

Abstract
A detrimental role for matrix metalloproteinase 8 (MMP8) has been identified in several pathological conditions, e.g., lethal hepatitis and the systemic inflammatory response syndrome. Since matrix MMP8-deficient mice are protected in the above-mentioned diseases, specific MMP8 inhibitors could be of clinical value. However, targeting a specific matrix metalloproteinase remains challenging due to the strong structural homology of matrix metalloproteinases, which form a family of 25 members in mammals. Single-domain antibodies, called nanobodies, offer a range of possibilities toward therapy since they are easy to generate, express, produce, and modify, e.g., by linkage to nanobodies directed against other target molecules. Hence, we generated small MMP8-binding nanobodies, and established a proof-of-principle for developing nanobodies that inhibit matrix metalloproteinase activity. Also, we demonstrated for the first time the possibility of expressing nanobodies systemically by in vivo electroporation of the muscle and its relevance as a potential therapy in inflammatory diseases.
Introduction
Matrix metalloproteinases (MMPs) are zinc-dependent endopeptidases that play different roles in various diseases, including ischemia/reperfusion injury,1 lung diseases,2,3 gut and brain inflammation,4,5,6 neurodegenerative disease,7 and vascular disease.8,9 Broad-spectrum MMP inhibitors have been tested in clinical trials for various cancers.10 However, all these trials have failed because broad-range MMP inhibition is associated with several serious side-effects.11 Therefore, selective or specific targeting of MMPs would be a more suitable therapeutic approach, especially when long-term treatment is needed.4
MMP8 has been implicated in several pathological conditions,12 including lung injury,13,14 the systemic inflammatory response syndrome (SIRS),15,16,17,18,19,20,21,22 cardiovascular disease,23 neuroinflammation,24,25,26 arthritis,27,28 hepatitis,29 and cancer.30,31 Some of these pathologies, namely SIRS, lung injury, hepatitis, and experimental autoimmune encephalitis, could benefit from MMP8 inhibition since mice deficient for MMP8 were found to be protected.14,20,25,32 In addition, increased serum levels of MMP8 were found in SIRS patients and were suggested to correlate with mortality.19,22 So, specific inhibition of MMP8 is a potential therapeutic strategy but the homology of the MMP family is high since they evolved via gene duplication in the mammalian genome, impeding the development of specific MMP inhibitors and reliable detection tools for specific MMP activity.4
Generally, the current MMP inhibitors are metabolically unstable, lack specificity, have a poor oral bioavailability and/or are associated with a dose-limiting toxicity.4,33 Specific antibodies could be an alternative for the existing chemical and peptidic MMP inhibitors.10,34 Nonetheless, developing classical antibodies inhibiting MMPs is difficult because their large size (150 kDa) complicates the binding to the catalytic pocket, which is small and difficult to access.
Alternatively, single-domain antibodies, called nanobodies (Nbs), have many advantages which might circumvent some of the problems associated with conventional antibodies. Members of the Camelidae mammal family (camels, alpacas, llamas) express both conventional antibodies (Abs) and “heavy-chain-only” Abs (HcAb).35,36 The latter lack the constant domain of the heavy chain (CH1), leading to the absence of the light chain.35,36 The variable domain of this HcAb is the smallest antigen-binding fragment (15 kDa) and is called the VHH domain or Nb.35,36 Nbs have many advantages compared to chemical inhibitors and conventional Abs that lack specificity and are expensive to produce, respectively. They are small, stable, and soluble, have a high affinity and specificity, and are easy and cheap to produce in prokaryotic systems.35,36,37 They can also be easily modified, for example by linking several Nbs targeting different molecules in order to enhance their therapeutic efficacy.36
In this paper, we describe the development of MMP8-binding Nbs. We identified and characterized an MMP8-inhibiting Nb of which we explored its therapeutic potential in systemic inflammation. Finally, we describe a new method to administer Nbs in mice, namely via in vivo electroporation.
Results
Sequencing and modeling of different MMP8-binding Nbs
Sequencing of 13 VHH genes from different VHH groups identified eight different Nbs belonging to three different groups based on the amino acid sequence sequences (Supplementary Figure S1a). Six of the Nbs, Nb14, Nb27, Nb25, Nb10, Nb21, and Nb39 show very high sequence similarities. This suggests that they are derived either from clonally-related B-cells as a result of somatic hypermutation or from the same B-cell that diversified due to polymerase chain reaction (PCR) errors during library construction. In contrast, Nb4 and Nb44 most likely belong to distinct families. SWISS-MODEL, a fully automated protein structure homology-modeling server,38 was used to determine the putative tertiary structures of the MMP8-binding Nbs. Based on a template homologous to the protein of interest, 3EZJ39 in case of the MMP8 Nbs, 3D models of each of the Nbs could be constructed (Supplementary Figure S1b).
