


Abstract
Formation of nucleosome free region (NFR) accompanied by specific histone modifications at flanking nucleosomes is an important prerequisite for enhancer and promoter activity. Due to this process, active regulatory elements often exhibit a distinct shape of histone signal in the form of a peak-valley-peak (PVP) pattern. However, different features of PVP patterns and their robustness in predicting active regulatory elements have never been systematically analyzed. Here, we present PARE, a novel computational method that systematically analyzes the H3K4me1 or H3K4me3 PVP patterns to predict NFRs. We show that NFRs predicted by H3K4me1 and me3 patterns are associated with active enhancers and promoters, respectively. Furthermore, asymmetry in the height of peaks flanking the central valley can predict the directionality of stable transcription at promoters. Using PARE on ChIP-seq histone modifications from four ENCODE cell lines and four hematopoietic differentiation stages, we identified several enhancers whose regulatory activity is stage specific and correlates positively with the expression of proximal genes in a particular stage. In conclusion, our results demonstrate that PVP patterns delineate both the histone modification landscape and the transcriptional activities governed by active enhancers and promoters, and therefore can be used for their prediction. PARE is freely available at http://servers.binf.ku.dk/pare.
INTRODUCTION
Enhancers and promoters are cis-regulatory elements (CREs) that control the spatiotemporal expression of genes in response to various external signals and across different cell types (1,2). While promoters are located at the 5′ end of genes, enhancers can be located at thousands of bases up- or downstream of their target gene(s) (1). An important prerequisite for the activity of CREs is the formation of a nucleosome free region (NFR) that is often reflected by the distinct positional distribution of histone modifications in the form of a peak-valley-peak (PVP) pattern (1,3,4). Indeed, PVP patterns have previously been associated with enhancer activity (5–7). However, they have never been systematically analyzed in terms of their efficacy to detect and elucidate distinctive properties of active CREs. Consequently, most standard analysis approaches use enrichment of histone modifications measured by ChIP-seq as the primary criteria to predict CREs. Prominent among these are the ‘signal-based’ and ‘machine learning-based’’ approaches.
The standard ‘signal-based’ approach is used in scenarios where limited histone modification data are available, for example, enrichment of H3K4me1 relative to H3K4me3 is used to predict enhancers, and the inverse ratio to predict promoters (1,8). Since enrichment of histone modifications does not always correlate with the activity of CREs (9,10), ‘machine learning-based’ methods trained on a large set of histone modifications data have been developed to increase the accuracy of predictions. Among these methods are ChromHMM (11) and Segway (12) that were used to predict enhancers and promoters across different cell lines studied during the ENCODE project. Other recently developed methods based on this approach are CSI-ANN (13), RFECS (14), EnhancerFinder (15) and DEEP (16).
The major drawback of both ‘signal’ and ‘machine learning’-based approaches is the low specificity of prediction, particularly of active CREs. This is due to the reason that enrichment of histone modifications can be observed at both ‘primed to be active’ (poised) and active CREs (1,3). Although the inclusion of H3K27ac enriches for active CREs (8), recent studies based on a more robust indicator of activity (divergent transcription) have shown low in vitro validation rates of histone-based predictions (17,18). Of note, these studies suggested that enrichment of H3K4me1 and H3K4me3 is neither a completely distinctive nor an exclusive feature of active enhancers and promoters (17,19,20). In fact, a recent study showed that enrichment of H3K4me3 correlates with the activity of CRE (both for enhancers and promoters) (20).
In view of the limitations mentioned above and the potential of a PVP pattern in detecting active regulatory elements, we developed a method that systematically analyzes a PVP pattern defined by H3K4me1 and H3K4me3 modifications to predict NFRs. We show that NFRs predicted by the H3K4me1 and H3K4me3 PVP patterns, characterize active enhancers and promoters, respectively. We show that the depth of PVP patterns (nfrDip score) is a reflection of active transcriptional regulation, measured by using complementary high-throughput sequencing data such as GRO-seq, CAGE, ChIA-PET, H3K27ac and Pol-II binding. Apart from the depth of the PVP pattern, we show that the asymmetry in this pattern can be used to predict the directionality of stable transcription at promoters. Also, we show a spatially distinct deposition pattern of H3K4me1 relative to H3K4me3 and H2A.Z histone marks at nucleosomes flanking enhancers and promoters. Finally, we use the method to identify hundreds of enhancers important in defining the identity of four ENCODE cell lines and four hematopoietic progenitor cells.
MATERIALS AND METHODS
Input data
We used ChIP-seq-based histone modification data (H3K4me1 and H3K4me3) for the prediction of NFRs in four human cell lines (GM12878, HeLa-S3, HepG2, K562). NFRs predicted using H3K4me1 and H3K4me3 modifications are annotated as enhancers (PVP based) and promoters (PVP based), respectively. The histone modification data for the four cell lines was downloaded in BAM format from the ENCODE project (21) (http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/).
Method to predict nucleosome free regions
To predict NFRs that include enhancers and promoters, the method analyzes H3K4me1 or H3K4me3 modification data on a genome wide scale. Various analysis steps in the method are summarized in Figure 1 and described as follows: (i) input to the method are mapped reads in BAM format corresponding to the two replicates of an H3K4me1/me3 ChIP-seq experiment (Figure 1A), (ii) identically mapped reads are collapsed into tags to remove polymerase chain reaction duplicates. This is followed by extending the 3′ end of tags to the actual fragment length as determined using Macs2 (22) (Figure 1B). Next, we normalize the tag counts with respect to the sequencing depth of each replicate (23). (iii) The computational method, blockbuster, is used to fit Gaussian distribution to the normalized tag density profile of H3K4me1/3 modifications across the whole genome (Figure 1C). blockbuster has been extensively applied on small RNA-seq data for a similar purpose (24). Regions where the tag density profile follows a Gaussian distribution and consists of a minimum number of tags (block threshold) are marked as containing a ‘tag block’ (Figure 1C; see next paragraph for details). (iv) Overlapping tag blocks are merged into a single block cluster, thus covering a much broader genomic region. All the regions ≥20 and <3 kb in length flanked by block clusters are defined as putative NFRs. We used an upper limit of 3 kb to define NFR as it corresponds to the 99% quartile limit for the width of both enhancers and promoters predicted by ENCODE (11,12). Further, only the NFRs that are reproducible between the two replicates are selected (Figure 1D). A score (Sn; nfrDip) is calculated for each putative NFR (n) as:
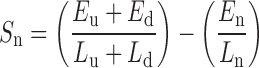 |
where; E is the total normalized tag count in block cluster upstream (u), block cluster downstream (d) and in the NFR (n), respectively. L represents the length in base pairs of the respective regions. The higher the nfrDip score (S), the better defined is the corresponding NFR. (v) We further test whether the score, Sn, for each putative NFR is significantly greater than that computed for the genomic background (Figure 1E). Specifically, a P-value is calculated for each putative NFR by counting the fraction of random genomic regions with a score greater than Sn. (vi) Putative NFRs with P-values <0.001 and Benjamini-Hochberg adjusted FDR < 0.05 are included in the final set of NFR predictions (Figure 1F). To compute the genomic background, we randomly sample 100 000 unique genomic regions of the same length distribution (as the putative NFRs) which overlap with the regions enriched for block clusters (used to define the putative NFRs), but not with putative NFRs.
Figure 1.

