

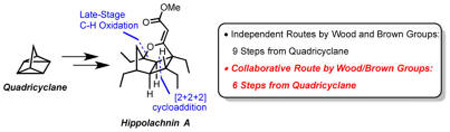
Abstract
Described herein are synthetic efforts toward the synthesis of hippolachnin A. Two independently devised routes from the Brown and Wood groups allowed for the synthesis of hippolachnin A from the unusual starting material, quadricyclane, by harnessing the power of late-stage C–H oxidation. Collaborative union of the best features of the two routes allowed for preparation of the molecule with improved efficiency.
Graphical Abstract
INTRODUCTION
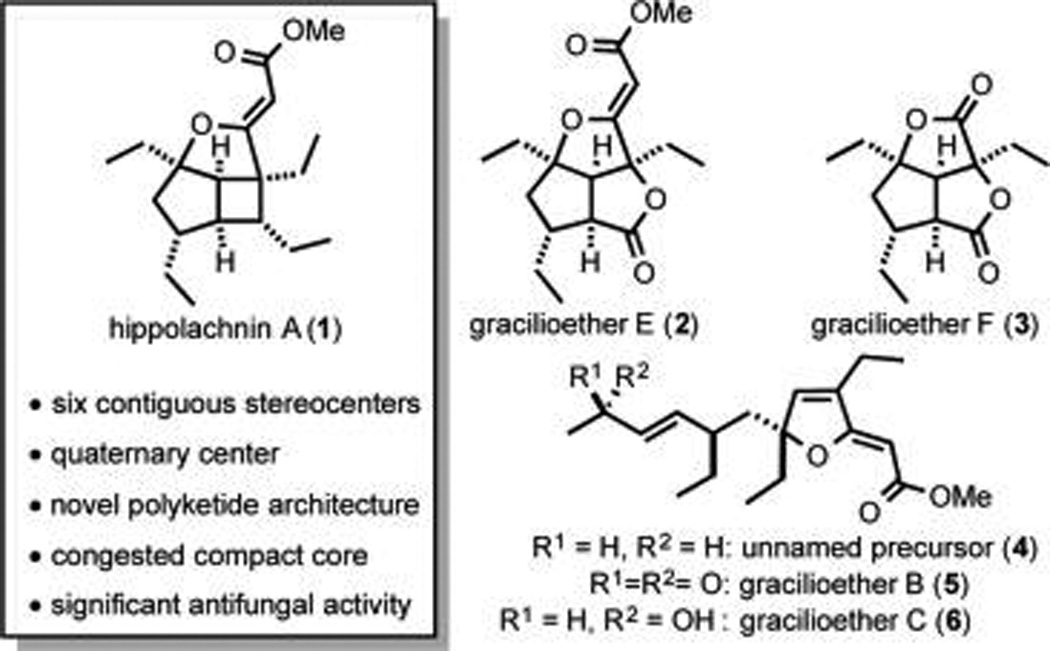
Hippolachnin A (1, Figure 1) is a complex natural product recently isolated from the marine sponge Hippospongia lachne (5.1 mg of hippolachnin A from 3.6 kg of sponge, 0.00014%) along with its proposed biogenic precursor (4).1 This polyketide-based molecule has an unprecedented carbon skeleton that bears six contiguous stereogenic centers. Preliminary biological evaluation of hippolachnin A (1) led to the discovery that it is a potent antifungal agent possessing activity against several species, including Cryptococcus neoformans (410 nM), Trichophyton rubrum (410 nM), and Candida glabrata (1.62 µM).1 Notably, treatments for C. neoformans infections are one of the most important unmet objectives in clinical mycology which, coupled with preliminary toxicity data illustrating inactivity against HCT-116, HeLa, and A549 human cell lines, renders hippolachnin A (1) an intriguing and medicinally important target for synthesis.2
Figure 1.
Hippolachnin A and related compounds.
