

Abstract
An oligoquinoline foldamer library was synthesized and screened for agonism of lipid bilayer catalysed assembly of islet amyloid polypeptide (IAPP). One tetraquinoline, ADM-116, showed exceptional potency not only in this assay, but also in secondary assays measuring lipid bilayer integrity and rescue of insulin secreting cells from the toxic effects of IAPP. Structure activity studies identified three additional oligoquiniolines, closely related to ADM-116, which also have strong activity in the primary, but not the secondary assays. This contrasts work using an oligopyrdyl foldamer scaffold in which all three assays are observed to be correlated. The results suggest that while there is commonality to the structures and pathways of IAPP conformational change, it is nevertheless possible to leverage foldamers to seperatly affect IAPP’s alternative gains-of-function.
Foldamers are chemical species defined by the presence of internal, non-covalent interactions that result in a limited set of conformations1,2. Foldamers, like polypeptides, typically take the form of polymers of defined length, with a common structured core and the potential for diverse side-chain derivatives. As such, they represent an opportunity to create nano-scale, definable surfaces with properties that are not confined to the conventional 20 or 4 for peptides and nucleic acids respectively.
This class of molecule has an established history in biological applications. Areas include antagonism of bioactive targets such as an agonist of GLP-1 receptor3, an inhibitor of HIV-cell fusion4, an antagonist of VEGF receptor5, antimicrobial agents6, cell penetrating delivery of cargo7, membrane curvature sensing8, molecular recognition9, and functional nanostructures10. Our group and others have expanded their usage as antagonists of amyloidogenic proteins including islet amyloid polypeptide (IAPP)11,12 and Aβ peptide13.
IAPP is a 37-residue peptide co-secreted with insulin by β-cells in the pancreatic islets of Langerhans. Amyloid fibrillation by IAPP is associated with Type 2 Diabetes, an endocrine disorder characterised by loss of glucose homeostasis, systemic insulin resistance, accumulation of amyloid deposits in the pancreas, and loss of insulin secreting β-cells14. Considerable evidence supports the involvement of membrane bound α-helical oligomers of IAPP as fundamental to these pathologies not only in IAPP15,16, but also Aβ from Alzheimer’s17 and α-synuclein from Parkinson’s18.
The sampling of α-helical states by IAPP has been associated with membrane permeation, penetration, catalysis of amyloid formation, and cytotoxicity in cell culture19. IAPP autoantibodies have also been reported in diabetic patients that are conformationally selective for α-helical oligomers16. Given the heterogeneity of observed species, it is plausible that alternative sub-structures are responsible for each of the different gains-of-function. Much of our own work, for example, could be accounted for by a mutation or small molecule diverting IAPP from one α-helical state to another. Consider that swapping the positions of residues 12 and 14 in IAPP does not change overall charge or hydrophobicity of the peptide. It does, however, diminish helicity20 and cytotoxicity21. In contrast, a similarly motivated change in which residues 15 and 16 are replaced by D-amino acids diminishes helicity and yet increases cytotoxicity22. These contrasts suggest the presence and functional importance of molecular scale interactions. Our working hypothesis is that differential binding to sub-populations of α-helical oligomers should serve as an effective approach to test for the presence of sub-states, and provide the mechanistic insights required to defining a therapeutic target.
IAPP is a member of a broad class of low-complexity proteins that do not adopt a single stable globular fold. Nevertheless, we have shown that small molecule foldamers can interact with exceptional specificity. In collaboration with Hamilton, we designed oligopyridylamide based α-helical mimetics to successfully target the α-helical conformation of IAPP23.
Structure-activity assessments in the above work focused on the variation of a side chain moiety of the central pyridyl group of a tripyridylamide. The most effective inhibitor, ADM-3, possessed an O-ethyl functionality at this position. Creation of a branch by addition of a single methyl group at the O-alkyl α-carbon, ADM-9, was strongly detrimental to antagonist activities23. This was a surprising result given the plastic nature of IAPP’s conformation. In this work, we report on a new modification of tripyridyl amide, and the systematic design and screening of a library of oligoquinolines using effects on IAPP fibrillation as our primary screen.