A schematic representation of the various steps of the PVP-based approach for the prediction of nucleosome free regions (NFRs). (A) The input to the method is a genome wide density profile of histone (H3K4me1 or H3K4me3) tags in the form of a BAM file (one per replicate). (B) The raw tag counts at genomic regions are normalized to account for any difference in sequencing depths between the two replicates. (C) Using blockbuster, closely spaced mapped tags are clustered into tag blocks at positions where the tag count is above threshold and follows a Gaussian distribution. (D) Tag blocks overlapping by at least 1 bp are merged into longer block clusters. Putative NFRs are defined as regions between consecutive tag blocks separated by a user defined range of distance (in base pairs). A score (nfrDip) is computed for each putative NFR (equation 1). (E) The false discovery rate is computed for each prediction by randomly shuffling the putative NFRs 100 000 times within the block clusters. (F) We select all putative NFRs at an FDR of <0.05 as robust NFRs. NFRs predicted using H3K4me1 and H3K4me3 marks are defined as active enhancers and promoters, respectively.
To compute the block threshold described above, we use the negative binomial distribution-based predictions of H3K4me1/3 enriched regions from Macs2 (22). Specifically, the block threshold is set to X such that 99.95% of H3K4me1/3 enriched regions have tag counts above X. The block threshold is computed individually for both replicates, and the mean of the two thresholds is chosen as the final threshold. To analyze datasets having low tag coverage caused by low input sample quantity, low sequencing depth or low antibody specificity, as inferred from initial quality checks (Supplementary Figures S1 and 2), we have set the minimum value of the block threshold (X) to five. For all the datasets analyzed in this study, the computed block threshold (X) clearly separated the density distributions of normalized tag counts at histone enriched and background regions, respectively (Supplementary Figures S3 and 4).
Benchmark dataset
To benchmark the performance of the PVP-based method, we compared the regulatory activity level of our predictions (9135 enhancers and 4946 promoters) with those predicted using the classic signal and ENCODE (machine learning)-based approaches in the HeLa cell line. For fair assessment, while comparing PVP-based predictions with ENCODE defined or signal-based predictions, we trim all the regions to 1000 bp, centered from the middle position. Specifically, we created the following datasets:
Signal-based predictions
We determined the genomic regions enriched for H3K4me1, H3K4me3 and H3K27ac histone modifications in HeLa cells using Macs2 (22). To increase the specificity of the enriched regions, we used a control sample prepared using a control antibody (‘IgG’ control). Data corresponding to the three histone marks and control in replicates was downloaded (http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/) from ENCODE in BAM format (21). Further, Irreproducible Discovery Rate (IDR) (25) was used at a false discovery rate of 0.05 to filter out irreproducible regions between the two replicates. This gave us a conservative and reproducible set of 20 803 and 18 199 regions enriched for H3K4me1 and H3K4me3, respectively. Next, we selected 9286 regions (1 kb) enriched for both H3K4me1 and H3K27ac, and not overlapping with TSS, TTS or exons of known genes from GENCODE (26) as active enhancers (signal based). Similarly, 8940 regions (1 kb) enriched for both H3K4me3 and H3K27ac (N = 14 746), but not enriched for H3K4me1, were selected as active promoters (signal based).
ENCODE defined predictions
We downloaded 41 844 enhancers and 21 741 promoters defined as part of the ENCODE project in HeLa cells. Specifically, these have been predicted using two machine learning-based methods, ChromHMM (11) and Segway (12), and we include only those predicted by both methods (http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeAwgSegmentation/). Next, we selected 20 019 enhancer regions (1 kb) enriched for H3K27ac modification as active enhancers (encode defined). Similarly, 18 497 promoter regions (1 kb) enriched for H3K27ac modification were selected as active promoters (encode defined).
Dataset used to study the regulatory activity of enhancers and promoters
To study the level of regulatory activity at enhancers and promoters, we downloaded BAM files corresponding to chromatin modification (H3K27ac, H2A.Z), transcription factor (TF) binding (P300, Pol2) long-range chromatin interaction (ChIA-PET), CpG methylation and gene expression (long RNA-seq) for all four cell lines from the ENCODE project (21). Similarly, nucleosome positioning (MNase-seq) data was downloaded for GM12878 and K562 cell lines. BAM files corresponding to both the replicates of experimental assays mentioned above were downloaded (http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/). Raw GRO-seq data in FastQ format, corresponding to HeLa cell line (27), was retrieved from GEO (GSM1558745) (28) and mapped to human reference genome (hg19) using bowtie2 (default parameters) (29). Genomic coordinates corresponding to CAGE defined regulatory elements, categorized based on the direction of stable transcription in HeLa cells, were retrieved from (18). This study also contained a dataset measuring the level of divergent transcription on exosome (hRRP40) KO which we also downloaded.
To study enhancer dynamics across different lineages of hematopoiesis, raw fastq files corresponding to two replicates of ChIP-seq (H3K4me1, H3K4me3 and H3K27ac) and ATAC-seq experiments performed on HSC, CLP, GMP and MEP cells were downloaded from GEO (GSE59636 and GSE59992) (28). Similarly, fastq files corresponding to all replicates of RNA-seq experiments in these four cells were downloaded from GEO (GSE60101) (28). Reads were mapped to mouse reference genome (mm9) using bowtie2 (default parameters) (29). The activities of enhancers were compared using the mean normalized tag count (TPM) from all replicates of each assay.
Association of enhancer activity with gene expression
We consider all the genes (5′ end) within a specified distance to an enhancer (<40 000 bp) or promoter (<500 bp) region as being associated with these elements. This means that to estimate the regulatory effect of an enhancer or promoter on expression, we sum the expression of all genes associated with these elements. We used cumulative gene expression and a distance of 40 kb to link enhancers with gene expression due to two reasons. First, enhancers tend to form a loop to interact with promoters, and most of these interactions have been shown to occur within a distance of ∼50 kb from the enhancer (30,31); and, second, enhancer–promoter interactions is a many-many relationship meaning that many enhancers can regulate the expression of a single gene (19,32,33) and vice versa (34). Furthermore, the closest gene may not be a target of an enhancer (34,35). All these factors are not taken into account when using the closest gene as the putative target of an enhancer. We also note that our distance thresholds (500 and 40 000 bp), although found suitable for human and mouse genomes, are not interchangeable between organisms and should vary depending upon their genome sizes.
Analysis of enhancer or promoter dynamics across ENCODE cell lines and hematopoietic cells
We used BEDTools (multiIntersectBed) (36) to determine enhancer or promoter regions, unique or shared between multiple cell types. In a few cases, we observed a region in a cell type overlapping with more than one region in another cell type leading to its duplicate representation in the output file. Therefore, we select a unique representative of such regions as the one overlapping with a region in later cell types having the highest nfrDip score. BEDTools was also used to analyze genomic regions in bed format throughout the study (36). ngs.plot (37) and bwtool (38) were used to plot histone enrichment relative to the center of enhancers and promoters as aggregation plots and heat maps.
Enrichment analysis of PVP-based enhancers and promoters at ENCODE-defined genome segments
Binomial test is used to compute the P-value as it was done in a previous study (39). Specifically, the frequencies by which 9135 enhancers overlap with seven ENCODE defined genome segments are compared with the frequency of overlaps that can be expected under the null model where each enhancer is a dart thrown randomly onto the genome. If an ENCODE genome segment covers a fraction P of the human genome (3 billion bases), then, under the simple binomial model; each of the 9135 enhancers has the probability P of overlapping with a genomic locus. For 9135 enhancers, the expected number of overlapping enhancers is μ = 9135 * P, with a standard deviation:
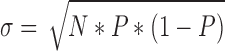 |
We then calculate the P-value using the normal approximation of the binomial distribution, pnorm function in R where;
 |
Similar analysis was performed for 4946 promoters to compute the significance of overlap.