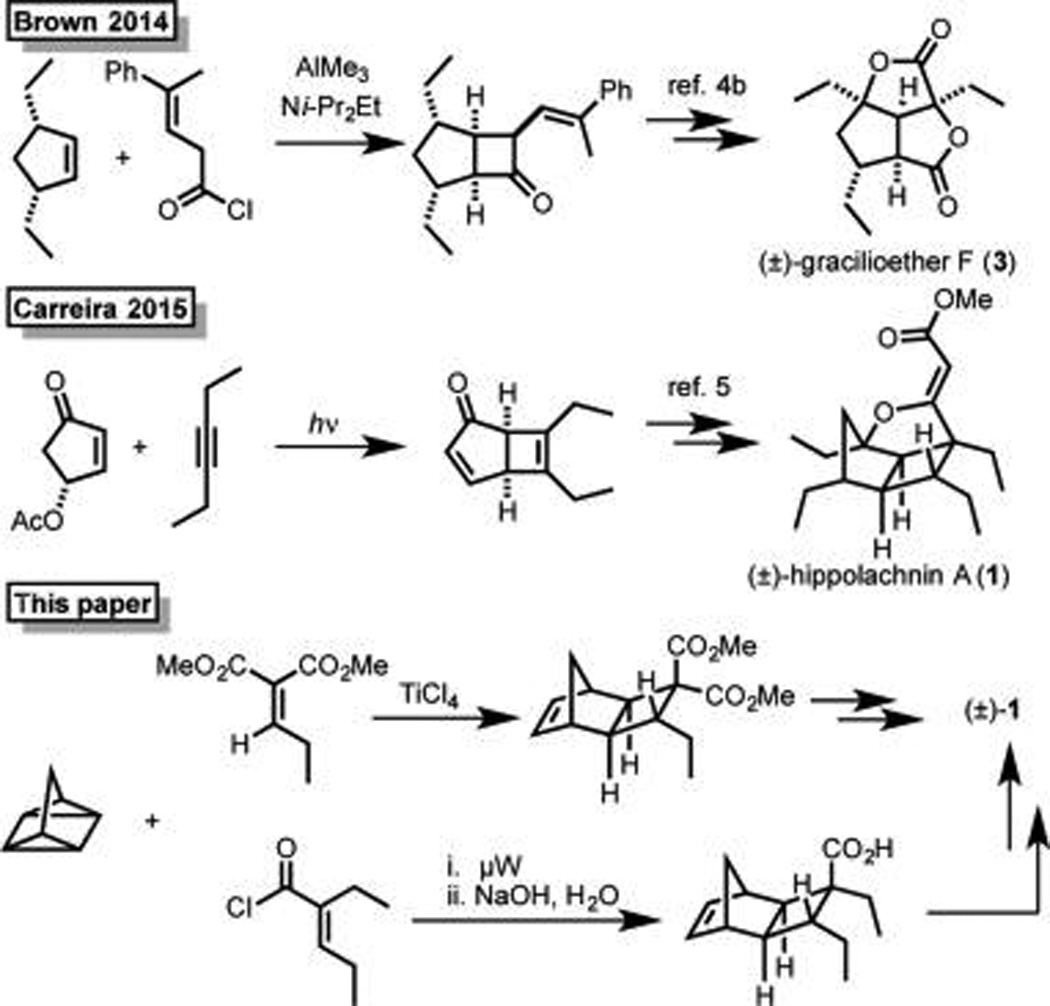
Hippolachnin A exhibits structural similarities to several other marine polyketides (2–6)3 which, given their intriguing structures and biological activity, have all attracted substantial attention from the synthetic community (Scheme 1). Early work from Ohira led to the synthesis of 4;4a more recently, syntheses of gracilioether F (3), and its proposed biogenic precursors gracilioethers B and C (5 and 6), were completed by two of us (C.M.R. and M.K.B.) and the Perkins group, respectively.4b,c In early 2015, Carreira and co-workers reported the first total synthesis of (±)-hippolachnin A utilizing a photochemical [2 + 2] reaction to establish the cyclobutane core (Scheme 1).5,6 Very recently, the Carreira group reported a synthesis of gracilioethers E (2) and F (3).7 Herein, we describe a short synthesis of (±)-hippolachnin A (1) that was devised in a collaborative effort based upon independent syntheses developed in the Wood and Brown groups. As outlined below, this synthetic strategy evolved from our recognition that each independently developed synthesis contains elements complementary to the other and that we could thereby provide better access to these important antifungal agents by combining our efforts.
Scheme 1.
Previous Synthetic Strategies
RESULTS AND DISCUSSION
Common Retrosynthesis
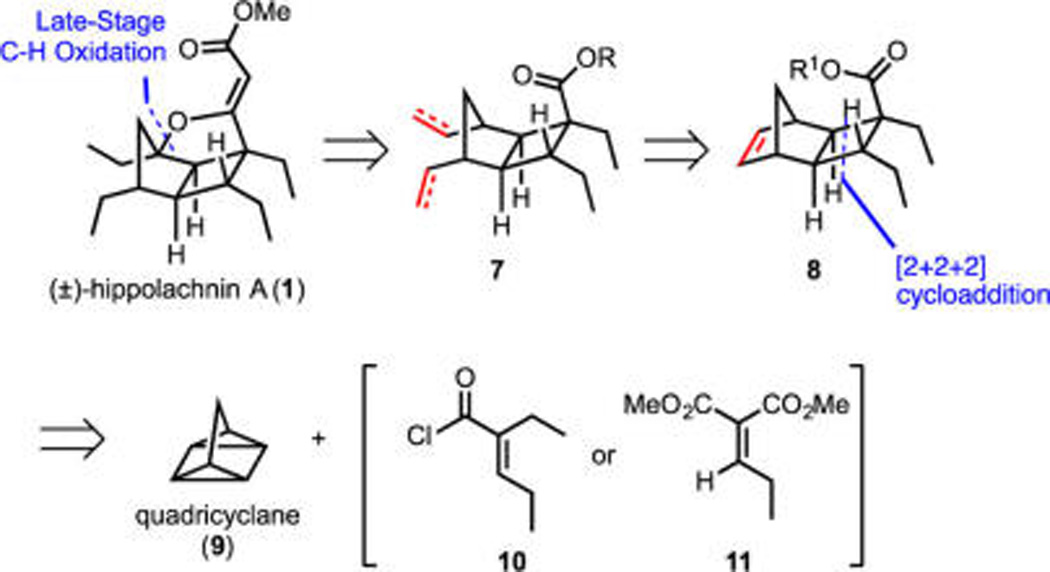
As alluded to above, the independently devised and combined routes described herein share several strategically simplifying disconnections. Illustrated retrosynthetically in Scheme 2 are key elements of these synthetic plans, both of which incorporate late-stage construction of the furan ring via C–H functionalization, a maneuver that8 allows propagation of a latent symmetry element via double processing of two terminal olefins (7 to 8) derived from ring-opening cross metathesis (ROCM) of a tricyclic intermediate (8). Variants of the latter were envisioned as arising from [2π + 2σ + 2σ] cycloadditions of the unusual hydrocarbon quadricyclane (9).9,10 To the best of our knowledge, prior to the current studies, quadricyclane had yet to be employed in complex molecule synthesis, despite being known for decades. Moreover, as this synthesis clearly exemplifies, recent advances in C–H activation chemistry have begun to impact synthetic strategy development to a point where the ubiquitous C–H bond can be viewed as functionalizable, thus rendering compounds like the quintessential symmetric hydrocarbon quadricyclane exceedingly rich in functionality.11
Scheme 2.