The kinetics of IAPP fibrillation are nucleation-dependent resulting in sigmoidal kinetic profiles (see Material and Methods for details in ESI†). Briefly, a 10 μM IAPP reaction was prepared by dilution of a 1 mM stock of IAPP in water into a buffer (100 mM KCl, 50 mM NaPi, pH 7.4) containing 630 μM large unilamellar vesicles (LUVs, DOPG:DOPC, 1:1, d = 100 nm) and 0.1 μM ThT as fluorescent reporter. IAPP fibrillation time is simplified to an expression of the reaction midpoint, t50, which is the time required for half of the monomeric IAPP precursor to convert into ThT-sensitive aggregates (Fig. 2a, inset and ESI† Fig. S1). The t50 for lipid-catalysed and aqueous IAPP fibrillation were 1.0±0.1 h and 19.1±2.1 h, respectively, consistent with our earlier published work23.
Fig. 2.

Effects of small molecules on processes mediated by IAPP. (a) Reaction mid-points, t50, of aqueous (white) and lipid-catalysed (black) fibrillation reactions. The t50 are expressed relative to reference reactions in which the indicated compound is absent. Inset: Representative lipid–catalysed kinetic profile (black) and sigmoidal fit (red) used to extract t50. Liposome catalysed reaction conditions: 10 μM IAPP, 10 μM oligoquinoline, DOPG:DOPC (1:1, 630 μM, d = 100 nm). Fully aqueous condition: 30 μM IAPP, 6 μM oligoquinoline. (b) Representative kinetic profiles for liposome leakage mediated by IAPP in the presence and absence of ADM-116. The dynamic range for renormalization is established using 20 μM magainin 2. Membrane leakage assayed using 100 nm, DOPG liposomes at 200 μM lipid in monomer units, 6 μM IAPP and stoichiometric oligoquinoline. (c) Representative burst profile of the diffusion of 20 nM fluorescently labelled ADM-116 through the excitation volume of a confocal microscope. Shown are 1 μM IAPP (black), 25 μM liposomes (magenta) or both (orange). Inset shows an expansion of the final 10 s of the trace. (d) Statistics for the effects observed using the indicated molecules in our primary, and two indicated secondary assays (see main text). All values are expressed as renormalized averages relative to IAPP-only controls. Cytotoxicity measured at 13 μM IAPP and stoichiometric small molecule. oligoquinoline. Cytotoxicity was measured using cell-titer blue colorimetric assay. Error bars for toxicity assay represent standard deviations from a minimum of three independent experiments each with four internal repeats (i.e. n ≥ 12).
Movement of the branch point of ADM-9 a single carbon away from the O-ethyl rescues activity. An O-isobutyl substitution was prepared, ADM-11, and assayed in our primary screen followed by orthogonal screening using one solution biophysical and one eukaryotic cell-based assay. In contrast to our report on ADM-9, ADM-11 is comparable if not superior to ADM-3. ADM-11 delays the kinetics of lipid associated IAPP fibrillation ~7 fold compared to ~3 fold for ADM-3 at a stoichiometric ratio of 1:1 (IAPP:small molecule) (Fig. 2d and ESI† Fig. S2). ADM-11 slows the kinetics of IAPP mediated membrane leakage slightly better than ADM-3. In toxic rescue experiments, ADM-11, like ADM-3 rescues IAPP mediated toxicity in a dose dependent manner with cell viability wholly restored upon reaching a stoichiometric ratio of 1:0.5, IAPP:small molecule (Fig. 2d and ESI† Fig. S3). This result clearly reinforces our observation of a sensitive role of the O-alkyl α-carbon in mediating interactions with IAPP.