Directionality score
The directionality of H3K4me3 at 4946 promoters is measured as proposed in a previous study (17). Specifically, the directionality score, D = (F − R)/(F + R), is computed for each promoter where F and R are the normalized tag count (TPM) of H3K4me3 1kb down- and upstream of the center of the promoter region (D = 0 means 50% reverse and 50% forward strand signal, while |D| close to 1 indicates an unidirectional H3K4me3 signal).
Enrichment analysis of transcription factor binding events
We downloaded ENCODE-defined ChiP-seq peaks, reflecting the binding of 52 TFs in GM12878, 56 TFs in Hela, 39 TFs in HepG2 and 99 TFs in K562. (http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeSydhTfbs/) (21). The total number of TF binding events within a region corresponds with the total number of peaks representing distinct TFs overlapping the region. Only the peaks belonging to the same cell line as the regions of interest were used for analysis.
Enrichment analysis of motifs at different enhancer dynamic classes
Motif enrichment was analyzed using HOMER (40). Sequences corresponding to each cell line specific category of enhancers were compared to an equal number of randomly selected genomic fragments of the average region size, matched for GC content and auto-normalized to remove bias from lower order oligo sequences. Motif enrichment was calculated on repeat masked sequences using the cumulative binomial distribution. One hundred motifs were searched for a range of motif lengths (7–14 bp), and after filtering for redundant motifs, the top 50 motifs resulting from each search were combined leading to a final set of 60 motifs. These were remapped and ranked according to enrichment (depletion) in the four cell lines.
Gene ontology analysis of genes proximal to different dynamic classes of enhancers
Gene ontology analysis was performed on genes proximal (<40 000 bp) to each cell line specific category of enhancers using clusterProfiler (41). Biological process terms from DAVID (42) were examined for each category of proximal genes and the top ten enriched terms (FDR < 0.01) from each cell line were reported.
RESULTS
Peak-valley-peak (PVP) patterns of histone modifications reveal active enhancers and promoters
We set out to compare H3K4me1 and H3K4me3 modifications patterns at 105 active and 81 inactive enhancers (experimentally validated) derived from HeLa cells (Figure 2A) (17). We observed an enrichment of an H3K4me1 PVP pattern at active enhancers that was absent at inactive enhancers (Figure 2A top and bottom). Similarly, comparison of promoters derived from the 500 highest and the 500 lowest expressed genes revealed an enrichment of an H3K4me3 PVP pattern at active promoters that was absent at inactive promoters (Figure 2B top and bottom). Thus, H3K4me1 and H3K4me3 PVP patterns distinctly characterize active enhancers and promoters, respectively. Visual inspection of the reads pileup in a genome browser further supports this observation as exemplified for enhancers in Figure 2C and for promoters in Supplementary Figure S5.
Figure 2.

Active enhancers and promoters are distinctively characterized by the H3K4me1 and H3K4me3 peak-valley-peak (PVP) pattern, respectively. (A and B) H3K4me1 and H3K4me3 PVP patterns at enhancers and promoters are categorized based on their activity in HeLa cells. (A) Active (N = 105) and inactive (N = 81) enhancers are defined based on their activity in luciferase assays (17). (B) Active (N = 500) and inactive (N = 500) promoters are defined based on the expression of the proximal gene. Normalized signal intensity (Tags Per Million; TPM) is plotted by centering all elements to their mid points, flanked by 1000 bp each. The valley defined by H3K4me1 and H3K4me3 at active enhancers and promoters, respectively, reflects the open chromatin state of the regulatory elements (NFR). (C) A representative example of two enhancer regions defined based on enrichment of the H3K4me1 signal in HeLa cells. While the one on the right is active as evident from the GRO-seq and Pol2 binding signal, the one on the left is inactive since it does not possess similar signs of regulatory activity. Also evident is the distinctive presence of the H3K4me1 defined PVP pattern at active enhancers while being absent at inactive enhancers.
To systematically analyze H3K4me1/me3 PVP patterns on a genome-wide scale, we developed a computational method for their efficient prediction. We reasoned that PVP patterns predicted using H3K4me1 and H3K4me3 modification data would reflect the genomic position of active enhancers and promoters, respectively (Figure 1). Briefly, our method scans the genome for regions of H3K4me1/me3 that follow a Gaussian density distribution. Next, regions flanked by Gaussian distribution(s) within a predefined range of width are selected as NFRs. Finally, NFRs exhibiting depletion of H3K4me1 and H3K4me3 signals, relative to the flanking regions (nfrDip score) at an FDR of <0.05, are defined as potential enhancers and promoters, respectively (see ‘Materials and Methods’ section). We note that two important assumptions are made during this study. First, enhancers are considered as entities that mediate activation of gene expression (classical definition), which may not hold true if the TF binding at an NFR (as defined by the H3K4me1 PVP pattern) acts as a repressor. Second, a region devoid of H3K4me1/me3 modification (valley in PVP pattern) is referred to as an NFR that more generally should be considered as depleted of nucleosomes and with boundaries that do not always correspond with those of an NFR.
The PVP-based approach distinctively identifies active enhancers and promoters
To assess the efficacy of our method, we analyzed H3K4me1/me3 PVP patterns in HeLa cells and predicted 9135 active enhancers (median width of 377 bp) and 4946 active promoters (median width of 163 bp). The DNase hypersensitivity signal reflecting an open chromatin state correlated well the width of NFRs defined at both enhancers (Figure 3A) and promoters (Figure 3B). Most predicted enhancers overlapped with ENCODE-defined enhancers (N = 5447; 60%; P-value < 5e-324; Binomial test) and were more than 1 kb away from the known transcription start sites (TSS) (7548; 83%; P-value < 5e-324; Fisher's exact test) (Figure 3C and D). Similarly, the majority of predicted promoters overlapped with ENCODE-defined TSS (N = 3087; 62%; P-value < 5e-324; Binomial test) and were within 1 kb to known TSS (4017; 81%; P-value = 3.2e-116; Fisher's exact test) (Figure 3C and D).
Figure 3.

Predictions based on H3K4me1 and H3K4me3 PVP patterns show characteristic features of active enhancers and promoters, respectively. (A) DNase hypersensitivity (DHS) signal at H3K4me1 PVP-based enhancers (N = 9.135). Heat map displays the DHS signal at enhancers sorted by the width of their central PVP pattern and flanked by 2000 bp to the mid point. (B) Same as A, but for H3K4me3 PVP-based promoters (N = 4946). (C) Percentage overlap of enhancers and promoters with seven distinct regulatory regions defined by ENCODE in HeLa cells (CTCF: CTCF enriched; E: enhancer; PF: promoter flanking; R: repressed; T: transcribed; TSS: transcription start site; WE: weak enhancer). (D) Proximity of PVP-based enhancers and promoters to TSS of GENCODE defined genes. P-values represent enrichment against a null hypothesis of equal overlap (50%) to both categories (x-axis) (26). (E) Exosome sensitivity of divergent transcripts from enhancers and promoters. Divergent transcription leading to the formation of exosome sensitive unstable transcripts is a characteristic feature of enhancers. (F) Percentage overlap of enhancers (N = 2104) and promoters (N = 3191) with regions classified based on the nature of divergent transcription (bidirectional stable, unidirectional stable or unstable). (G) Spearman rank correlation between the depth of the PVP pattern (nfrDip; see ‘Materials and Methods’ section) and the divergent transcription signal as measured by GRO-seq and CAGE tags.