Retrosynthesis
The Brown Route
As illustrated in Table 1, in their studies toward 1, C.M.R. and M.K.B. began by attempting the cycloaddition of quadricyclane (9) and electron deficient alkene 12.12 However, even at elevated temperatures and extended reaction times, <2% of the desired cycloadduct (13) was observed (Table 1, entry 1). It should be noted that thermal cycloreversion to norbornadiene is a competing process at higher temperature (t1/2 > 14 h at 140 °C);13 thus, application of more forcing conditions does not lead to improved results. On the basis of literature precedent indicating that more electron deficient alkenes are better substrates for cycloaddition with quadricyclane,12,14 efforts turned toward the use of acid chloride 10 which proved sufficiently electron poor to allow for formation of 14a in 73% yield and 3:1 dr (Table 1, entry 2). Further, reaction optimization revealed that use of microwave conditions at 140 °C for 4 h (vs conventional heating at 120 °C for 3 days) led to formation of 14a in 72% yield and 5:1 dr (Table 1, entry 5). The corresponding carboxylic acid (14b), which was obtained after quench of the reaction mixture with NaOH (aq), was easily recrystallized to provide 14b in >20:1 dr and 50% yield wherein five of the six contiguous stereocenters are set and access to the sixth is staged.15
Table 1.
Quadricyclane Cycloaddition
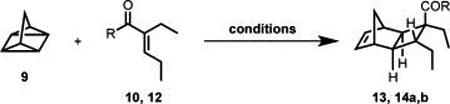 | ||||
|---|---|---|---|---|
| entry | R | conditions | product | yield (dr)a |
| 1 | OEt(12) | 140 °C, 4 equiv 9, 48 h | (OEt) 13 | <2% |
| 2 | Cl (10) | 120 °C, 4 equiv 9, 72 h | (Cl) 14a | 73% (3:1 dr) |
| 3 | Cl (10) | MW, 110 °C, 5 equiv 9, 2 h | (Cl) 14a | 31% (9:1 dr) |
| 4 | Cl (10) | MW, 110 °C, 5 equiv 9, 10 h | (Cl) 14a | 68% (6:1 dr) |
| 5 | Cl (10) | MW, 140 °C, 5 equiv 9, 4 h | (Cl) 14a | 74% (5:1 dr)b |
Determined by 1H NMR analysis with an internal standard.
A 50% isolated yield, >20:1 dr, recrystallization of the corresponding carboxylic acid (14b, R = OH) after quench with NaOH (aq).
With an efficient route to acid 14b in hand, the Brown group explored the ring opening cross metathesis (ROCM)/hydrogenation sequence. It was determined that treatment of 14b with the Grubbs first generation catalyst16 under an atmosphere of ethylene followed by H2 purge and addition of 10 mol % Pd/ C led to formation of 15 in >95% yield (Scheme 3). While it is known that the Grubbs catalyst can also promote hydrogenation,17 it proved easier to add a catalytic amount of Pd/C to complete the reduction.
Scheme 3.
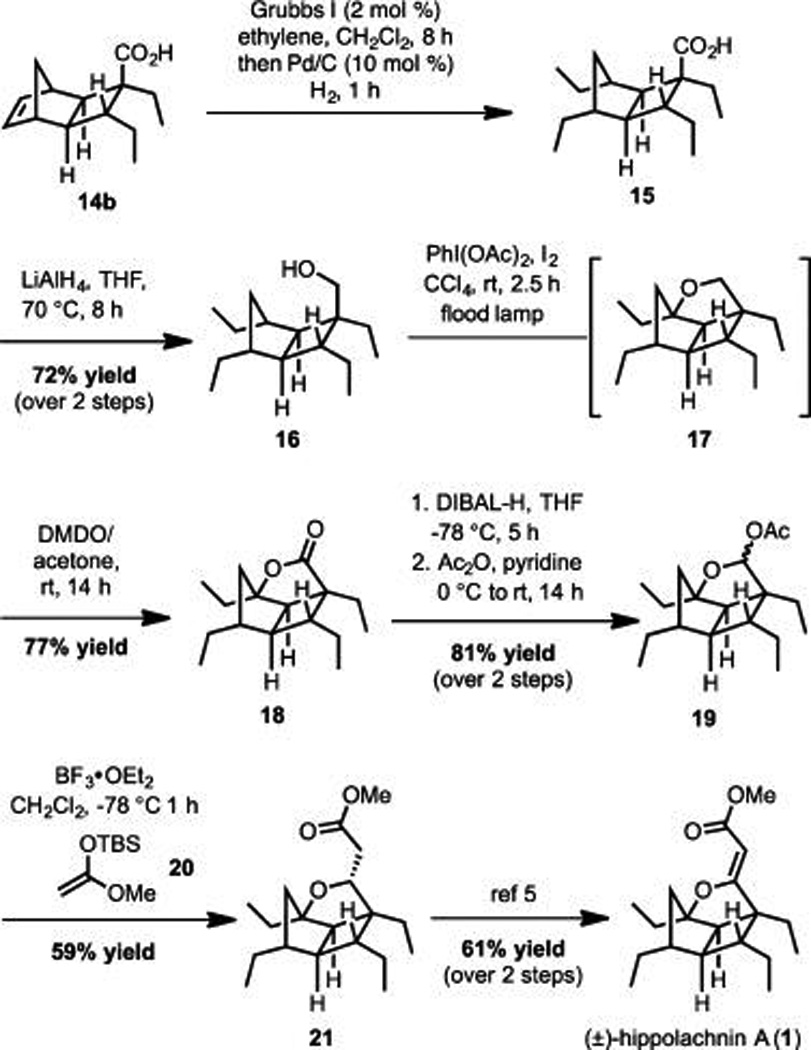
Brown Synthesis of Hippolachnin A
On the basis of previous work in the Brown group toward the synthesis of gracilioether F (3), it was envisioned that a carboxylic acid directed Csp3-H oxidation would be effective for introducing a lactone ring and setting the final stereogenic center.4a However, under the two sets of conditions that were applied in the gracilioether F synthesis, less than optimal results toward hippolachnin A (1) were observed (see the Supporting Information for details). Copper-promoted oxidation of 15, surprisingly, led to complete recovery of the starting material. Use of an Fe-catalyzed carboxylic acid directed C–H oxidation developed by the White group18 did lead to formation of the desired product (18); however, low yield, formation of over oxidation products, and complete consumption of the starting material led us to reevaluate our synthetic strategy.