A set of more than 40 oligoquinolines was synthesized and assessed using our primary assay. Within this library of compounds, the length of the scaffolds are monomer (5 compounds), dimer (6), trimer (8), tetramer (12), and pentamer (9). The dianionic tetraquinoline, ADM-116, exhibits the strongest inhibitory activity against lipid catalysed fibrillation (Fig. 1a, see chemical structures of the oligoquinolines in ESI†, Table S1). The kinetics for lipid catalysed IAPP fibrillation was completely suppressed over the time-frame of the experiment (45 h) in the presence of stoichiometric ADM-116 (Fig. 2a)24. ADM-116 is ≫10 fold more effective than our previously reported pentaquinoline, ADM-118 (earlier reported under the name OQ511) (Fig. 2a). In the absence of catalyst, ADM-116 delays the midpoint of IAPP fibrillation with a t50 that is ~6.5 fold slower than the control (Fig. 2a) at a stoichiometric ratio of 1:0.2 (IAPP:ADM-116). Overall, oligoquinolines observed to inhibit fiber formation under liposome catalysed conditions also inhibited under lipid-free conditions.
Fig. 1.
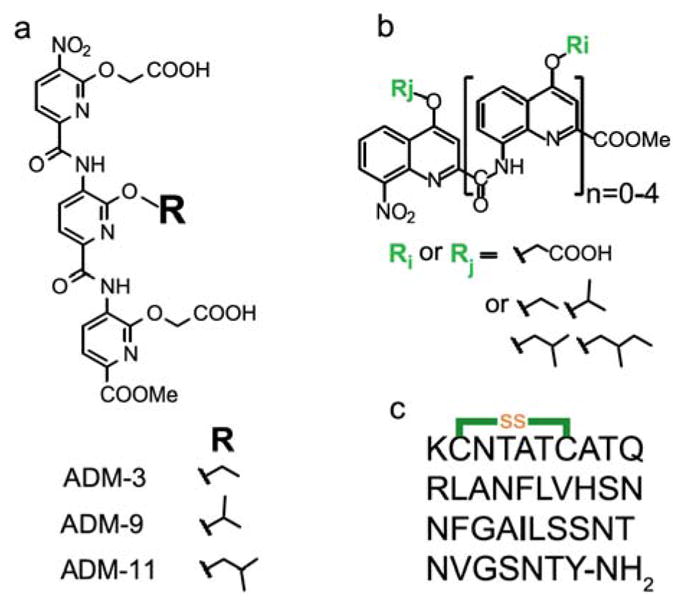
Chemical structures of the molecules used in this study. (a) Three related tripyridylamides. (b) Generic structure of the library of oligoquinolines. (c) Primary sequence of human IAPP, cyclized at C2 and C7 residues and with naturally occurring amidation at the C-terminal.
The inhibition of IAPP by ADM-116 is structure-specific. Analogues of ADM-116 in which the O-ethyl functionality of ADM-116 was replaced with O-isopropyl (ADM-129), O-isobutyl (ADM-130), and O-(2-methyl butane) (ADM-131) maintained strong activity in the primary assay. The relative t50 for the inhibition of lipid-catalysed IAPP self-assembly for stoichiometric ADM-129, ADM-130, and ADM-131 were 15 ± 0.5, 20 ± 0.8, and 17 ± 0.7 respectively (Fig. 2a). Compared to ADM-116, introduction of branched substituents at both the O-alkyl α-carbon or β-carbon can be seen to be detrimental, albeit weakly, to agonist activity. This suggests that both the α- and β- carbons of the O-alkyl side chain occupy positions necessary for IAPP activity.
