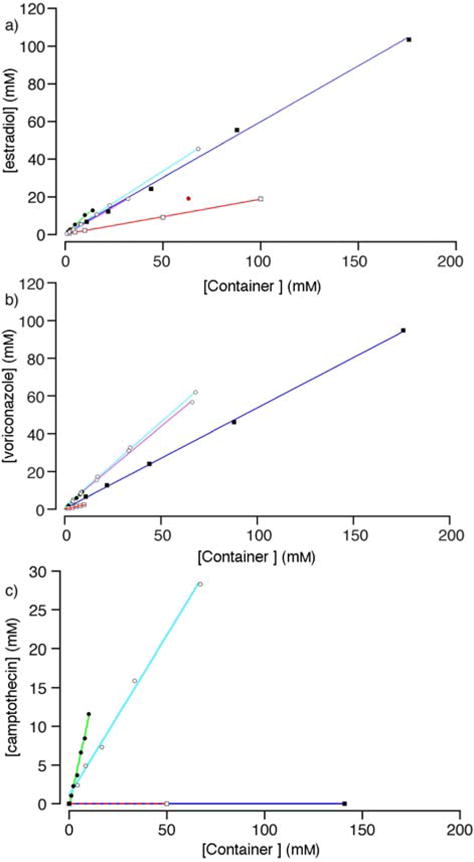
PSDs constructed from the solubilization data for: a) estradiol, b) voriconazole, c) camptothecin with different containers (M2, ●; M2C2, o; M2C4, ☉; HP-β-CD, □; SBE-β-CD, ■. Conditions: 20 mM sodium phosphate buffered D2O (pD = 7.4, rt). Data points colored red were not used in the linear regression.
Figure 5.
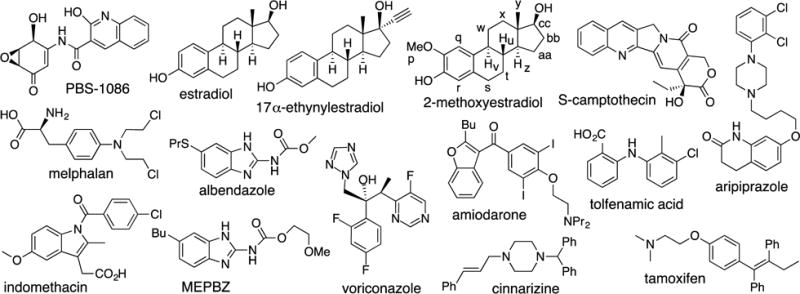
Chemical structures of the 15 insoluble drugs studied in this paper.
Table 1.
Parameters from the phase solubility diagrams.
| Initial slope of the PSD | Krel/Ka*104 (M−1) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||
| Drug | S0 (μM) | HP-β-CD | SBE-β-CD | M2 | M2C2 | M2C4 | HP-β-CD20a,21a | SBE-β-CD | M220a,21a | M2C2 | M2C4 |
| PBS-1086 | 4.5 | 0 | 0 | 0.89 | 0.70 | n.d. | – | – | 3.5/190 | 1/51 | n.d. |
| Ethynyl Estradiol | 24 | 0.47 | 0.93 | 1.1 | 0.57 | n.d. | 1/3.6 | 14/53 | TL/TL | 1.5/5.5 | n.d. |
| Camptothecin | 54 | 0 | 0 | 1.1 | 0.41 | n.d. | – | – | TL/TL | 1/1.3 | n.d. |
| Cinnarizine | 14 | 0.002 | 0.008 | 0 | 0.26 | n.d. | 1/0.016 | 4.0/0.058 | – | 170/2.5 | n.d. |
| Albendazole | 2.7 | 0.015 | 0.027 | 0.48 | 0.035 | n.d. | 1/0.54 | 1.9/1.1 | 62/34 | 2.4/1.4 | n.d. |
| Estradiol | 8.8 | 0.18 | 0.59 | 0.92 | 0.67 | 0.59 | 1/2.6 | 6.7/17 | 51/130 | 9.1/23 | 6.5/16 |
| Amiodarone | 66 | 0.089 | 0.60 | 1.03 | 0.65 | Gel | 1/0.15 | 15/2.3 | TL/TL | 19/2.8 | n.d. |
| Indomethacin | 5.7 | 0.013 | 0.23 | 0.47 | 0.13 | n.d. | 1/0.24 | 22/5.1 | 67/16 | 11/2.5 | n.d. |
| Aripiprazole | 23 | 0 | 0.048 | 0.31 | 0.28 | n.d. | – | 1.0/0.22 | 8.8/2.0 | 7.7/1.7 | n.d. |
| Melphalan | n.d. | 0.11 | 0.50 | 1.1 | 0.43 | n.d. | 1/– | 8/– | TL/– | 6.0/– | n.d. |
| Voriconazole | 38 | 0.22 | 0.53 | 1.0 | 0.89 | 0.85 | 1/0.76 | 4.0/3.0 | TL/TL | 29/21 | 19/14 |
| Tamoxifen | 12 | 0.028 | 0.63 | 0.10 | 0.058 | n.d. | 1/0.24 | 60/14 | 3.9/0.94 | 2.1/0.51 | n.d. |
| Tolfenamic acid | 1.9 | 0.0080 | 0.11 | 0.54 | 0.11 | n.d. | 1/0.41 | 15/6.4 | 150/63 | 16/6.6 | n.d. |
| 2-Methoxy estradiol | n.d. | 0.074 | 0.21 | 0.79 | 0.49 | n.d. | 1/– | 3.3/– | 47/– | 12/– | n.d. |
| MEPBZ | 1.4 | 0.024 | 0.046 | 0.53 | 0.28 | n.d. | 1/1.8 | 1.9/3.5 | 46/82 | 15/28 | n.d. |
n.d., not determined; –, cannot be calculated because slope = 0 or S0 was not determined; TL, too large to be determined because slope ≈ 1.