The typical β-pleated sheet immunoglobulin fold is conserved in Nbs.40 As indicated in Supplementary Figure S1a, two cysteines present in the VHH domain, Cys22 and Cys96, are responsible for the formation of an interdomain disulfide bridge between the β-sheets (Supplementary Figure S1a, full black line boxes). An additional interloop disulfide bridge linking CDR3 (Cys107) and CDR2 (Cys50) can be observed in Nb44, which might increase its stability significantly (Supplementary Figure S1a, red full line boxes). Nbs are also characterized by three complementarity determining regions (CDRs) (Supplementary Figure S1, black dotted line boxes), all contributing to antigen-binding specificity.41 Usually, the long and variable CDR3 loop accounts for most of the antigen binding interaction. As depicted in Supplementary Figure S1b, in particular, Nb44 is characterized by a long protruding CDR3 loop (red) and might therefore preferably target MMP8 in its active site cleft.
In vitro analysis of the different Nbs for MMP8 binding
To identify the Nbs with the highest affinity, binding efficiency of the purified monovalent MMP8_Nbs to mMMP8_CD was assessed by enzyme-linked immunosorbent assay (ELISA). Nb44 and Nb14 are depicted in Figure 1a and the KD values of all Nbs tested are shown in Table 1 (Figure 1a). A well-characterized Nb, the anti-β-lactamase Nb,42 was used as negative control. As expected, the control Nb (Nbctrl, against β–lactamase) did not bind mMMP8_CD. Calculation of the KD values of the different anti-MMP8 Nbs revealed that there are multiple Nbs with a high affinity for mMMP8_CD (Table 1). To investigate whether these Nbs recognize the 3D structure of mMMP8_CD or its primary amino acid sequence, binding affinity was also determined for denatured recombinant mMMP8_CD. In general, the binding capacity of the different Nbs for the denatured MMP8 was much lower (almost absent) compared to the native form, indicating that the 3D structure is essential for recognition (Figure 1b). In view of the potential clinical applications of Nb14, we confirmed its cross-reactivity with hMMP8_CD (KD, 158.4 nmol/l) (Figure 1c). Affinity for mMMP8_CD was also determined with surface plasmon resonance (SPR, Biacore) and gave a KD value for Nb14 of 240 nmol/l (Figure 1d). The differences in KD for binding of the Nbs determined by ELISA compared to SPR could be explained by technical differences.
Figure 1.

Analysis of binding and inhibitory constants of the different monomeric nanobodies (Nbs) for the catalytic domain of mouse matrix metalloproteinase 8 (mMMP8_CD). (a) Affinity of the anti-MMP8 Nbs for mMMP8_CD was determined by ELISA (KD Nb14, 1.3 nmol/l; KD Nb44, >100 nmol/l). (b) To determine whether the primary structure or the 3D conformation is recognized by the Nbs, binding analysis of the Nbs for both native (full line) and denatured (dotted line) mMMP8_CD was done by ELISA. (c) Cross-reactivity of Nb14 for the catalytic domain of human MMP8 (hMMP8_CD) was determined via ELISA (KD, 158 nmol/l). (d) Binding of Nb14 with mMMP8_CD was also determined by SPR. The full line shows the actual measurement, while the dotted line depicts the best theoretical fit. The best fit to determine the KD value for Nb14 is the two-state binding curve (KD, 228 nmol/l). (e,f) The EnzCheck gelatinase/collagenase assay was used to determine inhibitory capacity of the different Nbs for mMMP8_CD with DQ gelatin as substrate (IC50 Nb14 (e), 4.127 µmol/l; IC50 Nb44 (f), > 100 µmol/l). (g) The inhibitory capacity of Nb14 was also determined with DQ collagen type I as substrate and gave an IC50 value of 19.5 µmol/l. (n = 2). CD, catalytic domain; FL, full-length; h, human; IC50, half maximal inhibitory concentration; KD, binding affinity constant; m, mouse; MMP, matrix metalloproteinase; Nb, nanobody; SPR, surface plasmon resonance.