Due to the difficulties associated with carboxylic acid directed Csp3-H oxidation, it was reasoned that use of the corresponding alcohol and remote oxidation by established 1,5-hydrogen atom abstraction protocols might be more effective.19 Synthesis of the requisite alcohol (16) was accomplished by reduction of acid 15 with LiAlH4 (72% yield over two steps from acid 14b, Scheme 3).
The key late stage Csp3-H oxidation was accomplished by treatment of alcohol 16 with PhI(OAc)2 and I2 under flood lamp irradiation (Scheme 3). These conditions, originally identified by Suárez and co-workers,20 led to formation of furan 17. Since the lactone was desired, subsequent addition of DMDO/acetone directly to the reaction mixture allowed for further oxidation to generate 18 in 77% yield from alcohol 16 in a single step.
Conversion of lactone 18 to hippolachnin A (1) proved more challenging than expected. Initial efforts focusing on olefination and Clasien condensation procedures (including the method described in Scheme 4 for conversion of 26 to 1) proved unsuccessful. After significant experimentation, a protocol was devised that commenced with reduction of the lactone and subsequent acylation of the resultant lactol to form 19 (Scheme 3). Ensuing Lewis acid-promoted addition of silyl ketene acetal 20 to acetal 19 generated 21 in 59% yield. This compound intercepts Carreira’s penultimate intermediate, which was advanced to hippolachnin A (1) through a twostep sequence in 61% yield.5
Scheme 4.
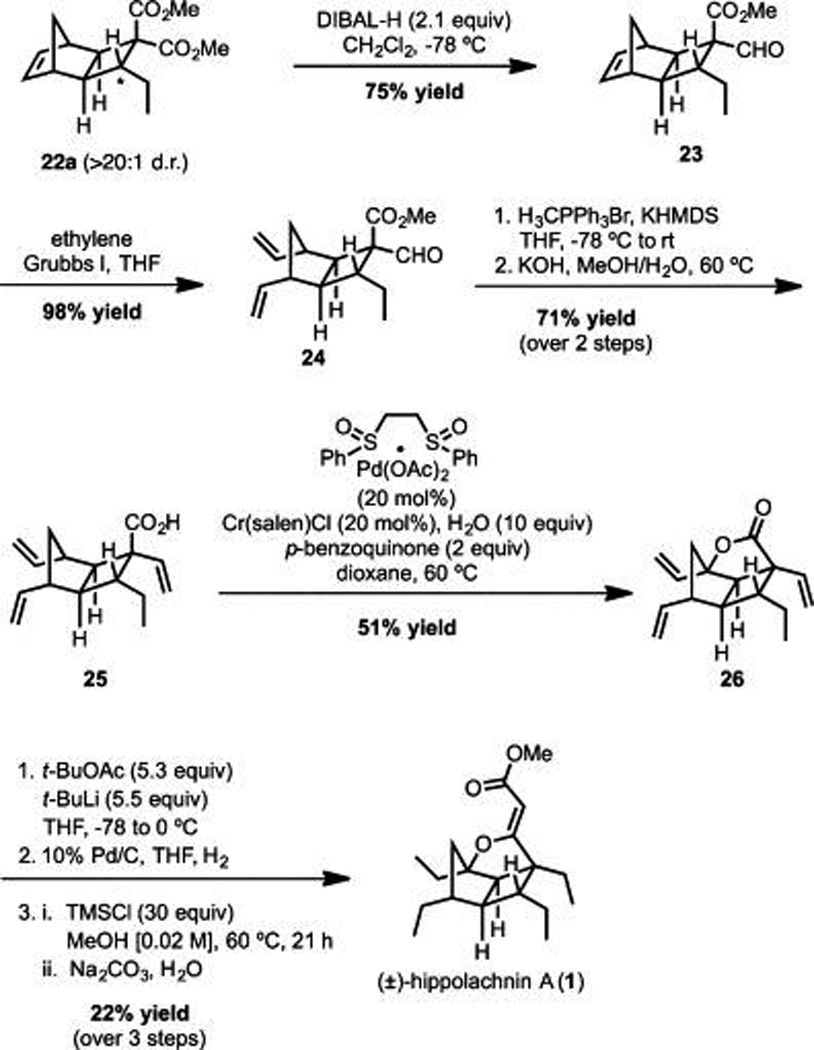
Wood Synthesis of Hippolachnin A
The Wood Route
As illustrated in Table 2, M.E.M. and J.L.W. initiated their synthetic studies by exploring the cycloaddition of known alkylidene malonate 11 with quadricyclane and found that under standard thermal conditions, long reactions times were required to achieve relatively low conversion.21 Although Lewis acid-promoted cycloadditions are well-known, they are unprecedented with quadricyclane; thus, a brief study was initiated into this possibility.22 In the event (Table 2), it was discovered that 10 mol % of TiCl4 effectively promotes the reaction of 11 and 9 to furnish a good yield of 22 in less than 4 h at 90 °C (Table 2, entry 7). Further exploration demonstrated that under these conditions the cycloaddition proceeds readily at room temperature, and even at 0 °C. Decreased loading of TiCl4 from 10 to 5 mol %, or even 2 mol % did not substantially diminish the yield (Table 2, entries 9 and 10). Ultimately, the best results were obtained when using 5 mol % of TiCl4 at 0 °C for 1 h (Table 2, entry 9).23
Table 2.
Development of [2π + 2σ + 2σ] with Trisubstituted Alkylidene Malonate
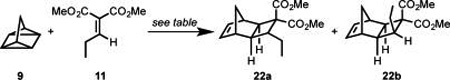 | |||||
|---|---|---|---|---|---|
| entrya | solvent | catalystb | time (h) |
dr (22a:22b)c |
% conversionc,d |
| 1 | EtOH | - | 144 | 9.1:1 | 52 (37)h |
| 2 | DCE | - | 4 | 2.3:1 | 11 |
| 3 | DCE | HCl | 4 | 1:1.1 | 20 |
| 4 | DCE | Cu(OTf)2 | 4 | - | - |
| 5 | DCE | ZnCl2 | 4 | 1.8:1 | 44 |
| 6 | DCE | AlCl3 | 4 | 2.1:1 | 67 |
| 7 | DCE | TiCl4 | 4 | 2.9:1 | >95 |
| 8e | CH2Cl2 | TiCl4f | 4 | 3.4:1 | >95 |