Container M2C4 forms Gels with Insoluble Drugs
Given that M2 is an excellent receptor and solubilizing agent for steroids,21a,26 we decided to first create PSDs for M2C2 and M2C4 with estradiol (Figure 4a). As can be seen, both M2C2 and M2C4 are excellent solubilizing agents for estradiol with slopes of 0.67 and 0.59 at lower concentrations of containers, respectively. M2C2 (68 mM) is capable of solubilizing 45 mM estradiol. For concentrations of M2C4 above 32 mM, the amount of estradiol solubilized plateaus at ≈ 20 mM. The presence of a plateau region in PSDs indicates the formation of container•drug complexes of limited solubility.23 In addition, when concentrations of M2C4 above 100 mM were used as a solubilizing agent for estradiol we observed the formation of gels. Similarly, the use of concentrations of M2C4 greater than 16 mM lead to the formation of gels when used as a solubilizing agent for amiodarone. Figure 4b shows the PSDs created for voriconazole. In this case, both M2C2 and M2C4 solubilize voriconazole efficiently with slopes of 0.89 and 0.85, respectively, with no evidence of gel formation at 68 mM container. Given the comparable PSD slopes for M2C2 and M2C4 at low concentrations and the observation of gels with M2C4 at higher concentrations – which are not appropriate for use as a solubilizing excipient – we decided to limit our studies of the remaining 12 drugs to M2C2 as a solubilizing agent.
Solubilizing Ability of M2C2 relative to M2
In a previous study of five acyclic CB[n]-type containers with different aromatic walls, we found that the M2 container often displayed the highest slope and Ka values toward the insoluble drugs.20a,21a As noted above, the maximum solubility of M2C2 (68 mM) is significantly higher than that of M2 (14 mM) which could translate into the ability to prepare formulations containing higher drug concentrations if the Ka values of M2C2 and M2 toward a given drug are equal. An examination of Table 1 reveals that M2 is the superior solubilizing agent (e.g. higher slope and Ka) in all cases. In some cases the difference in slope is small (e.g. 0.31 vs. 0.28 for aripiprazole) whereas in other cases the difference is substantial (e.g. 0.54 vs. 0.11 for tolfenamic acid). The Ka values for the 15 drugs toward M2 (M2C2) range from 0.94 – 190 × 104 M−1 (0.51 – 23 × 104 M−1); in several cases the slope of the PSD is unity and therefore, the Ka values are too large to be measured by PSD techniques. The average Krel value for M2 versus M2C2 toward a given drug is 6.6. The origins of this decrease in affinity of M2C2 compared to M2 is unclear although one can hypothesize that the location of the SO3Na solubilizing groups close to the cavity of M2C2 changes the aqueous solvation of the cavity which is known to provide a potent driving force in CB[n] complexation processes.7f,27 Despite the decrease in slope observed for M2C2 compared to M2, the concentration of drug that can be formulated with M2C2 is often higher than can be achieved with M2 as shown in Table 2. For example, the concentration of ethynylestradiol (33.6 versus 10.5 mM), camptothecin (28.3 versus 11.6 mM), and voriconazole (61.9 versus 9.23 mM) achieved with M2C2 are substantially higher than with M2. These data suggest that M2C2 has significant potential as a solubilizing excipient for insoluble drugs.
Table 2.
Highest drug concentrations (mM) achieved with each container.
| Drug | M2 | M2C2 | SBE-β-CD |
|---|---|---|---|
| PBS-1086 | 12.5 | 16.5 | 0 |
| Ethynylestradiol | 10.5 | 33.6 | 131 |
| Camptothecin | 11.6 | 28.3 | 0 |
| Cinnarizine | 0 | 4.40 | 1.71 |
| Albendazole | 4.48 | 2.78 | 3.94 |
| estradiol | 12.9 | 45.6 | 103 |
| amiodarone | 5.1 | 31.4 | 75.1 |
| Indomethacin | 6.70 | 6.69 | 9.92 |
| Aripiprazole | 3.20 | 9.26 | 8.44 |
| Melphalan | 16.1 | 38.2 | 87.8 |
| Voriconazole | 9.23 | 61.9 | 94.7 |
| Tamoxifen | 1.18 | 3.72 | 71.8 |
| Tolfenamic acid | 5.6 | 6.79 | 8.07 |
| 2-methoxyestradiol acid | 6.42 | 34.1 | 35.9 |
| MEPBZ | 5.07 | 20 | 5.54 |
Binding Preferences of M2C2
Previously, we elucidated the binding preferences of acyclic CB[n]-type container M2.21a As expected based on its aromatic-CB[n] hybrid structure M2 binds to hydrophobic or aromatic moieties in its cavity and cations at its portals. M2 displays a special affinity for steroids. Analysis of the complexation induced changes in chemical shift for the 15 drugs with M2C2 reveals similar preferences, namely cavity inclusion of the hydrophobic or aromatic moieties and portal binding of the cationic residues. Figure 6 shows the 1H NMR recorded for 2-methoxyestradiol alone in DMSO because it is insoluble in D2O, and in the presence of M2 or M2C2 in D2O. The similar pattern of significant upfield shifts observed for the protons on the steroidal nucleus of 2-methoxyestradiol (s-bb) establish a similar binding mode within the acyclic CB[n] cavity defined by the naphthalene sidewalls.
Figure 6.