Active enhancers are known to produce bidirectional unstable transcripts that are rapidly degraded by exosomes (18,43) and, in concordance, we observed a higher exosome sensitivity of divergent transcripts at predicted enhancers as compared to promoters (Figure 3E). Furthermore, 2104 out of 9135 enhancers (23%) showed signs of divergent transcription and 79% of these (1667 out of 2104) were enriched for bidirectional unstable transcripts (Figure 3F). Similarly, 3191 out of 4946 promoters (65%) showed signs of divergent transcription and 81% of these (2572 out of 3191) were enriched for bi- or unidirectional stable transcripts (Figure 3F). Most of our predictions were supported by at least two GRO-seq (7949 out of 9135 enhancers and 4682 out of 4946 promoters) or CAGE (5455 out of 9135 enhancers and 4657 out of 4946 promoters) tags. Strikingly, we also observed a significant correlation between the depth of the PVP pattern (nfrDip score; see ‘Materials and Methods’ section) and intensity of GRO-seq (ρ = 0.45; P-value < 5e-324) and CAGE (ρ = 0.63; P-value < 5e-324) signal at our combined predictions of GRO-seq or CAGE supported enhancers and promoters (Figure 3G). Thus, besides being a reflection of the active state of enhancers and promoters, the PVP pattern also signifies their level of activity.
PVP-based predictions are enriched for regulatory activity as compared to existing methodologies
To assess the advantage conferred by the PVP-based approach, we compared our predictions of enhancers (N = 9135) and promoters (N = 4946) with signal-based and ENCODE-defined (machine learning based) enhancers and promoters (all trimmed to 1000 bp; see the ‘Materials and Methods’ section). We observed an enrichment of positive regulatory activity at our enhancer predictions, as measured by the frequencies of TF binding events (Figure 4A), distant chromatin interactions (chromatin interaction analysis by paired-end tag sequencing (ChIA-PET); Figure 4B), reduced CpG methylation (Figure 4C) and higher level of active transcription as measured by GRO-seq (Figure 4D) and CAGE (Figure 4E). Similar enrichment of activity was observed at PVP based promoters (Supplementary Figure S6). Further, we also observed significantly higher expression levels of genes in proximity to PVP-based enhancers (<40 000 bp) and promoters (<500 bp) as compared to gene expression in proximity to ENCODE defined or signal-based enhancers and promoters (P-value < 0.01; Mann–Whitney test) (Figure 4F and Supplementary Figure S6), respectively. High expression of genes proximal to PVP-based predictions was also observed when enhancers and promoters were associated with the closest gene instead of with gene(s) located within 40 kb and 500 bp distance, respectively (see ‘Materials and Methods’ section) (Supplementary Figure S7).
Figure 4.

Predictions based on the PVP pattern are enriched for positive regulatory activity as compared to those predicted by the ENCODE and signal-based approaches. The activity of 9135 PVP-based enhancers are compared to 9286 signal based and 20 019 ENCODE defined enhancers by measuring (A) the number of transcription factor (TF) binding events determined based on the frequency of distinct ChIP-seq peaks representing 55 TFs at each prediction, (B) percentage of distal chromatin interactions as determined by ChIA-PET, (C) percentage of CpG methylated reads, (D and E) level of divergent transcription determined based on the number of GRO-seq and CAGE tags; and, (F) expression of gene(s) proximal to enhancers (<40 000 bp).
The majority of the PVP-based enhancers and promoters overlapped with ENCODE defined enhancers (66%; 5998 out of 9135) and promoters (79%; 3900 out of 4946), respectively (Supplementary Figure S8). Similar overlap was also observed for PVP-based predictions in three more ENCODE cell lines (GM12878, HepG2 and K562) (Supplementary Figure S9). Due to the reason that we use a fixed width of CREs (1000 bp), thus accounting for several proximal ENCODE predictions as overlap, we now observe a higher degree of overlap between PVP and ENCODE predictions as compared to the one reported in the previous section for HeLa cells (Supplementary Figure S10). Regardless of their overlap status, our predictions were consistently enriched for activity in comparison to ENCODE and/or signal-based predictions, as measured in terms of exosome sensitivity, divergent transcription, H3K27ac and Pol2 signal (Supplementary Figures S8 and 11). Several novel or misannotated promoters and enhancers showed characteristic features of active regulatory elements (Supplementary Figures S12–14). Furthermore, we observed similar enrichment levels of 55 TFs binding at ENCODE-defined and PVP-based enhancers (Supplementary Figure S15). Also, PVP-based enhancers and promoters are distributed across the genome (5′ UTR, intron, intergenic) in a proportion similar to that observed for ENCODE-defined enhancers and promoters (Supplementary Figure S16). These results suggest that the PVP-based approach identifies a subset of elements identified by other methods, with a high specificity for regions having active roles in transcriptional regulation.
Benchmark of enhancer predictions to experimentally validated enhancers
We benchmarked our method against 105 active and 81 inactive enhancers which were experimentally validated in the HeLa cell line using luciferase assay (17). Notably, positive activity in luciferase assays is not a definite proof of in vivo enhancer activity for reasons including (but not limited to) failure to recapitulate chromatin structure (9). However, we reasoned that this dataset is useful to benchmark the performance of our approach due to the fact that it was experimentally tested in the same cell line (HeLa) and predicted using a robust indicator of activity (divergent transcription). We recovered 40 out of 105 active enhancers (dark blue; true positive), while recovering nine out of 81 inactive enhancers (dark green; false positive) (Figure 5A). Importantly, recovered active enhancers (TP) showed significantly higher activity as compared to those not recovered (FP) by our approach (P-value < 0.01; Mann–Whitney test). This observation was consistent across diverse activity criteria such as Pol2 binding, GRO-seq, P300, CAGE and histone marks (Figure 5B). Strikingly, the nine enhancers that showed no activity in luciferase assay, but were predicted by the PVP-based approach (FP), also displayed high regulatory activity (Figure 5B). Interestingly, five out of nine enhancers have been annotated as TSS by ENCODE, perhaps due to a higher level of H3K4me3 as compared to the levels observed at the remaining four elements that were correctly annotated as enhancers. Indeed, the negligible polyA RNA-seq signal and lack of detectable gene structures further point toward a role of these five elements as enhancers (Supplementary Figure S14). As expected, inactive enhancers not recovered by the PVP-based approach (TN) showed a minimum level of activity.
Figure 5.

Benchmarking of PVP-based enhancer predictions using experimentally validated enhancers in HeLa cells. (A) Success rate by which experimentally validated active enhancers are recovered by the PVP-based approach. Shown as pie chart is the frequency of active (blue shade) and inactive (green shade) enhancers, categorized based on their recovery by the PVP-based approach. Blue and green color shades represent a total of 105 active (blue shade) and 85 inactive (green shade) enhancers, respectively. True positive (TP; dark blue) represents the 40 active enhancers correctly recovered, false negative (FN; light blue) represents the 65 active enhancers that were not recovered, false positive (FP; dark green) represents the nine inactive enhancers recovered as active and true negative (TN; light green) represents the 72 inactive enhancers that were not recovered. (B) Regulatory activity of 105 active and 85 inactive enhancers belonging to four prediction categories (TP, FN, FP and TN) measured in terms of signals from seven experimental assays (Pol2, GRO-seq, P300, CAGE, H3K4me1, H3K27ac and H3K4me3). (C) Sensitivity and specificity of the PVP-based approach and state of the art method, DEEP (16) measured based on their prediction performance on experimentally validated active and inactive enhancers.