In an orthogonal secondary assay, ADM-116 was the most effective antagonist of pre-amyloid IAPP mediated membrane leakage (Fig. 2b). Liposome leakage assays were conducted according to our earlier published method25. Briefly, liposomes (100% DOPG) are equilibrated in 100 mM KCl, 50 mM NaPi, pH 7.4 and then rendered unilamellar by extrusion. A 70 kDa fluorescein-labelled dextran is encapsulated into liposomes during this process. Here, kinetics are initiated by introducing 200 μM liposome into a solution of 4 μM IAPP and 6 mM of a fluorescent quencher, DPX (p-xylene-bis-pyridinium bromide). The kinetic profiles of leakage are single exponential with a decrease in fluorescence as a function of time (Fig. 2b). The leakage rate constant for 4 μM IAPP was 8.8 ± 1.6 × 10−4 s−1, which is in agreement with our previous reports23. ADM-116 reduces the leakage rate by a factor of ~4–5 with respect to the control reaction (only IAPP) at equimolar protein:compound. This inhibition does not appear to be a simple consequence of displacing IAPP from the liposome surface. Using a previously characterized fluorescein tagged variant of ADM-11624, we observe that the IAPP:ADM-116 complex and not ADM-116 readily associates with the liposome (Fig. 2c). Analogues of ADM-116 showed greatly reduced leakage activity (Fig. 2d) with a rank order of ADM-116≫ADM-130>ADM-129>ADM-131.
ADM-116 was the most effective inhibitor of IAPP mediated cytotoxicity in INS-1 cells. IAPP readily induces cytotoxicity in INS-1 cells, a well-established, immortal model of pancreatic β-cells. The viability of INS-1 cells is reduced to 42±3% (58±3% when expressed as toxicity) 48 h after addition of 13 μM IAPP to the culture media. For simplicity of presentation, we present toxicity effects relative to this compound-free control. At 1:1, ADM-116 completely restores cell viability. Under matched conditions, the close analogues, ADM-129, ADM-130, and ADM-131, rescue only weakly with cell toxicities of 94±7%, 91±8%, and 96±8 respectively (n=16).
The best performing tripyridylamide, ADM-3, and the best performing tetraquinoline, ADM-116, are dianionic. Moreover, both induce α-helical structure in IAPP. This is consistent with our goals of using structured foldamers to capture transiently sampled structures or alternatively, to induce structure in wholly disordered systems11,12,23,24. NMR studies on related compounds suggest that the binding site for both compounds is the N-terminal domain of IAPP that runs from residue 1-2212,26. A perhaps unsurprising result given the cationic nature of this region of the peptide. It is facile to model the carboxylates of ADM-116 and ADM-3 to separate charge by similar distances (Fig. 3). Importantly, these distances are comparable to the rise of two winds of an α-helix. Two winds separate, for example, Arg 11 and His 18 within IAPP’s helical subdomain.
Fig. 3.
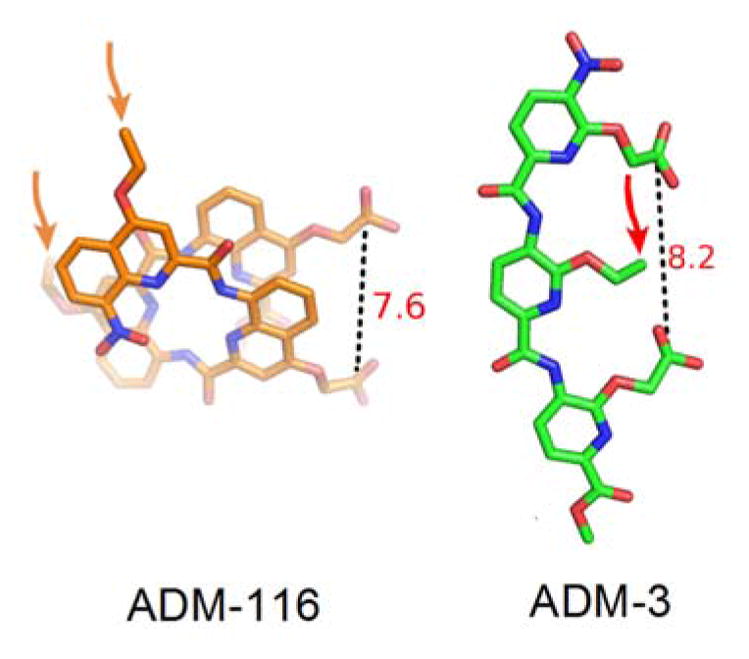
Atomic models of ADM-116 and ADM-3 based on crystal structures23,24. Models of ADM-116 and ADM-3 are readily created in which each show two exposed carboxylates held at comparable distances apart. Arrows indicate the location of ether-linked aliphatic moieties.