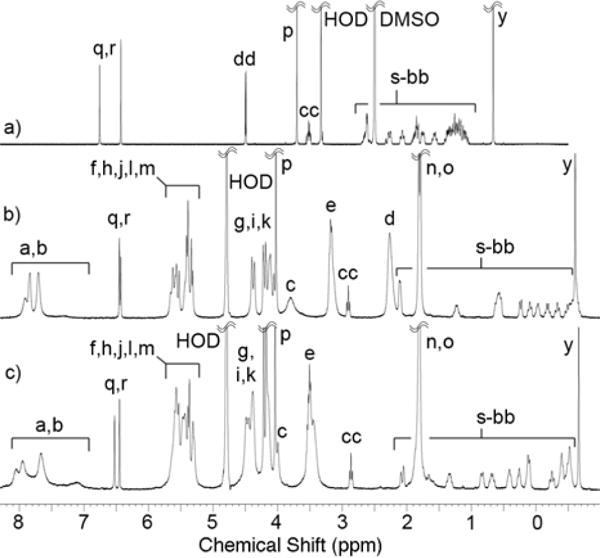
1H NMR spectra recorded (400 MHz, RT) for 2-methoxyestradiol: a) alone in DMSO-d6, or b) with M2, or c) with M2C2 in 20 mM sodium phosphate buffered D2O at pD 7.4.
Comparisons Among M1, M2, M2C2, HP-β-CD, and SBE-β-CD
Previously, we established that M2 was a better solubilizing agent than M1 and HP-β-CD according to PSD slope and Krel values.21a Inspection of the PSDs for estradiol, voriconazole, and camptothecin (Figure 4) visually confirms that M2C2 performs better as a solubilizing agent than either HP-β-CD or SBE-β-CD. The data given in Table 1 show that M2C2 also performs better than HP-β-CD as a solubilizing agent for the remaining drugs in the test set with Krel values ranging from a low of 1.5 to a high of 170 with an average of 10. The Ka values for HP-β-CD range from 0.016 – 3.6 × 104 M−1 whereas for M2C2 they range from 0.51 – 51 × 104 M−1. Several drugs (PBS-1086, camptothecin, aripiprazole) are nicely fomulated with M2C2 where HP-β-CD is unsuccessful. It is also relevant to compare the solubilizing ability of M2C2 versus highly soluble container M120a,21a toward the insoluble drugs. Solubilization data is not available for 2-methoxyestradiol or MEPBZ with M1. Among the remaining 13 containers, M2C2 is a better solubilizing agent than M1 as judged by PSD slope toward 8 drugs, whereas M2C2 and M1 are comparable solubilizing agents for PBS-1086 and tamoxifen. In only three cases (melphalan, cinnarizine, albendazole) is M1 more efficient than M2C2. The results given in Table 1 also establish that SBE-β-CD is a better solubilizing agent than HP-β-CD for all of the drugs tested in terms of PSD slope and Ka.13e The Krel value for SBE-β-CD versus HP-β-CD ranges from a low of 1.9 to a high of 60. The anionic nature of SBE-β-CD makes it a better host for the drugs in the test set, many of which are cationic.13e A comparison between the solubilizing abilities of M2C2 and SBE-β-CD is more subtle. In some cases (e.g. ethynylestradiol, tamoxifen) the slope of the PSD is substantially higher for SBE-β-CD whereas in other cases (cinnarizine, aripiprazole, voriconazole, 2-methoxyestradiol, MEPBZ) M2C2 has a substantially larger slope. In several cases, the slopes are comparable (e.g. albendazole, amiodarone, estradiol). Significantly, M2C2 – but not SBE-β-CD or HP-β-CD – was able to solubilize PBS-1086 and camptothecin. Table 2 gives the highest concentrations of drug that we achieved with M2, M2C2, and SBE-β-CD. For drugs where the slope of the PSD is significantly larger for SBE-β-CD (ethynylestradiol, tamoxifen), it is possible to prepare much higher drug concentrations due to the higher inherent solubility of SBE-β-CD (> 175 mM) relative to M2C2. Even in the case of voriconazole and amiodarone where the PSD slope is larger with M2C2, more concentrated solutions are ultimately obtained by using very high concentrations of SBE-β-CD. There are, however, situations where M2C2 clearly performs better than SBE-β-CD (e.g. PBS-1086, camptothecin, MEPBZ) both in terms of PSD slope and maximum [drug].
In Vitro Cytotoxicity Assays
Given the very good performance of M2C2 as a solubilizing agent for insoluble drugs, we wanted to take additional steps toward demonstrating the potential of M2C2 in practical applications. Accordingly, we decided to perform a series of in vitro cytotoxicity assays to assess its biocompatibility. We elected to perform complementary cell viability (MTS) and cell cytolysis (Adenylate Kinase (AK) release) assays in human cell lines of kidney (HEK 293), human liver (HepG2), and human monocyte (THP-1) origin. As can be seen in Figure 7a,b,c the cells show decreases in cell viability (metabolic activity) at concentrations greater than 1 mM. In the complementary AK release assay no significant differences were noted between the untreated control cells and cells treated with up to 10 mM of M2C2. This result demonstrates that although at doses of 1 mM M2C2 started to affect cell metabolism this did not lead to cell death. In combination, these results suggest that M2C2 shows relatively low cytotoxicity toward the investigated cell lines.
Figure 7.
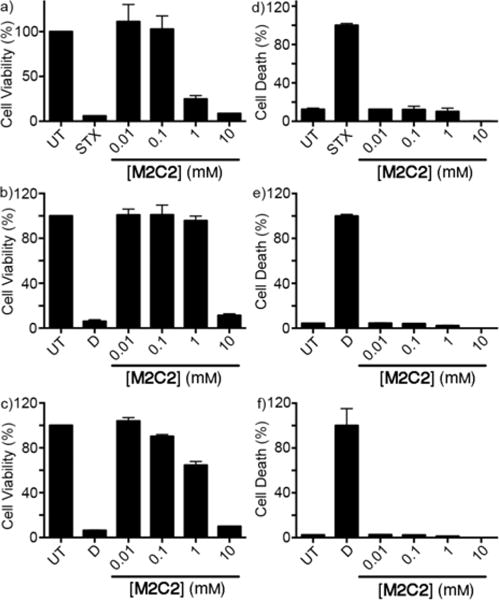
In vitro toxicology of M2C2. MTS (a, b, c) and AK release assay (d, e, f) were performed to evaluate the cell viability and the cell death of three cell lines treated with M2C2. THP-1 (a, d), HepG2 (b, e) and HEK293 (c, f). UT = untreated cells, STX = Staurosporine, D = Distilled water. Representative data from two independent experiments performed in triplicate. Error bars represent the standard deviation. Cells were treated with solutions of M2C2 for 24 hours.