Quantitatively, our method showed a low sensitivity of 0.39, but a high specificity of 0.89. (Figure 5C). To put this into perspective, we compared the performance of our method with that of a state of the art method, DEEP (16), using the same benchmark dataset (Figure 5C). In total, DEEP recovered 78 out of 105 active enhancers while recovering 39 out of 81 inactive enhancers, thus showing a high sensitivity of 0.74 but a low specificity of 0.52 (Figure 5C). The high sensitivity of DEEP, as compared to the PVP-based approach, can invariably be attributed to the large number of putative enhancers (N = 133 914) predicted by DEEP, which is ∼15-fold higher than 9135 enhancers predicted by our approach. This highlights the primary drawback of many current approaches as they predict a large number of putative enhancers without taking into account any definite activity criteria which is more important when the goal is to compare the activity of regulatory elements across different time points or physiological conditions. By predicting active regulatory elements with high specificity, we have addressed this drawback. Taken together, these results suggest that the PVP pattern-based approach identifies high-confidence active regulatory elements.
The differential ratio of H3K4me1/me3 up- and downstream to promoters reflects their directionality toward stable transcription
After establishing the depth of the PVP pattern (nfrDip) as an indicator of the activity of regulatory elements, we analyzed the additional two characteristics of the PVP pattern (asymmetry and width). We observed distinct H3K4me1/me3 profiles at 24 007 DNase hypersensitive sites (DHS) categorized based on the directionality of stable and unstable transcription (divergent transcription) in the HeLa cell line (18) (Figure 6A). Specifically, H3K4me3 modification was enriched toward the direction of stable transcription and H3K4me1 modification was enriched toward the direction of unstable transcription, although at a relatively small scale (Figure 6A). Upon plotting the two histone modifications (H3K4me1/3) and CAGE tags (stable transcription) at 4946 PVP-based promoters, we observed a similar trend where directionality of stable transcription correlated with that of H3K4me3 enrichment (Figure 6B). Further, upon using RNA-seq (polyA) read count as a measure for proximal gene expression, we also observed higher gene expression in the direction of H3K4me3 enrichment (Figure 6B). However, we did not observe a correlation between the directionality score of H3K4me3 and the proximal gene expression that is almost constant (Figure 6B).
Figure 6.

The distinct ratio of H3K4me3 and H3K4me1 up- and downstream to promoters is useful for the prediction of directionality of stable transcription. (A) The normalized H3K4me1 and H3K4me3 signal (TPM) at regulatory elements categorized based on the presence and direction of stable divergent transcripts (18). The signal is plotted by centering to the mid point of elements, flanked by 1000 bp each. The arrows above the plots represent the direction of stable (black) and unstable (gray) transcription. The histone profile at bi- (Stable-Stable; SS) and unidirectional (Stable-Unstable; SU and Unstable-Stable; US) stable transcribed elements was used to train a random forest classifier that was later used to predict directionality of stable transcription. (B) The normalized H3K4me1/me3 and polyA RNA-seq signal (TPM) at 4946 PVP-based promoters sorted by their directionality score (see ‘Materials and Methods’ section). The signal is plotted by centering to the mid point of promoters, flanked by 3000 bp each. Also shown is the fold change (log2) in normalized CAGE and polyA RNA-seq tag counts (TPM) between forward (pos.) and reverse (neg.) strands, along with total RNA-seq signal as a measure for proximal gene expression.
In order to explore the potential of the H3K4me1/me3 profile for predicting the directionality of stable transcripts, we first excluded the 10 999 DHS associated with bidirectional unstable transcription (UU in Figure 6A; mostly enhancers) and subsequently analyzed the remaining 13 008 DHS associated with stable transcription from at least one end (SS, SU, US in Figure 6A; mostly promoters). We used the normalized signal (tags per million; TPM) of two histone marks (H3K4me1 and H3K4me3) at 2682 SS (stable-stable), 4838 SU (stable-unstable) and 5488 US (unstable-stable) DHS binned into four 250 bp windows (two up- and two downstream to the mid-point) to train a random forest classifier. On classifying our 4946 PVP-based promoter predictions in HeLa cells, we observed 389, 2255 and 2302 promoters exhibiting SS, SU and US divergent transcription, respectively. As expected, we observed a higher enrichment of unstable transcripts upon exosome (hRRP40) KO at SU and US promoters, more specifically toward the direction of unstable transcription than toward the direction of stable transcription (Supplementary Figure S17). This suggests that reasonably accurate predictions of the directionality of stable transcription can be made using the H3K4me1/me3 modification signal profiles at promoters.
H3K4me1/me3 modifications are deposited in spatially distinct patterns at nucleosomes flanking regulatory elements
We next asked whether the PVP patterns at enhancers and promoters have different width. We observed that H3K4me1 PVP patterns at enhancers are significantly broader than H3K4me3 PVP patterns at promoters (median ∼400 versus ∼150 bp, P-value < 0.01; Mann–Whitney test) across four human cell lines analyzed (GM12878, HeLa, HepG2, K562) (Supplementary Figure S18). To examine this difference in relation to the positioning of nucleosomes, we overlaid nucleosome positioning (MNase-seq) and the H2A.Z variant signals with H3K4me1 and H3K4me3 signals (all scaled between 0 and 1) at predicted enhancers and promoters in the GM12878 (Figure 7A) and K562 (Figure 7B) cell lines. We analyzed these two cell lines due to the availability of MNase-seq data.
Figure 7.

Deposition patterns of histone modifications at nucleosomes flanking enhancers and promoters are spatially distinct. (A) Scaled signal reflecting the H2A.Z (black), H3K4me1 (blue) and H3K4me3 (green) signals at nucleosomes (red; MNase-seq) flanking 8215 enhancers (left; top panel) and 8441 promoters (right; top panel) predicted in the GM12878 cell line, respectively. Blue and green arrows represent the approximate position of flanking nucleosomes that are most enriched for H3K4me1 and H3K4me3/H2A.Z modifications, respectively. (B) Same as A, but for 3183 enhancers (left) and 8435 promoters (right) predicted in the K562 cell line.
First, at both enhancers and promoters, nucleosomes flanking the NFR are positioned at around the same distance (red lines) which is in agreement with a recent study (20) and suggests that the widths of NFRs at both enhancers and promoters is similar. Second, the highest enrichments (peak point) for H2A.Z and H3K4me3 modifications are consistently observed at the nucleosomes relatively close to the center of the NFR as compared with the nucleosomes enriched for H3K4me1 modification at both enhancers and promoters (Figure 7A and B). This may point toward a mechanistic correspondence in the pattern by which the H2A.Z variant and the H3K4me3 mark are deposited at nucleosomes which appears to be different from the pattern by which the H3K4me1 mark is deposited at the flanking nucleosomes. Further, similar to the H3K4me3 modification, we observed a significantly higher enrichment of H2A.Z modification at promoters as compared with enhancers (P-value < 0.01; Mann–Whitney test). This suggests that, similar to the H3K4me3 mark (20), the presence of the H2A.Z variant could also reflect Pol2 driven divergent transcription at regulatory elements which is much higher at promoters as compared with enhancers.