Table 1. Dissociation constants (KD) of the different nanobodies for the catalytic domain of mouse matrix metalloproteinase 8 (nmol/l).

To investigate the Nanobodies' capacity to inhibit MMP8 activity, we used the gelatin and collagen type I cleavage properties of MMP8. The EnzCheck Gelatinase/Collagenase assay kit was used to determine the IC50 values of the different MMP8-binding Nanobodies. Inhibition of mMMP8_CD was quantitatively determined by measuring changes in fluorescence (relative light units) over time in which a strong inhibition is correlated with a slower (or even absent) increase in fluorescence. Again, Nbctrl was used as a negative control. Figure 1 demonstrates the strongest inhibition of mMMP8_CD by Nb14 (IC50, 4.359 µmol/l) (Figure 1e) in comparison to Nb44 (Figure 1f) and the other MMP8-binding nanobodies (Supplementary Figure S2; Table 2). This was done with DQ gelatin as a substrate. However, the inhibitory capacity of Nb14 for mMMP8_CD was also determined using a more relevant substrate, namely DQ collagen type I, which resulted in an IC50 value of 19.5 µmol/l (Figure 1g).
Table 2. Half maximal inhibitory concentrations (IC50) of the different nanobodies for the catalytic domain of mouse matrix metalloproteinase 8 (µmol/l).

Members of the metzincin superfamily, including MMPs and ADAMs (a disintegrin and metalloproteinase), are structurally related.4 Therefore, cross-reactivity was tested for several human MMPs (hMMPs) and two well-studied members of the ADAM family, namely ADAM10 and ADAM17 (TACE), of which the latter is the closest known MMP analogue.11,43 Inhibition of these ADAM family members by broad-spectrum MMP inhibitors is believed to cause side-effects and should thus be avoided with new generation MMP blockers such as our Nanobody.11 Cross reactivity for hMMP1, -2, -3, -7, -9, -10, -12, -13, and -14 was tested by ELISA and this revealed that Nb14 does not bind with other hMMPs (Supplementary Figure S3a). Additionally, no cross-reactivity was found of Nb14 for ADAM 10 and ADAM 17 showing the specificity of this Nb (Supplementary Figure S3b,c).
Optimization and characterization of the modified Nb14
Based on the KD (0.24 nmol/l) and IC50 (4.359 µmol/l) values, Nb14 was selected for further optimization. Monovalent Nbs are about 15 kDa in size and since the cut-off value of the kidney is 60 kDa, this leads to a rapid clearance from the body. Indeed, intraperitoneal (i.p.) injection of 100 µg monomeric Nb14, followed by serum collection at different time points, revealed that Nb14 has a half-life (T1/2) of ~2 hours (Figure 2a). For that reason, we linked Nb14 via a flexible [Gly4Ser]3 linker to an anti-albumin Nb (NbAlb), resulting in a bispecific Nb14_NbAlb, and to NbAlb followed by a second Nb14 in order to create a trispecific Nb14_NbAlb_Nb14 (Figure 2b). The presence of the anti-Alb moiety in these Nb constructs will increase the molecular weight and thus the serum half-life, but will also lead to extra retention in the serum due to binding to albumin (67 kDa).44 Additionally, since albumin extravasates from the blood to inflammatory sites, binding of a Nb to albumin will lead to accumulation of the Nb at inflammatory sites.44,45 To produce recombinant Nbs in the periplasmic space of Escherichia coli, Nb sequences were cloned from the pUC57 into the pHEN6c vector. Nb14_NbAlb_Nb14 and Nb14_NbAlb were produced and purified as a protein but the yield of Nb14_NbAlb_Nb14 was very low in the prokaryotic WK6 E. coli system. Therefore, this Nb14_NbAlb_Nb14 sequence was cloned into the pAOXZalfa vector to transform the yeast Pichia pastoris in order to obtain a higher yield (304 mg from 160 ml culture). Cloning was done in a similar way and production gave a higher yield in Pichia compared to E. coli. Correspondingly, the P. pastoris expression system was also used to express a trivalent control Nb, NbAlb_NbCtrl_NbCtrl.
Figure 2.