Maximum Tolerated Dose Study
To assess the in vivo toxicity of M2C2 we performed a maximal tolerated dose (MTD) study using outbred Swiss Webster mice. Mice were dosed daily via bolus injection of solvent, 475 mg kg−1 M2C2, or 950 mg kg−1 M2C2 intraperitoneally for 14 consecutive days and then monitored for an additional two weeks for signs of sickness and weight change. Even at the maximal dose (950 mg/kg), the mice appeared healthy with no adverse reactions upon injection of the compound. Throughout the course of the study, the M2C2 treated mice did not differ significantly in weight from the mice treated with solvent alone (20 mM phosphate buffer, pH 7.4). We conclude that M2C2 displays no significant toxicity under the conditions of the MTD study.
Conclusions
In summary, we have reported the synthesis and characterization of M2C2 and M2C4 that are analogues of our previously reported M2 container. Relative to M2, both M2C2 and M2C4 containers possess superior solubility in aqueous solution and do not undergo strong self-association processes that would impinge upon their function as solubilizing excipients for insoluble drugs. Container M2C4 has a propensity to form gels both on its own at high concentrations and in the presence of drugs at lower concentrations. We constructed PSDs for the drugs in the test set for M2C2 and SBE-β-CD to compare their solubilizing abilities. We find that M2C2 and SBE-β-CD are complementary in that each container solubilizes some drugs better than the other in terms of both PSD slope and the maximum [drug]. In several cases M2C2 and SBE-β-CD are comparable solubilizing agents. The results of in vitro cytotoxicity tests conducted using human kidney, liver, and monocyte cells demonstrate the good biocompatibility of M2C2. In vivo maximum tolerated dose studies in mice show that the animals tolerate M2C2 at very high (950 mg kg−1) doses. As a whole, this paper demonstrates that M2C2 is an excellent solubilizing excipient for insoluble drugs that is in some cases comparable to SBE-β-CD which is used to formulate several drugs for administration to humans. In other cases (e.g. PBS-1086, camptothecin, MEPBZ) M2C2 is clearly the better solubilizing agent. Comparison of the solubilizing ability of M2C2 with M1 shows that M2C2 is as good or better than M1 in 10 out of 13 cases. The work suggests that M2C2 is a useful addition to the toolbox of solubilizing excipients to solve formulation challenges encountered by the pharmaceutical industry.
Experimental Section
Animal Studies
Animal studies were conducted at the University of Maryland, College Park, Maryland, USA, under the approval and guidance of Institutional Animal Care and Use Committee (IACUC; protocol #R-14-02).
Compound 2
1,4-Dihydroxynaphthalene (72 g, 0.45 mol), dibromoethane (400 mL, 4.64 mol), K2CO3 (250 g, 1.81 mol) and 18-crown-6 (5.77 g, 0.022 mol) were stirred in acetonitrile (400 mL) at 70°C, under nitrogen, for 3.5 days. The mixture was cooled to RT and filtered. The filtrate was concentrated by rotary evaporation. The crude solid was triturated with acetone (100 mL) and the solid was obtained by filtration. The solid was washed with the filtrate on the Büchner funnel, then with three portions of acetone (15 mL). The product was obtained as a white solid after drying under high vacuum (67 g, 40%). M.p. 126–128°C. IR (ATR, cm−1): 3438w, 2892w, 1592m, 1270s, 1099s, 766s. 1H NMR (500 MHz, CDCl3): 8.28–8.27 (m, 2H), 7.56–7.54 (m, 2H), 6.68 (s, 2H), 4.41 (t, J = 6 Hz, 4H), 3.76 (t, J = 6 Hz, 4H). 13C NMR (125 MHz, CDCl3): 148.6, 126.7, 126.4, 122.0, 105.1, 68.7, 29.6.
Compound 3
A solution of 2 (40.2 g, 107.5 mmol) and Na2SO3 (29.8 g, 236.4 mmol) in H2O/DMF (1:1, 400 mL) under N2 were stirred at 100°C for 24 h. The mixture was cooled to RT and filtered. The filtrate was poured into acetone (2 L) and the precipitate was obtained by filtration and dried on the filter funnel overnight. The crude solid was dissolved in boiling water (86 mL) and then EtOH (250 mL) was added to precipitate the product. The mixture was cooled to room temperature, filtered and the product was obtained as a white solid after drying under high vacuum (36.7 g, 81%). M.p.>300°C (dec.). IR (ATR, cm−1): 3438w, 2941w, 1595w, 1200s, 1043s, 767m. 1H NMR (400 MHz, D2O): 8.28–8.26 (m, 2H), 7.62–7.59 (m, 2H), 7.00 (s, 2H), 4.54 (t, J = 6 Hz, 4H), 3.50 (t, J = 6 Hz, 4H). 13C NMR (125 MHz, D2O, dioxane as reference): 151.0, 129.3, 129.0, 124.6, 109.2, 69.5, 53.4. Low-Resolution MS (ESI): m/z 375 ([M+H-2Na] −, calculated for C14H15O8S2−: 375).
