
Figure 7-1.
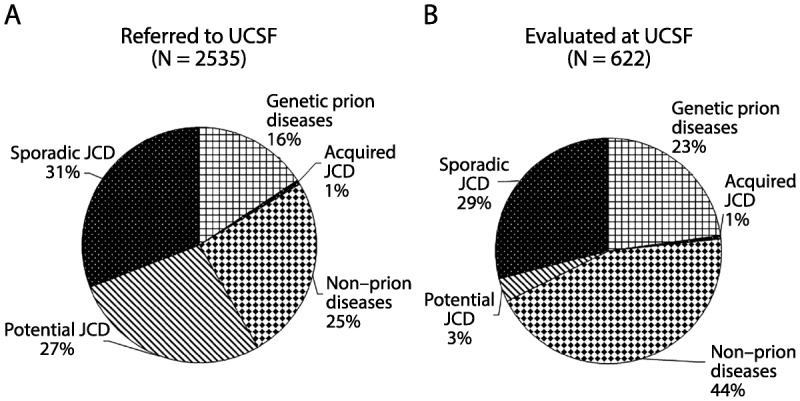
Major diagnostic categories of patients with rapidly progressive dementia (RPD) referred to, versus evaluated at, the University of California, San Francisco (UCSF) rapidly progressive dementia program over 13 years. A, Diagnostic distribution of patients with RPD referred to UCSF over about a 13-year period, most of whom had extensive medical record review, but only about one-fourth of whom were evaluated in person at UCSF. Almost one-third of cases referred to (as well as evaluated at) UCSF were diagnosed with sporadic Jakob-Creutzfeldt disease. In more than one-fourth of referred cases, although a sporadic Jakob-Creutzfeldt disease diagnosis (potential sporadic Jakob-Creutzfeldt disease) was suspected, not enough information existed to make a probable Jakob-Creutzfeldt disease diagnosis.5,6 Acquired Jakob-Creutzfeldt disease includes iatrogenic and infectious forms of prion disease. The genetic prion diseases category included patients who had confirmed mutations (autosomal dominant) in the prion protein gene, PRNP, or were from families with genetic prion disease. Whereas many of the genetic prion diseases presented similarly to sporadic Jakob-Creutzfeldt disease, as an RPD, a significant minority had clinical presentations more similar to other more slowly progressive diseases, such as Alzheimer disease or atypical parkinsonian or ataxic syndromes.2 One-fourth of cases were diagnosed with a nonprion etiology for their RPD. B, Diagnostic distribution of patients with RPD evaluated in person at UCSF. A larger percentage of nonprion RPDs and genetic prion diseases is evident; the latter is a bias partly because of the UCSF research program in genetic prion diseases and antibody-mediated encephalopathies.
JCD = Jakob-Creutzfeldt disease.