Pharmacokinetic properties of the MMP8-binding Nb14 and binding capacity of the modified multivalent Nb14. (a) Serum Nb levels at different time points after intraperitoneal injection (i.p.) injection of 100 µg of the monovalent Nb14 in mice (n = 10) (T1/2 Nb14, 2 hours). (b) A schematic overview of the different Nb14 constructs shows that all constructs contain a C-terminal His6 tag for purification and detection purposes and an N-terminal signal sequence (ss) that is removed after secretion. (1) The monovalent Nb14 is cloned into the pHEN6C vector and contains a pelB signal sequence for Nb protein secretion in the periplasmic space of bacteria. (2) The bispecific Nb14_NbAlb contains an anti-albumin binding Nb connected to Nb14 via a flexible linker ([G4S]3). It was cloned in the pHEN6C (pelB) for protein production in bacteria and in the pCAGGs vector for electroporation as cDNA. The pCAGGs vector contains a sigal sequence that leads to secretion of the Nb out of the electroporated cells. (3) The trispecific Nb14_NbAlb_Nb14 contains two flexible hinges to connect an anti-albumin Nb with two Nb14 domains. This construct was cloned in the pAOXZalfa vector, containing the α-mating factor pre-pro-signal sequence as a signal sequence, for the production of Nb proteins in yeast. (c) Affinity of Nb14, Nb14_NbAlb and Nb14_NbAlb_Nb14 (black) and their control Nb counterparts (gray) for mMMP8_CD was determined by ELISA (KD Nb14, 0.24 nmol/l; KD Nb14_NbAlb, 0.1 nmol/l; Nb14_NbAlb_Nb14, 0.016 nmol/l). (d) ELISA was performed to determine binding of the Nbs for mMMP8_FL (black) and results in KD values of 1.69, 0.63, and 0.057 nmol/l for Nb14, Nb14_NbAlb and Nb14_NbAlb_Nb14, respectively. The affinity of all Nbs was compared to mMMP8_CD in the same test (gray). (e) Binding capacity of all Nb conjugates for hMMP8_CD (gray) compared to mMMP8_CD (black) gave KD values of 158.4, 40.4, and 0.158 nmol/l for Nb14, Nb14_NbAlb and Nb14_NbAlb_Nb14, respectively. (n = 2) (f) Binding for albumin was determined via ELISA with Nb14 and Nbctrl as negative controls. Affinity of the monovalent NbAlb and the modified Nbs gives the following KD values: NbAlb, 0.32; Nb14_NbAlb, 8.14 nmol/l; Nb14_NbAlb_Nb14, 1.48 nmol/l; Nbctrl_NbAlb, 39.61 nmol/l; NbAlb_Nbctrl_Nbctrl, 2.013 nmol/l. (g,h) Binding affinities of the bispecific (n = 2) (g) and trispecific Nb14 (h) for mMMP8_CD were determined with surface plasmon resonance (SPR, Biacore). SPR analysis shows the on-rate, when the Nb binds the chip, in the first part of the graph, while the second phase represents the off-rate, when the Nb dissociates from its substrate (arrows). The full line shows the actual measurement, while the dotted line depicts the best theoretical fit. The best fit to determine KD values for Nb14_NbAlb is the two-state binding curve, while the best fit for Nb14_NbAlb_Nb14 is the bivalent binding model. This bivalent binding model describes the interaction of a monovalent ligand with a molecule carrying two identical and independent binding sites (KD Nb14_NbAlb, 240 nmol/l; KD Nb14_NbAlb_Nb14, 3.7 nmol/l). CD, catalytic domain; FL, full-length; h, human; KD, binding affinity constant; m, mouse; MMP, matrix metalloproteinase; Nb, Nanobody; SPR, surface plasmon resonance; T1/2, half-life.