Container M2C2
To a solution of 628 (7.49 g, 9.59 mmol) in TFA/Ac2O (1:1, 90 mL) was added 3 (12.1 g, 28.78 mmol) and the mixture was stirred at 70°C for 2 h, under nitrogen. The mixture was cooled to RT and poured into 700 mL of EtOH. The solid was filtered and washed on the filter funnel with three portions of EtOH (150 mL) and then dried on the filter funnel. The crude solid was dissolved in 250 mL of water and the pH was adjusted to pH 7 with a concentrated NaOH solution. Acetonitrile (1.1 L) was added to the aqueous solution to precipitate the product. The solid was filtered. The solid was dissolved in boiling water (100 mL), then acetonitrile (300 mL) was added and the mixture was cooled to RT. The product was obtained by filtration after drying under high vacuum as a white solid (8.77 g, 58%). M.p.>300°C. IR (ATR, cm−1): 3349w, 2941w, 1722s, 1468s, 1172s, 1033s, 798m. 1H NMR (600 MHz, D2O): 7.72–7.71 (m, 4H), 7.04–7.02 (m, 4H), 5.51 (d, J = 16 Hz, 4H), 5.50 (d, J = 15 Hz, 2H), 5.37 (d, J = 16 Hz, 4H), 5.35 (d, J = 9 Hz, 2H), 5.24 (d, J = 9 Hz, 2H), 4.54 (d, J = 16 Hz, 4H), 4.46–4.42 (m, 4H), 4.19 (d, J = 16 Hz, 4H), 4.19–4.15 (m, 4H), 3.94 (d, J = 15 Hz, 2H), 3.49 (t, J = 7 Hz, 8H), 1.78 (s, 6H), 1.77 (s, 6H). 13C NMR (125 MHz, D2O, dioxane as reference): 158.5, 157.6, 150.0, 129.5, 129.4, 128.6, 124.5, 80.4, 79.1, 73.4, 73.2, 72.1, 55.0, 52.7, 50.5, 38.0, 17.4, 16.8. HR-MS (ESI): m/z 769.1329 ([M-2Na]2−, calculated for C58H60N16Na2O24S42−: 769.1328).
Compound 5
A solution of 1,4-butanesultone (13.04 g, 96 mmol) in 1,4-dioxane (79 mL) was added to a solution of 1,4-dihydroxynaphtalene (6.14 g, 38 mmol) and NaOH (4.9 g, 123 mmol) in water (49 mL). The mixture was stirred at RT for 12 h. MeOH (200 mL) was added to the solution and the precipitate was isolated by filtration. The solid was dissolved in water (35 mL) and re-precipitated by addition of EtOH (70 mL). After filtration and drying under high vacuum, the product (5) was obtained as a light gray solid (4.69 g, 26%). M.p.>300°C (dec.). IR (ATR, cm−1): 3417w, 2922w, 1594w, 1185s, 1040s, 768m. 1H NMR (400 MHz, D2O): 8.25–8.22 (m, 2H), 7.62–7.59 (m, 2H), 6.97 (s, 2H), 4.22 (t, J = 5.5 Hz, 4H), 3.02 (t, J = 7.5 Hz, 4H), 2.02–1.99 (m, 8H). 13C NMR (125 MHz, D2O, dioxane as reference): 148.7, 126.9, 126.6, 122.1, 106.8, 69.2, 51.5, 28.2, 21.9. Low-Resolution MS (ESI): m/z 215 ([M-2Na]2−, calculated for C18H22O8S22−: 215).
Container M2C4
To a solution of 628 (1.14 g, 1.46 mmol) in TFA/Ac2O (1:1, 14 mL) was added 5 (2.40 g, 5.04 mmol) and the mixture was stirred at 70 °C for 3 h, under nitrogen. The reaction mixture was cooled to RT and poured into EtOH (100 mL). The crude solid was obtained by filtration and dried on the filter funnel. The crude solid was dissolved in water (10 mL) and the pH was adjusted to pH 7 with a concentrated NaOH solution. EtOH (40 mL) was added to the boiling aqueous solution to precipitate the product and the mixture was cooled to RT. The solid was obtained by filtration. The crude solid was dissolved in boiling water (5 mL) and then reprecipitated by the addition of EtOH (5 mL) and the mixture was cooled to RT and the solid was obtained by filtration. A second re-precipitation under the same conditions delivered the product as a white solid after drying under high vacuum (1.06 g, 43%). M.p.>300°C. IR (ATR, cm−1): 3438w, 2942w, 1722s, 1468s, 1177s, 1036s, 798m. 1H NMR (400 MHz, D2O): 7.90–7.88 (m, 4H), 7.47–7.45 (m, 4H), 5.54 (d, J = 15 Hz, 2H), 5.51 (d, J = 16 Hz, 4H), 5.40 (d, J = 9 Hz, 2H), 5.38 (d, J = 16 Hz, 4H), 5.29 (d, J = 9 Hz, 2H), 4.42 (d, J = 16 Hz, 4H), 4.24 (d, J = 16 Hz, 4H), 4.10–4.05 (m, 4H), 4.00 (d, J = 15 Hz, 2H), 3.85–3.80 (m, 4H), 2.93–2.77 (m, 8H), 1.96–1.86 (m, 8H), 1.86–1.71 (m, 8H), 1.76 (s, 12H). 13C NMR (125 MHz, D2O, dioxane as reference): 158.5, 157.7, 150.2, 129.7, 129.2, 128.5, 124.5, 80.5, 79.2, 77.3, 73.4, 73.1, 54.9, 52.8, 50.5, 38.1, 30.4, 23.0, 17.4, 16.9. HR-MS (ESI): m/z 825.1954 ([M-2Na]2−, calculated for C66H76N16Na2O24S42−: 825.1954).
Supplementary Material
Figure 8.