| 9e | CH2Cl2 | TiCl4f | 1 | 3.7:1 | >95 (82)h |
| 10e | CH2Cl2 | TiCl4g | 4 | 4.4:1 | 73 |
90 °C.
10 mol %
Determined by 1H NMR in CDCl3.
Conversion of alkylidene to cycloadducts A and B.
0 °C
5 mol %
2 mol %
Isolated yield
Although the diastereoselectivity of the accelerated reaction was poor, recrystallization furnished the desired diastereomer in >20:1 dr, which allowed for the ready preparation of 22 on gram scale. With ample material in hand, the approach continued by next reducing 22a to aldehyde 23 employing a procedure described by Yuan and co-workers (Scheme 4).24 Exposure of 23 to ethylene in the presence of Grubbs first generation catalyst furnished diene 24 which,15 upon Wittig methylenation and ester saponification, furnished the desired C–H activation-oxidation substrate 25.
On the basis of studies in a simplified system, initial efforts to advance 25 to 26 focused on conditions closely aligned with those appearing in the seminal publications from the White group utilizing a palladium bis-sulfoxide catalyst; however, these conditions proved unsatisfactory with the more heavily functionalized substrate (25).25 Fortunately, upon screening a variety of additives and varying the catalyst and oxidant loadings (see Supporting Information), we found that the addition of superstoichiometric quantities of water to the reaction mixture was crucial for obtaining reproducibly good yields of lactone 26.26,27
Having completed construction of the tricyclic lactone core of (±)-hippolachnin A, the Wood effort turned to installation of the remaining vinylogous carbonate moiety (Scheme 4). To this end, a number of methyl acetate equivalents were surveyed which led to the finding that condensation of 26 with tert-butyl acetate proceeds readily in the presence of excess tert-butyllithium to furnish, upon workup, the cyclized vinylogous carbonate as a mixture of diastereomers, which can be used without purification in the subsequent step.28 Hydrogenation of the terminal alkenes proceeded smoothly in the presence of Pd/C and exposure of the derived product to methanolic HCl induced transesterification of the t-butyl ester to give (±)-hippolachnin A (1).
Collaborative Route
With the independent synthetic routes essentially complete, the Wood and Brown Groups became aware of each other’s work during a poster session at the National Organic Symposium.29 Given the similarities between the respective approaches and the fact that each had complementary strengths, rather than compete, we launched a collaborative effort designed to fuse the best features of each route into an overall shorter and more efficient synthesis. With regard to the Brown route, we recognized that the cycloaddition with 10 provides improved access to much of the stereochemical complexity of hippolachnin A (cf., 14b and 22a), but undesired redox manipulations near the end of the synthesis lowered efficiency (18 to 1). In contrast, analysis of the Wood route reveals a relatively efficient, end-game and a less than optimal approach to carboxylic acid 25, which is largely due to use of alkylidene malonate 11 in the initial cycloaddition step. Overall, it appeared that a route devised from the early stages of the Brown synthesis and the endgame of the Wood route would be optimal.