Activity dynamics of regulatory elements across four ENCODE cell lines and four hematopoietic stem/progenitor cells
We next asked whether PVP patterns could be used to study spatiotemporal dynamics in the activity of regulatory elements, and therefore analyzed H3K4me1 PVP patterns in four human ENCODE cell lines (GM12878, HeLa, HepG2 and K562) to predict enhancers. Due to the distinct phenotypes of these four cell lines, the majority of the enhancers (28 940 out of 30 969; 93%) were specific to a single cell line (44) (Figure 8 and Supplementary Table S1). We observed a strong correlation between the cell line specificity and activity of enhancers as measured in terms of the H3K27ac PVP pattern, Pol2-binding and proximal gene expression (Figure 8). Interestingly, cell line specific enhancers were also enriched for motifs that are known to play important roles in defining the identities of these four cell lines (Supplementary Figure S19A). Examples are HNF4A and POU2F2 motifs, critical in hepatocytes and immune cells, that were enriched in HepG2 and GM12878 cell lines, respectively (45,46). Similar specificity in disease ontology was also observed for genes proximal to cell line specific enhancers such as leukemia in GM12878, reproductive organ cancer in HeLa and lipid storage disease in HepG2 (Supplementary Figure S19B). Unlike enhancers, promoters are less cell line specific (11 098 out of 16 954; 66%) (Supplementary Figure S20), and we observed 900 promoter regions that were active in all four cell lines. These promoters were significantly enriched (369 out of 900; P-value = 1.4e-13; Fisher's exact test) in proximity (<500 bp) to the TSS of housekeeping genes (N = 3919) (47).
Figure 8.

Activity dynamics of enhancers predicted across four human cell lines. (A) Frequency of PVP-based enhancers classified based on their activity dynamics across four cell lines. Filled circles represent the cell line in which the enhancers are active. (B) Normalized H3K27ac modification signal (TPM) at enhancers centered to their mid points (flanked by 1000 bp each) across four cell lines. (C) Normalized Pol2 binding signal (TPM) at enhancers across four cell lines. (D) Normalized expression (TPM) of genes proximal (<40 000 bp) to enhancers across four cell lines.
Due to the distinct phenotype of cell lines analyzed above, we observed a clear cell line specificity in the activity of most enhancers. To explore the potential of the PVP-based approach in detecting enhancer specificity in much more closely related cells, we analyzed H3K4me1 PVP patterns using a recently published ChIP-seq dataset from four different hematopoietic stem/progenitor populations (HSC: hematopoietic stem cells, CLP: common lymphoid progenitors, GMP: granulocyte monocyte progenitor and MEP: megakaryocyte erythroid progenitor) (48). We predicted a total of 43 999 enhancers (Supplementary Table S2). Out of these, 29 975 enhancers are novel (PVP-only) and showed significantly higher activity (H3K27ac signal and proximal gene expression) as compared with the remaining 14 024 enhancers that we recovered (common), and also against 27 983 enhancers that were only predicted in the original study (48) (Lara-A et al.) for which we observed the lowest activity (Supplementary Figure S21A–C). We hypothesized that many of the more active novel enhancers were missed previously because they are also enriched for the H3K4me3 modification (Supplementary Figure S21D), a filter used in the original study to exclude promoters. Recent studies have shown that this criterion is not necessarily correct (17–19,43) because H3K4me3 is correlated with the activity of both enhancers and promoters (43). As expected, PVP-based enhancers were enriched for the H3K4me3 mark but this enrichment was lower than that observed at known TSS (Supplementary Figure S21D). We observed several enhancers whose activity is specific to each of the four cell types (HSC, CLP, GMP and MEP), and is supported by cell type specific chromatin accessibility and proximal gene expression patterns (Supplementary Figure S21E–H). Furthermore, these specificity patterns were reproducible for all the three classes of enhancers (PVP-only, common and Lara-A et al. only) (Supplementary Figures S21–23). Finally, the classification of enhancer dynamics is also supported by an enrichment of binding motifs for lineage specific TFs in their respective classes (Supplementary Figure S21I). Hence, GATA motifs are enriched in MEPs while CEBPA and PU.1 motifs are enriched in GMPs, reflecting the importance of GATA1 and CEBPA/PU.1, respectively, in these lineages. Taken together, analysis of these two datasets suggests that the PVP approach is robust in classifying distinct classes of regulatory elements based on dynamics in their activity, thus enabling effective analysis of their spatiotemporal activity.
DISCUSSION
Several studies have shown PVP histone modification patterns at regulatory elements as a manifestation of their active state (5,6,49,50). However, the different characteristic features of these patterns and their robustness in predicting active regulatory elements have never been systematically analyzed. Due to the unavailability of computational approaches to effectively capture this pattern, most standard analyses of histone modification data are based on enrichments of their signals. As such enrichment can be observed at both active and poised (‘primed to be active’) elements, it invariably predicts a collection of both active and currently inactive regulatory elements (3,19). This may result in an over-estimation of the regulatory complexity of the cell.
In this study, we systematically analyze the PVP patterns of H3K4me1 and H3K4me3 modifications in order to detect and characterize distinct properties of active enhancers and promoters. Specifically, we show that, besides being highly efficient in distinguishing between enhancers and promoters, the depth of the valley in the PVP pattern (nfrDip) also reflects the activity of these regulatory elements. This allows for measuring the activity of regulatory elements across different spatiotemporal conditions by using only single histone marks. Indeed, we predict several enhancer elements that showed strong ENCODE cell line specificity in their activity as determined by their positive correlation with the H3K27ac signal, Pol2 binding and proximal gene expression. We observed a higher percentage of cell-line specific enhancers (∼90%) as compared to that reported previously (∼80%) (44). This may be due to the reason that by enriching for activity, our approach regards enhancers that are active in one cell line but poised or silent in other cell line(s) as cell line specific, which otherwise (using the signal-based approach) are considered common between cell lines (44). Most of our enhancer and promoter predictions overlap with regulatory elements identified by ENCODE and show consistently high activity. Conversely, many ENCODE predictions were not recovered by our approach, primarily due to their low activity level suggesting that they might be silent or in a poised state. Similarly, we recover some regions that are annotated as TSS by ENCODE, presumably due to a relatively high H3K4me3 signal, as enhancers. Indeed, a low in vitro validation rate of ENCODE defined enhancers and enrichment of H3K4me3 as a mark for the activity of both enhancers and promoters, as opposed to it being a distinguishing feature of promoters, has also been shown in recent studies (17,19,20). Finally, we also benchmark our approach by recovering experimentally validated enhancers at a high specificity (89%). Intriguingly, we also recovered nine enhancers that showed characteristic signatures of activity but were previously annotated as inactive in HeLa cells (17). Apart from limitations of luciferase assay, extensive genomic rearrangements in HeLa cells potentially leading to loss of necessary genomic context for enhancers, can be probable reasons behind this observation (51).