Similarities between ADM-3 and ADM-116 end once consideration is given to the position of the sensitive aliphatic moieties. In our primary assay, substitution of ethyl with isopropyl or isobutyl on the quinoline scaffold is detrimental to activity. In contrast, on the pyridyl scaffold, only the ethyl to isopropyl23 and not the ethyl to isobutyl group substitution is detrimental. Moreover, modifications of ADM-116 that resulted in modest loss of fiber formation inhibition activity gave a near total loss of both leakage and toxic rescue activities (Fig. 2d). In contrast, changes in primary and secondary assays are wholly correlated for ADM-3 variants23 (Fig. 2d). Note that relative to the carboxylates, the structural location of the aliphatic group targeted for variation in these efforts is not comparable between the two classes of scaffold (Fig. 3). Whereas in tripyridyl amide, the aliphatic group is colinear and bracketed by the carboxylates, in the tetraquinoline amide, the aliphatic moieties and two carboxylate groups are roughly equidistant from one another. There is therefore little reason to believe that the aliphatic groups in the two compound classes interact with the same site on IAPP. Alternatively, if they do interact with the same groups on IAPP, then the induced protein structures are unlikely to be identical.
In summary, we have shown that two different foldamer scaffolds with similar surface moieties can both rescue INS-1 cells from IAPP induced toxicity. However, whereas the oligopyridylamide scaffold reveals correlation between effects on two solution biophysical observables (leakage and amyloid formation) and toxic rescue, the oligoquinoline scaffold shows correlation only between leakage and toxicity. This suggests the presence of sub-populations relevant to amyloid formation that are not relevant to toxicity (and vice versa). Delineation between membrane bound, IAPP substates should therefore be possible by contrasting use of both scaffolds. The broader impact of such insight extends to several other diseases, notably Alzheimer’s where a partially ordered 42 residue peptide, Aβ, undergoes structural transitions to α-helical states upon interaction with biological membranes.
Supplementary Material
Acknowledgments
This research was supported by NIH GM094693 and an American Diabetes Association mentor-based postdoctoral fellowship to MB.
Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
Notes and references
- 1.Gellman SH. Acc Chem Res. 1998;31:173–180. [Google Scholar]
- 2.Goodman CM, Choi S, Shandler S, DeGrado WF. Nat Chem Biol. 2007;3:252–262. doi: 10.1038/nchembio876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Denton EV, Craig CJ, Pongratz RL, Appelbaum JS, Doerner AE, Narayanan A, Shulman GI, Cline GW, Schepartz A. Org Lett. 2013;15:5318–5321. doi: 10.1021/ol402568j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Horne WS, Johnson LM, Ketas TJ, Klasse PJ, Lu M, Moore JP, Gellman SH. Proc Natl Acad Sci. 2009;106:14751–14756. doi: 10.1073/pnas.0902663106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Udugamasooriya DG, Dineen SP, Brekken RA, Kodadek TA. J Am Chem Soc. 2008;130:5744–5752. doi: 10.1021/ja711193x. [DOI] [PubMed] [Google Scholar]