In accord with this plan, we began the combined route with cycloaddition of acid chloride 10 to access 14b as delineated by Brown. Subsequent ring-opening cross metathesis with ethylene provided 27 in excellent yield. We were pleased to find that the Pd-catalyzed allylic oxidation conditions used in the Wood route functioned well to generate lactone 28 in 70% yield. Installation of the unsaturated methyl ester was accomplished utilizing the same end-game approach employed by the Wood group and furnished Hippolachnin A in 15% yield over three steps (26% yield brsm) (Scheme 5).
Scheme 5.
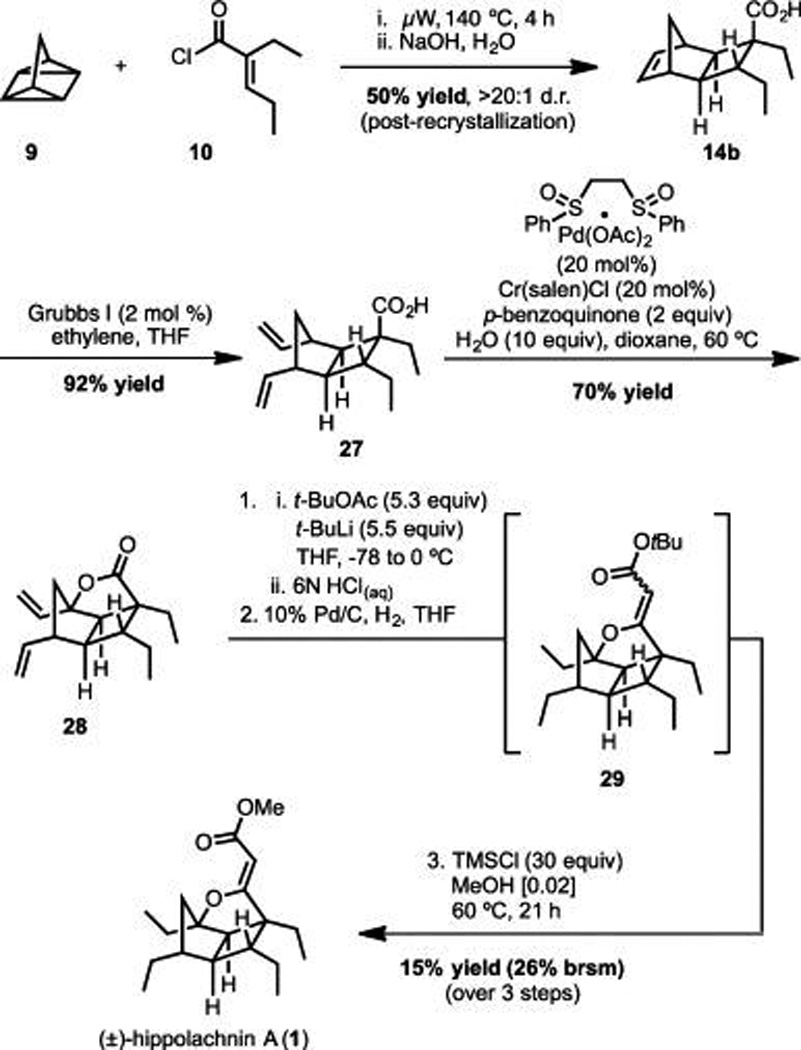
Collaborative Total Synthesis of (±)-Hippolachnin A
CONCLUSIONS
As outlined above, three syntheses of (±)-hippolachnin A have been developed that employ quadricyclane as point of departure and take advantage of the inherent reactivity of this highly symmetrical hydrocarbon. To the best of our knowledge, this is the first time a [2π + 2σ + 2σ] cycloaddition of quadricyclane has been employed in complex molecule synthesis, and its use here not only allows the rapid and controlled introduction of six contiguous stereocenters, but also highlights the power of directed C–H oxidation chemistry in enabling the use of simple hydrocarbons as precursors in the production of highly functionalized compounds.
Importantly, this combined effort exemplifies the synergism that can arise through collaborations among groups that traditionally may have been viewed as competitors. In this instance, advances in synthetic methods, as well as the recognition that synthetic efficiency arises from convergent strategies exploiting latent elements of symmetry, led the Brown and Wood groups to independently develop routes to (1) that were strategically aligned but differed in the employed tactics. As a result of the latter, each effort produced advances in different areas of synthesis. For example, the Brown route provides greater insight into the electronics required for quadricyclane cycloaddition chemistry and further expands the scope of C–H oxidations involving alcohols, whereas the Wood effort illustrates the potential utility of Lewis acid-promoted reactions of quadricyclane and brought to fore the importance of adventitious water in some applications of the White C–H oxidation chemistry. When combined, these efforts resulted in an improved synthetic strategy that delivers the target molecule in a longest linear sequence of only seven steps. Clearly, there are benefits that arise from both independent and collaborative investigations. In this case, advances in the science of synthesis were broadened by the former and the ability to efficiently access the target were eventually improved by the latter. It is our belief that this clearly illustrates that a productive scientific community is one that recognizes the importance of research driven by the interests and passion of individual investigators while at the same time fostering collaborative efforts.