More direct evidence of enhancer and promoter activities is the level of divergent transcription at these elements (17,20) as detected by GRO-seq and CAGE technologies. However, due to the low level of transcription and high exosome sensitivity of divergent transcripts, especially at enhancers, both these methods require relatively high levels of input material to effectively capture these elements (17,18,35). Due to the comparatively less stringent requirement of sample amounts, histone ChIP-seq is therefore the method of choice in order to capture regulatory elements for low-abundant cell types such as stem and progenitor cells. Here we have shown that this can be done with high specificity by employing the PVP approach. Furthermore, we show that H3K4me3 and H3K4me1 modifications flanking the central promoter are enriched toward the direction of stable and unstable transcription, respectively. This differential histone enrichment correlates with asymmetry in the direction of stable transcription at promoters, which in turn has been implicated with the evolution of new protein coding RNAs from unstable RNAs (43,52). However, whether differential enrichment of histone marks is a consequence or determinant of divergent transcription is yet not clear. We show that histone asymmetry can be used to predict the directionality of stable transcription at promoters. Further studies of the H3K4me1/me3 PVP pattern can lead to a higher accuracy in the predictions. Intriguingly, H3K4me1 and H3K4me3 modifications are deposited in a spatially distinct pattern with respect to the center of the PVP pattern (NFR). We confirm this distinct pattern in two ENCODE cell lines (GM12878 and K562) using nucleosome positioning and H2A.Z variant data, suggesting that H3K4me3 and H2A.Z modifications are enriched at nucleosomes positioned relatively closer to the center of the NFR than the ones enriched for H3K4me1 modification. One possible explanation for this distinct spatial arrangement has been suggested recently in the form of a unique role of H3K4me1 in establishing boundaries at promoters, thus restricting the recruitment of chromatin-modifying enzymes to a defined region (53). In view of our observations, this role of H3K4me1 is also plausible at enhancers.
Based solely on modifications of a single histone tail residue, we anticipate that our approach will be of general interest for the prediction of active regulatory elements in tissue samples where limited ChIP-seq data is available. Also, due to its simple formulation, the proposed method is applicable to virtually any ChIP-based assay where it has the potential to greatly increase the specificity of the predicted CREs.
AVAILABILITY
An implementation of the PVP-based approach as a computational method, PARE (Predict Active Regulatory Elements) is freely available at http://servers.binf.ku.dk/pare.
Supplementary Material
Acknowledgments
We thank Janus Schou Jakobsen, Albin Sandelin and Chirag Nepal for critical reading of this manuscript. We also thank the ENCODE and FANTOM consortium for making their data publicly available.
Footnotes
Present address: Frederik Otzen Bagger, EMBL-EBI, Welcome Trust Genome Campus, Hinxton and Department of Haematology, University of Cambridge, Cambridge, UK.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
NovoNordisk Foundation; Danish Research Council for Strategic Research; The Lundbeck Foundation; NovoNordisk Foundation centre grant (The Novo Nordisk Foundation section for Stem Cell biology in Human Disease). Funding for open access charge: NovoNordisk Foundation; Danish Research Council for Strategic Research.
Conflict of interest statement. None declared.
REFERENCES
- 1.Shlyueva D., Stampfel G., Stark A. Transcriptional enhancers: from properties to genome-wide predictions. Nat. Rev. Genet. 2014;15:272–286. doi: 10.1038/nrg3682. [DOI] [PubMed] [Google Scholar]
- 2.Butler J.E.F., Kadonaga J.T. The RNA polymerase II core promoter: a key component in the regulation of gene expression. Genes Dev. 2002;16:2583–2592. doi: 10.1101/gad.1026202. [DOI] [PubMed] [Google Scholar]
- 3.Calo E., Wysocka J. Modification of enhancer chromatin: what, how, and why? Mol. Cell. 2013;49:825–837. doi: 10.1016/j.molcel.2013.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pundhir S., Poirazi P., Gorodkin J. Emerging applications of read profiles towards the functional annotation of the genome. Front. Genet. 2015;6:1–11. doi: 10.3389/fgene.2015.00188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoffman B.G., Robertson G., Zavaglia B., Beach M., Cullum R., Lee S., Soukhatcheva G., Li L., Wederell E.D., Thiessen N., et al. Locus co-occupancy, nucleosome positioning, and H3K4me1 regulate the functionality of FOXA2-, HNF4A-, and PDX1-bound loci in islets and liver. Genome Res. 2010;20:1037–1051. doi: 10.1101/gr.104356.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaikkonen M.U., Spann N.J., Heinz S., Romanoski C.E., Allison K.a., Stender J.D., Chun H.B., Tough D.F., Prinjha R.K., Benner C., et al. Remodeling of the enhancer landscape during macrophage activation is coupled to enhancer transcription. Mol. Cell. 2013;51:310–325. doi: 10.1016/j.molcel.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bonn S., Zinzen R.P., Girardot C., Gustafson E.H., Perez-Gonzalez A., Delhomme N., Ghavi-Helm Y., Wilczyński B., Riddell A., Furlong E.E.M. Tissue-specific analysis of chromatin state identifies temporal signatures of enhancer activity during embryonic development. Nat. Genet. 2012;44:148–156. doi: 10.1038/ng.1064. [DOI] [PubMed] [Google Scholar]
- 8.Heintzman N.D., Stuart R.K., Hon G., Fu Y., Ching C.W., Hawkins R.D., Barrera L.O., Van Calcar S., Qu C., Ching K.A., et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- 9.Arnold C.D., Gerlach D., Stelzer C., Boryń Ł.M., Rath M., Stark A. Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science. 2013;339:1074–1077. doi: 10.1126/science.1232542. [DOI] [PubMed] [Google Scholar]
- 10.Pekowska A., Benoukraf T., Zacarias-Cabeza J., Belhocine M., Koch F., Holota H., Imbert J., Andrau J.-C., Ferrier P., Spicuglia S. H3K4 tri-methylation provides an epigenetic signature of active enhancers. EMBO J. 2011;30:4198–4210. doi: 10.1038/emboj.2011.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ernst J., Kellis M. ChromHMM: automating chromatin-state discovery and characterization. Nat. Methods. 2012;9:215–216. doi: 10.1038/nmeth.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hoffman M.M., Buske O.J., Wang J., Weng Z., Bilmes J.A., Noble W.S. Unsupervised pattern discovery in human chromatin structure through genomic segmentation. Nat. Methods. 2012;9:473–476. doi: 10.1038/nmeth.1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Firpi H.A., Ucar D., Tan K. Discover regulatory DNA elements using chromatin signatures and artificial neural network. Bioinformatics. 2010;26:1579–1586. doi: 10.1093/bioinformatics/btq248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rajagopal N., Xie W., Li Y., Wagner U., Wang W., Stamatoyannopoulos J., Ernst J., Kellis M., Ren B. RFECS: a random-forest based algorithm for enhancer identification from chromatin state. PLoS Comput. Biol. 2013;9:e1002968. doi: 10.1371/journal.pcbi.1002968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu Y., Sun L., Chen Z., Whitaker J.W., Wang T., Wang W. Predicting enhancer transcription and activity from chromatin modifications. Nucleic Acids Res. 2013;41:10032–10043. doi: 10.1093/nar/gkt826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kleftogiannis D., Kalnis P., Bajic V.B. DEEP: a general computational framework for predicting enhancers. Nucleic Acids Res. 2014;43:e6. doi: 10.1093/nar/gku1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andersson R., Gebhard C., Miguel-Escalada I., Hoof I., Bornholdt J., Boyd M., Chen Y., Zhao X., Schmidl C., Suzuki T., et al. An atlas of active enhancers across human cell types and tissues. Nature. 2014;507:455–461. doi: 10.1038/nature12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Andersson R., Refsing Andersen P., Valen E., Core L., Bornholdt J., Boyd M., Heick Jensen T., Sandelin A. Nuclear stability and transcriptional directionality separate functionally distinct RNA species. Nat. Commun. 2014;5:5336. doi: 10.1038/ncomms6336. [DOI] [PubMed] [Google Scholar]