- 6.Choi S, Isaacs A, Clements D, Liu D, Kim H, Scott RW, Winkler JD, DeGrado WF. Proc Natl Acad Sci. 2009;106:6968–6973. doi: 10.1073/pnas.0811818106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Douat C, Aisenbrey C, Antunes S, Decossas M, Lambert O, Bechinger B, Kichler A, Guichard G. Angew Chem Intl Ed. 2015;54:11133–11137. doi: 10.1002/anie.201504884. [DOI] [PubMed] [Google Scholar]
- 8.Gunasekara RW, Zhao Y. Langmuir. 2015;31:3919–3925. doi: 10.1021/acs.langmuir.5b00379. [DOI] [PubMed] [Google Scholar]
- 9.Chandramouli N, Ferrand Y, Lautrette G, Kauffmann B, Mackereth CD, Laguerre M, Dubreuil D, Huc I. Nat Chem. 2015;7:334–41. doi: 10.1038/nchem.2195. [DOI] [PubMed] [Google Scholar]
- 10.Collie GW, Pulka-Ziach K, Lombardo CM, Fremaux J, Rosu F, Decossas M, Mauran L, Lambert O, Gabelica V, Mackereth CD, Guichard G. Nat Chem. 2015;7:871–878. doi: 10.1038/nchem.2353. [DOI] [PubMed] [Google Scholar]
- 11.Kumar SS, Miranker AD. Chem Commun. 2013;49:4749–4751. doi: 10.1039/c3cc41452c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kumar S, Brown M, Nath A, Miranker A. Chem Biol. 2014;21:775–781. doi: 10.1016/j.chembiol.2014.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seither KM, McMahon HA, Singh N, Wang H, Cushman-Nick M, Montalvo GL, DeGrado WF, Shorter J. Biochem J. 2014;464:85–98. doi: 10.1042/BJ20131609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Westermark P, Andersson A, Westermark GT. Physiol Rev. 2011;91:795–826. doi: 10.1152/physrev.00042.2009. [DOI] [PubMed] [Google Scholar]
- 15.Hebda J, Miranker AD. Ann Rev Biophys. 2009;38:125–152. doi: 10.1146/annurev.biophys.050708.133622. [DOI] [PubMed] [Google Scholar]
- 16.Bram Y, Frydman-Marom A, Yanai I, Gilead S, Shaltiel KR, Amdursky N, Gazit E. Sci Rep. 2015;4:4267, 1–9. doi: 10.1038/srep04267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kirkitadze MD, Condron MM, Teplow DB. J Mol Biol. 2001;312:1103–1119. doi: 10.1006/jmbi.2001.4970. [DOI] [PubMed] [Google Scholar]
- 18.Bartels T, Choi JG, Selkoe DJ. Nature. 2011;477:107–110. doi: 10.1038/nature10324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Last N, Schlamadinger DE, Miranker AD. Prot Sci. 2013;22:870–882. doi: 10.1002/pro.2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koo BW, Hebda JA, Miranker AD. Protein Eng Des Sel. 2008;21:147–54. doi: 10.1093/protein/gzm076. [DOI] [PubMed] [Google Scholar]
- 21.Magzoub M, Miranker AD. FASEB J. 2012;26:1228–38. doi: 10.1096/fj.11-194613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De Carufel CA, Quittot N, Nguyen PT, Bourgault S. Angew Chem Int Ed. 2015;54:14383–14387. doi: 10.1002/anie.201507092. [DOI] [PubMed] [Google Scholar]
- 23.Kumar S, Schlamadinger DE, Brown MA, Dunn JM, Mercado B, Hebda JA, Saraogi I, Rhoades E, Hamilton AD, Miranker AD. Chem Biol. 2015;22:369–378. doi: 10.1016/j.chembiol.2015.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kumar S, Birol M, Schlamadinger DE, Wojcik SP, Rhoades E, Miranker AD. Nat Commun. doi: 10.1038/ncomms11412. accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Last NB, Rhoades E, Miranker AD. Proc Natl Acad Sci USA. 2011;108:9460–9465. doi: 10.1073/pnas.1102356108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hebda JA, Saraogi I, Magzoub M, Hamilton AD, Miranker AD. Chem Biol. 2009;16:943–950. doi: 10.1016/j.chembiol.2009.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.