Supplementary Material
Acknowledgments
C.M.R. and M.K.B. gratefully acknowledge financial support from Indiana University, the Indiana University McCormick Science Grant, and the NIH (R01GM110131-01). C.M.R. is grateful for an Indiana University Paget Organic Fellowship. M.E.M. and J.L.W. are grateful for generous funding from Baylor University, the Welch Foundation (Chair, AA-006) and the Cancer Research & Prevention Institute of Texas (CPRIT, R1309). M.E.M. is grateful for the support of the NSF GRFP (2013156410).
Footnotes
ASSOCIATED CONTENT
Supporting Information
The authors declare no competing financial interest.
REFERENCES
- 1.Piao S-J, Song Y-L, Jiao W-H, Yang F, Liu X-F, Chen W-S, Han B-N, Lin H-W. Org. Lett. 2013;15:3526–3529. doi: 10.1021/ol400933x. [DOI] [PubMed] [Google Scholar]
- 2.(a) Butts A, Krysan DJ. PLoS Pathog. 2012;8:e1002870. doi: 10.1371/journal.ppat.1002870. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Roemer T, Krysan DJ. Cold Spring Harbor Perspect. Med. 2014;4:a019703. doi: 10.1101/cshperspect.a019703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Ueoka R, Nakao Y, Kawatsu S, Yaegashi J, Matsumoto Y, Matsunaga S, Furihata K, van Soest RWM, Fusetani N. J. Org. Chem. 2009;74:4203. doi: 10.1021/jo900380f. [DOI] [PubMed] [Google Scholar]; (b) Festa C, De Marino S, D’Auria MV, Deharo E, Gonzalez G, Deyssard C, Petek S, Bifulco G, Zampella A. Tetrahedron. 2012;68:10157. [Google Scholar]; (c) Festa C, Lauro G, De Marino S, D’Auria MV, Monti MC, Casapullo A, D’Amore C, Renga B, Mencarelli A, Petek S, Bifulco G, Fiorucci S, Zampella A. J. Med. Chem. 2012;55:8303. doi: 10.1021/jm300911g. [DOI] [PubMed] [Google Scholar]; (d) Festa C, D’Amore C, Renga B, Lauro G, Marino S, D’Auria M, Bifulco G, Zampella A, Fiorucci S. Mar. Drugs. 2013;11:2314. doi: 10.3390/md11072314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Akiyama M, Isoda Y, Nishimoto M, Kobayashi A, Togawa D, Hirao N, Kuboki A, Ohira S. Tetrahedron Lett. 2005;46:7483. (b) Gracilioether F: Rasik CM, Brown KM. Angew. Chem. Int. Ed. 2014;53:14522–14526. doi: 10.1002/anie.201408055. (c) Gracilioethers B and C: Norris MD, Perkins MV, Sorensen EJ. Org. Lett. 2015;17:668–671. doi: 10.1021/ol503695j.
- 5.Ruider SA, Sandmeier T, Carreira EM. Angew. Chem. Int. Ed. 2015;54:2378–2382. doi: 10.1002/anie.201410419. [DOI] [PubMed] [Google Scholar]
- 6.For studies toward Hippolachnin A, see: Datta R, Dixon RJ, Ghosh S. Tetrahedron Lett. 2016;57:29.
- 7.Ruider SA, Carreira EM. Org. Lett. 2016;18:220. doi: 10.1021/acs.orglett.5b03356. [DOI] [PubMed] [Google Scholar]
- 8.For reviews regarding C–H functionalization, see: Godula K. Science. 2006;312:67. doi: 10.1126/science.1114731. Gutekunst WR, Baran PS. Chem. Soc. Rev. 2011;40:1976. doi: 10.1039/c0cs00182a. Newhouse T, Baran PS. Angew. Chem. Int. Ed. 2011;50:3362. doi: 10.1002/anie.201006368.
- 9.Quadricyclane is commercially available (Exciton, Inc.: ~$0.40/mmol) or can be readily prepared by photochemical irradiation of norbornadiene, see: Smith CD. Org. Synth. 1988;6:962.
- 10.Fleck M, Yang C, Wada T, Inoue Y, Bach T. Chem. Commun. 2007:822–824. doi: 10.1039/b613985j. [DOI] [PubMed] [Google Scholar]
- 11.(a) Arndtsen BA, Bergman RG, Mobley TA, Peterson TH. Acc. Chem. Res. 1995;28:154–162. [Google Scholar]; (b) Crabtree RH. J. Chem. Soc., Dalton Trans. 2001;17:2437–2450. [Google Scholar]; (c) Crabtree RH. J. Organomet. Chem. 2004;689:4083–4091. [Google Scholar]; (d) Chen X, Engle KM, Wang D-H, Yu J-Q. Angew. Chem. Int. Ed. 2009;48:5094–5115. doi: 10.1002/anie.200806273. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Lyons TW, Sanford MS. Chem. Rev. 2010;110:1147–1169. doi: 10.1021/cr900184e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Newhouse T, Baran PS. Angew. Chem. Int. Ed. 2011;50:3362–3374. doi: 10.1002/anie.201006368. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Brückl T, Baxter RD, Ishihara Y, Baran PS. Acc. Chem. Res. 2012;45:826–839. doi: 10.1021/ar200194b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) White MC. Science. 2012;335:807. doi: 10.1126/science.1207661. [DOI] [PubMed] [Google Scholar]
- 12.For seminal reports regarding cycloadditions with quadricyclane, see: Smith CD. J. Am. Chem. Soc. 1966;88:4273–4274. Tabushi I, Yamamura K, Yoshida Z. J. Am. Chem. Soc. 1972;94:787–792. For a review, see: Petrov VA, Vasil’ev NV. Curr. Org. Synth. 2006;3:215.