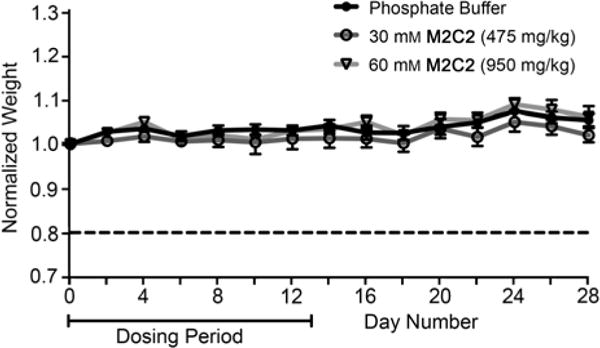
Female Swiss Webster mice (n = 7 per group) were dosed intraperitoneally with solvent (phosphate buffer), 475 mg/kg M2C2, or 950 mg/kg M2C2 for 14 consecutive days. Weight change is represented as the group average (n = 7) of mouse weight at day n to weight at day 0. Error bars represent standard error of the mean.
Acknowledgments
We thank the National Institutes of Health (CA168365) for financial support of this work. Shane Falcinelli was supported by an HHMI undergraduate research fellowship. We thank Dr. Xiaoyong Lu for performing mass spectrometric measurements.
Footnotes
Supporting information for this article is given via a link at the end of the document.
Contributor Information
Volker Briken, Email: vbriken@umd.edu.
Lyle Isaacs, Email: LIsaacs@umd.edu.
References
- 1.a) Pluth MD, Raymond KN. Chem Soc Rev. 2007;36:161–171. doi: 10.1039/b603168b. [DOI] [PubMed] [Google Scholar]; b) Northrop BH, Zheng YR, Chi KW, Stang PJ. Acc Chem Res. 2009;42:1554–1563. doi: 10.1021/ar900077c. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Diederich F. Angew Chem. Intl Ed Engl. 1988;27:362–386. [Google Scholar]; d) Cram DJ. Angew Chem. Int Ed Engl. 1988;27:1009–1020. [Google Scholar]; e) Lehn JM. Angew Chem. Int Ed Engl. 1988;27:89–112. [Google Scholar]; f) Ogoshi T, Kanai S, Fujinami S, Yamagishi TA, Nakamoto Y. J Am Chem Soc. 2008;130:5022–5023. doi: 10.1021/ja711260m. [DOI] [PubMed] [Google Scholar]; g) Masson E, Ling X, Joseph R, Kyeremeh-Mensah L, Lu X. RSC Adv. 2012;2:1213–1247. [Google Scholar]; h) Gutsche CD. Acc Chem Res. 1983;16:161–170. [Google Scholar]; i) Boehmer V. Angew Chem. Int Ed Engl. 1995;34:713–745. [Google Scholar]; j) Rekharsky MV, Inoue Y. Chem Rev. 1998;98:1875–1917. doi: 10.1021/cr970015o. [DOI] [PubMed] [Google Scholar]; k) Barnes JC, Juricek M, Strutt NL, Frasconi M, Sampath S, Giesener MA, McGrier PL, Bruns CJ, Stern CL, Sarjeant AA, Stoddart JF. J Am Chem Soc. 2012;135:183–192. doi: 10.1021/ja307360n. [DOI] [PubMed] [Google Scholar]; l) Rebek J. Acc Chem Res. 2009;42:1660–1668. doi: 10.1021/ar9001203. [DOI] [PubMed] [Google Scholar]; m) Xue M, Yang Y, Chi X, Zhang Z, Huang F. Acc Chem Res. 2012;45:1294–1308. doi: 10.1021/ar2003418. [DOI] [PubMed] [Google Scholar]; n) Kay ER, Leigh DA. Angew Chem. Int Ed. 2015;54:10080–10088. doi: 10.1002/anie.201503375. [DOI] [PMC free article] [PubMed] [Google Scholar]; o) Hermann K, Ruan Y, Hardin AM, Hadad CM, Badjic JD. Chem Soc Rev. 2015;44:500–514. doi: 10.1039/c4cs00140k. [DOI] [PubMed] [Google Scholar]; p) Lin Z, Emge TJ, Warmuth R. Chem - Eur J. 2011;17:9395–9405. doi: 10.1002/chem.201100527. [DOI] [PubMed] [Google Scholar]; q) Zarra S, Wood DM, Roberts DA, Nitschke JR. Chem Soc Rev. 2015;44:419–432. doi: 10.1039/c4cs00165f. [DOI] [PubMed] [Google Scholar]; r) Yoshizawa M, Klosterman J, Fujita M. Angew Chem. Int Ed. 2009;48:3418–3438. doi: 10.1002/anie.200805340. [DOI] [PubMed] [Google Scholar]
- 2.Mal P, Breiner B, Rissanen K, Nitschke JR. Science. 2009;324:1697–1699. doi: 10.1126/science.1175313. [DOI] [PubMed] [Google Scholar]
- 3.Juricek M, Strutt NL, Barnes JC, Butterfield AM, Dale EJ, Baldridge KK, Stoddart JF, Siegel JS. Nat Chem. 2014;6:222–228. doi: 10.1038/nchem.1842. [DOI] [PubMed] [Google Scholar]
- 4.Pluth MD, Bergman RG, Raymond KN. Science. 2007;316:85–88. doi: 10.1126/science.1138748. [DOI] [PubMed] [Google Scholar]
- 5.You L, Zha D, Anslyn EV. Chem Rev. 2015;115:7840–7892. doi: 10.1021/cr5005524. [DOI] [PubMed] [Google Scholar]
- 6.a) Liu S, Russell DH, Zinnel NF, Gibb BC. J Am Chem Soc. 2013;135:4314–4324. doi: 10.1021/ja310741q. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ajami D, Liu L, Rebek J., Jr Chem Soc Rev. 2015;44:490–499. doi: 10.1039/c4cs00065j. [DOI] [PubMed] [Google Scholar]