- 19.Andersson R. Promoter or enhancer, what's the difference? Deconstruction of established distinctions and presentation of a unifying model. Bioessays. 2014;37:314–323. doi: 10.1002/bies.201400162. [DOI] [PubMed] [Google Scholar]
- 20.Core L.J., Martins A.L., Danko C.G., Waters C.T., Siepel A., Lis J.T. Analysis of nascent RNA identifies a unified architecture of initiation regions at mammalian promoters and enhancers. Nat. Genet. 2014;46:1311–1320. doi: 10.1038/ng.3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Consortium E.P., others An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anders S., Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Langenberger D., Bermudez-Santana C., Hertel J., Hoffmann S., Khaitovich P., Stadler P.F. Evidence for human microRNA-offset RNAs in small RNA sequencing data. Bioinformatics. 2009;25:2298–2301. doi: 10.1093/bioinformatics/btp419. [DOI] [PubMed] [Google Scholar]
- 25.Li Q., Brown J., Huang H., Bickel P. Measuring reproducibility of high-throughput experiments. Ann. Appl. Stat. 2011;5:1752–1779. [Google Scholar]
- 26.Derrien T., Johnson R., Bussotti G., Tanzer A., Djebali S., Tilgner H., Guernec G., Martin D., Merkel A., Knowles D.G., et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22:1775–1789. doi: 10.1101/gr.132159.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duttke S.H.C., Lacadie S.A., Kadonaga J.T., Ohler U., Duttke S.H.C., Lacadie S.A., Ibrahim M.M., Glass C.K., Corcoran D.L. Article human promoters are intrinsically directional. Mol. Cell. 2015;57:1–11. doi: 10.1016/j.molcel.2014.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barrett T., Wilhite S.E., Ledoux P., Evangelista C., Kim I.F., Tomashevsky M., Marshall K.A., Phillippy K.H., Sherman P.M., Holko M., et al. NCBI GEO: archive for functional genomics data sets - update. Nucleic Acids Res. 2013;41:D991–D995. doi: 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chepelev I., Wei G., Wangsa D., Tang Q., Zhao K. Characterization of genome-wide enhancer-promoter interactions reveals co-expression of interacting genes and modes of higher order chromatin organization. Cell Res. 2012;22:490–503. doi: 10.1038/cr.2012.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ing-Simmons E., Seitan V., Faure A., Flicek P., Carroll T., Dekker J., Fisher A., Lenhard B., Merkenschlager M. Spatial enhancer clustering and regulation of enhancer-proximal genes by cohesin. Genome Res. 2015;25:504–513. doi: 10.1101/gr.184986.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hong J.-W., Hendrix D.A., Levine M.S. Shadow enhancers as a source of evolutionary novelty. Science. 2008;321:1314. doi: 10.1126/science.1160631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ferretti E., Cambronero F., Tümpel S., Longobardi E., Wiedemann L.M., Blasi F., Krumlauf R. Hoxb1 enhancer and control of rhombomere 4 expression: complex interplay between PREP1-PBX1-HOXB1 binding sites. Mol. Cell. Biol. 2005;25:8541–8552. doi: 10.1128/MCB.25.19.8541-8552.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pennacchio L.A., Bickmore W., Dean A., Nobrega M.A., Bejerano G. Enhancers: five essential questions. Nat. Rev. Genet. 2013;14:288–295. doi: 10.1038/nrg3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Whitaker J.W., Nguyen T.T., Zhu Y., Wildberg A., Wang W. Computational schemes for the prediction and annotation of enhancers from epigenomic assays. Methods. 2015;72:86–94. doi: 10.1016/j.ymeth.2014.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shen L., Shao N., Liu X., Nestler E. ngs.plot: quick mining and visualization of next-generation sequencing data by integrating genomic databases. BMC Genomics. 2014;15:284. doi: 10.1186/1471-2164-15-284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pohl A., Beato M. bwtool: a tool for bigWig files. Bioinformatics. 2014;30:1618–1619. doi: 10.1093/bioinformatics/btu056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pundhir S., Gorodkin J. Differential and coherent processing patterns from small RNAs. Sci. Rep. 2015;5:12062. doi: 10.1038/srep12062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu G., Wang L.-G., Han Y., He Q.-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. Omi. A J. Integr. Biol. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang D.W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 43.Core L., Waterfall J., Lis J. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science. 2008;322:1845–1848. doi: 10.1126/science.1162228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Heintzman N.D., Hon G.C., Hawkins R.D., Kheradpour P., Stark A., Harp L.F., Ye Z., Lee L.K., Stuart R.K., Ching C.W., et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–112. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jiang G., Nepomuceno L., Hopkins K., Sladek F.M. Exclusive homodimerization of the orphan receptor hepatocyte nuclear factor 4 defines a new subclass of nuclear receptors. Mol. Cell. Biol. 1995;15:5131–5143. doi: 10.1128/mcb.15.9.5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.García-Cosío M., Santón A., Martín P., Camarasa N., Montalbán C., García J.F., Bellas C. Analysis of transcription factor OCT.1, OCT.2 and BOB.1 expression using tissue arrays in classical Hodgkin's lymphoma. Mod. Pathol. 2004;17:1531–1538. doi: 10.1038/modpathol.3800227. [DOI] [PubMed] [Google Scholar]
- 47.Eisenberg E., Levanon E.Y. Human housekeeping genes, revisited. Trends Genet. 2013;29:569–574. doi: 10.1016/j.tig.2013.05.010. [DOI] [PubMed] [Google Scholar]
- 48.Lara-Astiaso D., Weiner A., Lorenzo-Vivas E., Zaretsky I., Jaitin D.A., David E., Keren-Shaul H., Mildner A., Winter D., Jung S., et al. Chromatin state dynamics during blood formation. Science. 2014;345:943–949. doi: 10.1126/science.1256271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang J., Zhuang J., Iyer S., Lin X., Whitfield T.W., Greven M.C., Pierce B.G., Dong X., Kundaje A., Cheng Y., et al. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 2012;22:1798–1812. doi: 10.1101/gr.139105.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fleming J.D., Pavesi G., Benatti P., Imbriano C., Mantovani R., Struhl K. NF-Y coassociates with FOS at promoters, enhancers, repetitive elements, and inactive chromatin regions, and is stereo-positioned with growth-controlling transcription factors. Genome Res. 2013;23:1195–1209. doi: 10.1101/gr.148080.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Landry J.J.M., Pyl P.T., Rausch T., Zichner T., Tekkedil M.M., Stütz A.M., Jauch A., Aiyar R.S., Pau G., Delhomme N., et al. The genomic and transcriptomic landscape of a HeLa cell line. G3 (Bethesda). 2013;3:1213–1224. doi: 10.1534/g3.113.005777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu X., Sharp P.A. Divergent transcription: a driving force for new gene origination? Cell. 2013;155:990–996. doi: 10.1016/j.cell.2013.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cheng J., Blum R., Bowman C., Hu D., Shilatifard A., Shen S., Dynlacht B.D. A role for H3K4 monomethylation in gene repression and partitioning of chromatin readers. Mol. Cell. 2014;53:979–992. doi: 10.1016/j.molcel.2014.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.