- 13.Hammond GS, Turro NJ, Fischer A. J. Am. Chem. Soc. 1961;83:4674. [Google Scholar]
- 14.Hermeling D, Schaefer HJ. Chem. Ber. 1988;121:1151–1158. [Google Scholar]
- 15.X-ray structure of 14b is available. See the Supporting Information for details.
- 16.Schwab PE, Grubbs RH, Siller JW. J. Am. Chem. Soc. 1996;118:100. [Google Scholar]
- 17.Louie J, Bielawski CW, Grubbs RH. J. Am. Chem. Soc. 2001;123:11312. doi: 10.1021/ja016431e. [DOI] [PubMed] [Google Scholar]
- 18.(a) Chen MS, White MC. Science. 2007;318:783. doi: 10.1126/science.1148597. [DOI] [PubMed] [Google Scholar]; (b) Bigi MA, Reed SA, White MC. J. Am. Chem. Soc. 2012;134:9721. doi: 10.1021/ja301685r. [DOI] [PubMed] [Google Scholar]
- 19.For a review regarding 1,5-remote functionalization involving alkoxy radicals (e.g. Barton and hypohalite reactions), see: Čeković Ž. Tetrahedron. 2003;59:8073.
- 20.Concepción JI, Francisco CG, Hernández R, Suárez E. Tetrahedron Lett. 1984;25:1953. [Google Scholar]
- 21.(a) Wang M, Wang Z, Shi Y-H, Shi X-X, Fossey JS, Deng W-P. Angew. Chem. Int. Ed. 2011;50:4897–4900. doi: 10.1002/anie.201007960. [DOI] [PubMed] [Google Scholar]; (b) Tietze LF, Beifuss U, Rawson D, Meyers AI. Org. Synth. 1993;71:167–171. [Google Scholar]
- 22.(a) Yamauchi M, Aoki T, Li M-Z, Honda Y. Tetrahedron: Asymmetry. 2001;12:3113–3118. [Google Scholar]; (b) Evans DA, Olhava EJ, Johnson JS, Janey JM. Angew. Chem. Int. Ed. 1998;37:3372– 3375. doi: 10.1002/(SICI)1521-3773(19981231)37:24<3372::AID-ANIE3372>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 23.While this brief exploration of Lewis acid catalysts was sufficient for our synthesis of hippolachnin A, we are currently expanding the scope to accommodate more electron rich olefins as well as asymmetric variants.
- 24.Yuan C, Jiao L, Yu Z-X. Tetrahedron Lett. 2010;51:5674–5676. [Google Scholar]
- 25.Gormisky PE, White MC. J. Am. Chem. Soc. 2013;135:14052–14055. doi: 10.1021/ja407388y. [DOI] [PubMed] [Google Scholar]; (a) Osberger TJ, White MC. J. Am. Chem. Soc. 2014;136:11176–11181. doi: 10.1021/ja506036q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ammann SE, Rice GT, White MC. J. Am. Chem. Soc. 2014;136:10834–10837. doi: 10.1021/ja503322e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Malik M, Witkowski G, Jaroz S. Org. Lett. 2014;16:3816–3819. doi: 10.1021/ol501730p. [DOI] [PubMed] [Google Scholar]
- 26.Our initial C–H activations with an old bottle of palladium bissulfoxide catalyst yielded consistently 40% of desired product. However, upon switching to a new bottle, this dropped to a consistent 29% and prompted a search for more reproducible conditions that was guided by the use of various additives listed in publications from the M.C. White group.25
-
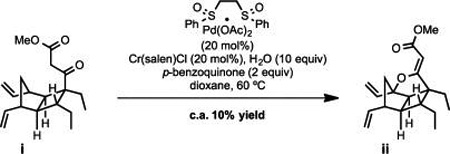
27.Intrigued by the possibility of directly accessing the vinylogous carbonate moiety by application of the White chemistry, we performed a brief study employing beta-keto ester i as substrate. Although the reaction proved feasible and the derived product (ii) was converted to (±)-hippolachnin A, optimization efforts were met with only limited success.
- 28.Attempts to condense methyl acetate with the lactone led predominately to the formation of methyl acetoacetate. Further investigations into Wittig olefinations, Horner-Wadsworth-Emmons olefinations, and aldol variations were also unsuccessful. While we were able to successfully condense acetonitrile, we were unable to convert the nitrile into the necessary methyl ester in appreciable yields.
- 29.(a) Rasik CM, Brown MK. Total Synthesis of Gracilioether F and (±)-Hippolachnin A; Poster presentation at the 44th National Organic Chemistry Symposium, University of Maryland; June 28th, 2015; College Park, MD, USA. [Google Scholar]; (b) McCallum ME, Wood JL. Total Synthesis of Hippolachnin A; Poster presentation at the 44th National Organic Chemistry Symposium, University of Maryland; June 28th, 2015; College Park, MD, USA. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.