- 7.a) Mock WL, Shih NY. J Org Chem. 1986;51:4440–4446. [Google Scholar]; b) Liu S, Ruspic C, Mukhopadhyay P, Chakrabarti S, Zavalij PY, Isaacs L. J Am Chem Soc. 2005;127:15959–15967. doi: 10.1021/ja055013x. [DOI] [PubMed] [Google Scholar]; c) Cao L, Šekutor M, Zavalij PY, Mlinaricć-Majerski K, Glaser R, Isaacs L. Angew Chem. Int Ed. 2014;53:988–993. doi: 10.1002/anie.201309635. [DOI] [PubMed] [Google Scholar]; d) Isaacs L. Acc Chem Res. 2014;47:2052–2062. doi: 10.1021/ar500075g. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Moghaddam S, Yang C, Rekharsky M, Ko YH, Kim K, Inoue Y, Gilson MK. J Am Chem Soc. 2011;133:3570–3581. doi: 10.1021/ja109904u. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Biedermann F, Uzunova VD, Scherman OA, Nau WM, De Simone A. J Am Chem Soc. 2012;134:15318–15323. doi: 10.1021/ja303309e. [DOI] [PubMed] [Google Scholar]; g) Shetty D, Khedkar JK, Park KM, Kim K. Chem Soc Rev. 2015;44:8747–8761. doi: 10.1039/c5cs00631g. [DOI] [PubMed] [Google Scholar]
- 8.a) Mock WL, Irra TA, Wepsiec JP, Adhya M. J Org Chem. 1989;54:5302–5308. [Google Scholar]; b) Pemberton BC, Raghunathan R, Volla S, Sivaguru J. Chem Eur J. 2012;18:12178–12190. doi: 10.1002/chem.201202083. [DOI] [PubMed] [Google Scholar]; c) Zheng L, Sonzini S, Ambarwati M, Rosta E, Scherman OA, Herrmann A. Angew Chem. Int Ed. 2015;54:13007–13011. doi: 10.1002/anie.201505628. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Jon SY, Ko YH, Park SH, Kim HJ, Kim K. Chem Commun. 2001 doi: 10.1039/b105153a. 1938–1939. [DOI] [PubMed] [Google Scholar]
- 9.a) Dsouza R, Hennig A, Nau W. Chem Eur J. 2012;18:3444–3459. doi: 10.1002/chem.201103364. [DOI] [PubMed] [Google Scholar]; b) Ghale G, Lanctot AG, Kreissl HT, Jacob MH, Weingart H, Winterhalter M, Nau WM. Angew Chem. Int Ed. 2014;53:2762–2765. doi: 10.1002/anie.201309583. [DOI] [PubMed] [Google Scholar]; c) Gong B, Choi BK, Kim JY, Shetty D, Ko YH, Selvapalam N, Lee NK, Kim K. J Am Chem Soc. 2015;137:8908–8911. doi: 10.1021/jacs.5b05385. [DOI] [PubMed] [Google Scholar]
- 10.a) Barrow SJ, Kasera S, Rowland MJ, del Barrio J, Scherman OA. Chem Rev. 2015;115:12320–12406. doi: 10.1021/acs.chemrev.5b00341. [DOI] [PubMed] [Google Scholar]; b) Dang D, Nguyen H, Merkx M, Brunsveld L. Angew Chem. Int Ed. 2013;52:2915–2919. doi: 10.1002/anie.201208239. [DOI] [PubMed] [Google Scholar]; c) Walsh Z, Janecek ER, Hodgkinson JT, Sedlmair J, Koutsioubas A, Spring DR, Welch M, Hirschmugl CJ, Toprakcioglu C, Nitschke JR, Jones M, Scherman OA. Proc Natl Acad Sci U S A. 2014;111:17743–17748. doi: 10.1073/pnas.1406037111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Lim S, Kim H, Selvapalam N, Kim KJ, Cho SJ, Seo G, Kim K. Angew Chem Int Ed. 2008;47:3352–3355. doi: 10.1002/anie.200800772. [DOI] [PubMed] [Google Scholar]; b) Tian JA, Ma SQ, Thallapally PK, Fowler D, McGrail BP, Atwood JL. Chem Commun. 2011;47:7626–7628. doi: 10.1039/c1cc12689j. [DOI] [PubMed] [Google Scholar]; c) Miyahara Y, Abe K, Inazu T. Angew Chem. Int Ed. 2002;41:3020–3023. doi: 10.1002/1521-3773(20020816)41:16<3020::AID-ANIE3020>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 12.a) Hauss DJ. Adv Drug Delivery Rev. 2007;59:667–676. doi: 10.1016/j.addr.2007.05.006. [DOI] [PubMed] [Google Scholar]; b) Lipinski CA. J Pharmacol Toxicol Methods. 2000;44:235–249. doi: 10.1016/s1056-8719(00)00107-6. [DOI] [PubMed] [Google Scholar]
- 13.a) Leuner C, Dressman J. Eur J Pharmaceut Biopharmaceut. 2000;50:47–60. doi: 10.1016/s0939-6411(00)00076-x. [DOI] [PubMed] [Google Scholar]; b) Muller RH, Keck CM. J Biotechnol. 2004;113:151–170. doi: 10.1016/j.jbiotec.2004.06.007. [DOI] [PubMed] [Google Scholar]; c) Serajuddin ATM. Adv Drug Delivery Rev. 2007;59:603–616. doi: 10.1016/j.addr.2007.05.010. [DOI] [PubMed] [Google Scholar]; d) Stella VJ, Nti-Addae KW. Adv Drug Delivery Rev. 2007;59:677–694. doi: 10.1016/j.addr.2007.05.013. [DOI] [PubMed] [Google Scholar]; e) Okimoto K, Rajewski RA, Uekama K, Jona JA, Stella VJ. Pharm Res. 1996;13:256–264. doi: 10.1023/a:1016047215907. [DOI] [PubMed] [Google Scholar]; f) Stella VJ, Rajewski RA. Pharm Res. 1997;14:556–567. doi: 10.1023/a:1012136608249. [DOI] [PubMed] [Google Scholar]