After expression in P. pastoris and purification, Nb14, Nb14_NbAlb and Nb14_NbAlb_Nb14 were compared for binding to mMMP8_CD (Figure 2c), mMMP8_FL (Figure 2d), hMMP8_CD (Figure 2e), and albumin via ELISA (Figure 2f). Dissociation constants (KD) of the monovalent, bispecific and trispecific Nb14 for the different proteins were compared (Figure 2c–f) and are shown in Table 3. KD values were calculated by a nonlinear regression model via the saturation binding equation and yielded values of 0.24, 0.1, and 0.016 nmol/l for Nb14, Nb14_NbAlb and Nb14_NbAlb_Nb14, respectively, for binding to mMMP8_CD. Addition of a second Nb14 in the trispecific construct improved avidity for MMP8. Since there is a possibility that conjugation of Nb14 (or Nbctrl) to NbAlb interferes with its albumin binding properties, the affinity of the modified Nbs for albumin was determined. Nb14 was used as negative control since binding to albumin is absent. Binding of the modified constructs gave KD values of 0.32, 8.14, and 1.48 nmol/l for NbAlb, Nb14_NbAlb, Nb14_NbAlb_Nb14, respectively, showing that conjugation of the different Nbs affected albumin binding, but this was rather limited since KD values were still in a similar low nM range. As the Nbs were raised against mMMP8_CD, cross reactivity for mMMP8_FL had to be verified (Figure 2d). KD values of the monovalent, bispecific and trispecific Nb14 were found to be 1.69, 0.63, and 0.057 nmol/l for mMMP8_FL, respectively, based on ELISA. These results show that the Nbs bind both the catalytic domain of MMP8 as well as the full-length version. Again, affinities were determined with SPR and generated KD values of 240 and 3.7 nmol/l for Nb14_NbAlb and Nb14_NbAlb_Nb14, respectively (Figure 2g,h). Due to the better (lower) off rate, the trispecific construct has a better binding capacity than the monovalent Nb14 (Table 4). In addition, in view of future clinical purposes, cross-reactivity for hMMP8_CD was confirmed (Figure 2e) and revealed the following KD values: 158.4 nmol/l for Nb14, 40.4 nmol/l for Nb14_NbAlb and 0.158 nmol/l for Nb14_NbAlb_Nb14, which again shows better binding in case of the trispecific tool.
Table 3. Dissociation constants (KD) of Nb14, Nb14_NbAlb and Nb14_NbAlb_Nb14 for MMP8 and albumin.
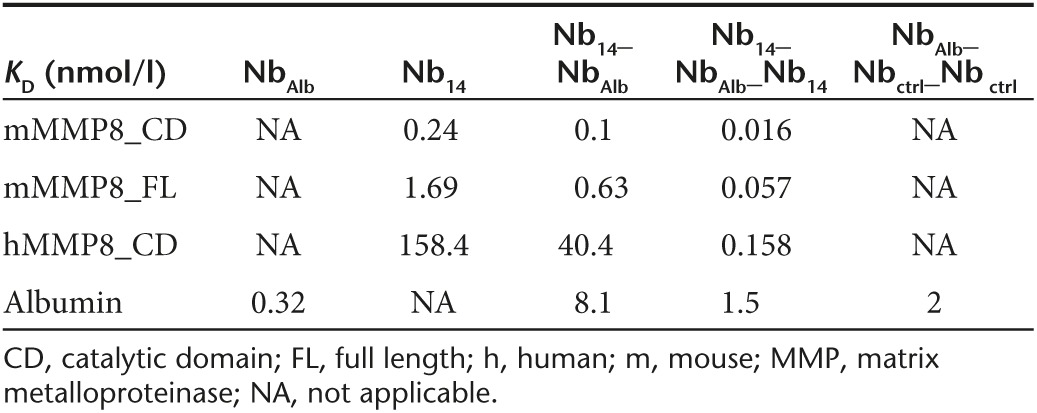
Table 4. Off-rate constants of Nb14, Nb14_NbAlb and Nb14_NbAlb_Nb14 for the catalytic domain of mouse MMP8 (s−1).
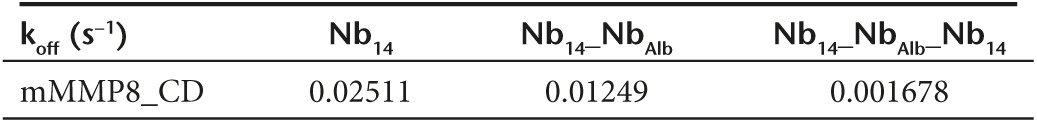
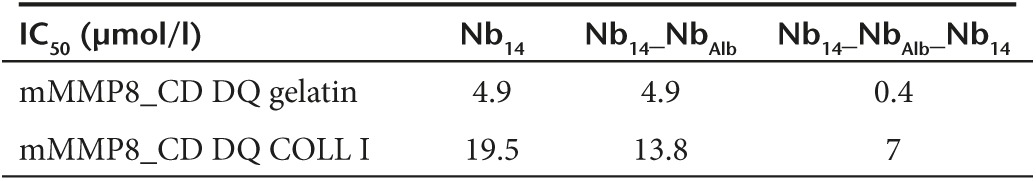
The inhibitory capacity of the different Nb constructs was determined by two different fluorescent substrates namely DQ gelatin (Figure 3a,b) and DQ collagen type I (Figure 3c,d). Both are substrates for MMP8 and contain a quenched fluorophore which is released when the substrate is cleaved. Nbs were incubated with mMMP8_CD for 30 minutes after which the substrate was added. The reduced increase in fluorescence measured over time represents the inhibitory capacity of the Nbs. This revealed an IC50 value for the gelatin substrate of 4.9, 4.9, and 0.4 µmol/l for the monomeric Nb14, bispecific Nb14_NbAlb and trispecific Nb14_NbAlb_Nb14, respectively. This shows that the increased binding of Nb14_NbAlb_Nb14 for mMMP8 compared to Nb14 is also reflected in a better inhibition (Table 5). A similar trend was observed for inhibition of collagen I but in general higher IC50 values were observed (Table 5). Next, we determined the pharmacokinetic properties of the trispecific Nb14_NbAlb_Nb14. Mice were injected i.p. with 100 µg and serum levels were determined by ELISA. We found a half-life of 28 hours after one i.p. injection which is significantly longer than the 2 hours half-life of Nb14 (Figure 3e).