- 14.a) Ghosh I, Nau WM. Adv Drug Delivery Rev. 2012;64:764–783. doi: 10.1016/j.addr.2012.01.015. [DOI] [PubMed] [Google Scholar]; b) Dong N, Wang X, Pan J, Tao Z. Acta Chim Sinica. 2011;69:1431–1437. [Google Scholar]; c) Walker S, Oun R, McInnes FJ, Wheate NJ. Isr J Chem. 2011;51:616–624. [Google Scholar]; d) Zhao Y, Buck DP, Morris DL, Pourgholami MH, Day AI, Collins JG. Org Biomol Chem. 2008;6:4509–4515. doi: 10.1039/b813759e. [DOI] [PubMed] [Google Scholar]
- 15.a) Uzunova VD, Cullinane C, Brix K, Nau WM, Day AI. Org Biomol Chem. 2010;8:2037–2042. doi: 10.1039/b925555a. [DOI] [PubMed] [Google Scholar]; b) Hettiarachchi G, Nguyen D, Wu J, Lucas D, Ma D, Isaacs L, Briken V. PLoS One. 2010;5:e10514. doi: 10.1371/journal.pone.0010514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.a) Chen H, Y-W CJ, Li S, Liu JJ, Wyman I, Lee SM-Y, Macartney DH, Wang R. RSC Adv. 2015;5:63745–63752. [Google Scholar]; b) Li S, Chen H, Yang X, Bardelang D, Wyman IW, Wan J, Lee SMY, Wang R. ACS Med Chem Lett. 2015;6:1174–1178. doi: 10.1021/acsmedchemlett.5b00372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang J, Coulston RJ, Jones ST, Geng J, Scherman OA, Abell C. Science. 2012;335:690–694. doi: 10.1126/science.1215416. [DOI] [PubMed] [Google Scholar]
- 18.a) Cao L, Hettiarachchi G, Briken V, Isaacs L. Angew Chem. Int Ed. 2013;52:12033–12037. doi: 10.1002/anie.201305061. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kim E, Kim D, Jung H, Lee J, Paul S, Selvapalam N, Yang Y, Lim N, Park CG, Kim K. Angew Chem. Int Ed. 2010;49:4405–4408. doi: 10.1002/anie.201000818. [DOI] [PubMed] [Google Scholar]
- 19.a) Huang WH, Zavalij PY, Isaacs L. J Am Chem Soc. 2008;130:8446–8454. doi: 10.1021/ja8013693. [DOI] [PubMed] [Google Scholar]; b) Lucas D, Minami T, Iannuzzi G, Cao L, Wittenberg JB, Anzenbacher P, Isaacs L. J Am Chem Soc. 2011;133:17966–17976. doi: 10.1021/ja208229d. [DOI] [PubMed] [Google Scholar]
- 20.a) Ma D, Hettiarachchi G, Nguyen D, Zhang B, Wittenberg JB, Zavalij PY, Briken V, Isaacs L. Nat Chem. 2012;4:503–510. doi: 10.1038/nchem.1326. [DOI] [PubMed] [Google Scholar]; b) Hettiarachchi G, Samanta S, Falcinelli S, Zhang B, Moncelet D, Isaacs L, Briken V. Mol Pharmaceut. 2016 doi: 10.1021/acs.molpharmaceut.1025b00723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.a) Zhang B, Isaacs L. J Med Chem. 2014;57:9554–9563. doi: 10.1021/jm501276u. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gilberg L, Zhang B, Zavalij PY, Sindelar V, Isaacs L. Org Biomol Chem. 2015;13:4041–4050. doi: 10.1039/c5ob00184f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.CCDC-1452394 (M2C4) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from the cambridge crystallographic data centre via http://www.Ccdc.Cam.Ac.Uk/data_request/cif.
- 23.a) Higuchi T, Connors KA. Adv Anal Chem Inst. 1965;4:117–212. [Google Scholar]; b) Connors KA. Binding constants. John Wiley & Sons; New York: p. 1987. [Google Scholar]
- 24.Fabre C, Mimura N, Bobb K, Kong SY, Gorgun G, Cirstea D, Hu J, Minami J, Ohguchi H, Zhang J, Meshulam J, Carrasco RD, Tai YT, Richardson PG, Hideshima T, Anderson KC. Clin Cancer Res. 2012;18:4669–4681. doi: 10.1158/1078-0432.CCR-12-0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mandadapu V. PhD dissertation. University of New South Wales; Canberra, Australia: 2014. [Google Scholar]
- 26.a) Ma D, Zhang B, Hoffmann U, Sundrup MG, Eikermann M, Isaacs L. Angew Chem. Int Ed. 2012;51:11358–11362. doi: 10.1002/anie.201206031. [DOI] [PubMed] [Google Scholar]; b) Hoffmann U, Grosse-Sundrup M, Eikermann-Haerter K, Zaremba S, Ayata C, Zhang B, Ma D, Isaacs L, Eikermann M. Anesthesiology. 2013;119:317–325. doi: 10.1097/ALN.0b013e3182910213. [DOI] [PubMed] [Google Scholar]; c) Haerter F, Simons JCP, Foerster U, Duarte IM, Diaz-Gil D, Eikermann-Haerter K, Ayata C, Ganapati S, Zhang B, Blobner M, Isaacs L, Eikermann M. Anesthesiology. 2015;123:1337–1349. doi: 10.1097/ALN.0000000000000868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Biedermann F, Nau WM, Schneider HJ. Angew Chem. Int Ed. 2014;53:11158–11171. doi: 10.1002/anie.201310958. [DOI] [PubMed] [Google Scholar]
- 28.Ma D, Zavalij PY, Isaacs L. J Org Chem. 2010;75:4786–4795. doi: 10.1021/jo100760g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.