Figure 3.

Inhibitory and pharmacokinetic properties of the modified bispecific and trispecific anti-MMP8 Nb. (a,b) Inhibitory capacity of Nb14_NbAlb (a) and Nb14_NbAlb_Nb14 (b) for mMMP8_CD with gelatin as substrate gives IC50 values of 4.8 and 0.4 µmol/l, respectively. (n = 2) (c,d) Inhibitory capacity with DQ collagen type I as substrate for mMMP8_CD gives 13.8 and 7 µmol/l as IC50 values for Nb14_NbAlb and Nb14_NbAlb_Nb14, respectively. (n = 2) (e) Nb levels in the serum of mice (n = 10) after a single i.p. injection of 100 µg Nb14_NbAlb_Nb14, were determined by ELISA and gives a T1/2 value of 28 hours. IC50, half maximal inhibitory concentration; i.p., intraperitoneal; Nb, nanobody; T1/2, half-life.
Table 5. Half maximal inhibitory concentrations (IC50) of Nb14, Nb14_NbAlb and Nb14_NbAlb_Nb14 for the catalytic domain of mouse MMP8 (µmol/l) determined with the fluorescent substrates DQ gelatin and DQ collagen type I.
Docking model of Nb14 with MMP8
In silico analysis was performed to predict the binding of Nb14 with both mMMP8_CD and hMMP8_CD. Therefore, both homology and docking modeling was conducted. Structural information on hMMP8_CD is accessible in the Protein DataBank (PDB: 2OY4) but since no experimental structure of mMMP8_CD is available, a homology model was built based on 2OY4 and 1FBL, which is pig MMP1. Additionally, to generate a homology model of Nb14 (two models, Nb14_m1 and Nb14_m2), multiple templates from PDB, namely 4LAJ, 3EZJ, 3TPK, and 4M3J, were used. Docking models were obtained for the best two Nb14 models in combination with hMMP8_CD (one model) or mMMP8_CD (two models, mMMP8_CD_m1 and mMMP8_CD_m2). All models were validated by RAMPAGE46 and the best models were used for docking by ClusPro47 to predict binding of Nb14 to MMP8. Homology models and docking results were analyzed and figures rendered using PyMOL.
Figure 4 displays the top-10 conformations for one of these six different combinations in which the three preferable binding sites are depicted by 1, 2, and 3 (Figure 4). Since the ELISA data showed that Nb14 has the same affinity for activated mMMP8_FL compared to nonactivated (data not shown), binding site 2 is less likely to be the binding site of Nb14 to MMP8 as it is located in the active site. Therefore, binding sites 1 and 2, which are located outside the active site, are the best candidates (Figure 5a). This might also explain the good binding (nmol/l) but disappointing inhibitory capacity (µmol/l) of Nb14. Indeed, if the active site of MMP8 would be the Nb14-binding site, this would result in direct inhibition. In contrast, when Nb14 would bind to another site, this can induce indirect inhibition by steric hindrance or by the induction of conformational changes. In addition, the presence of Lysine residues (Lys) (Figure 5a, blue surface) was inspected with the in silico docking models because the SPR data indicated a lower affinity of all the Nb constructs forMMP8 compared to the binding results obtained by ELISA (Figure 5a). In SPR, Lys residues are involved in the covalent coupling of MMP8 to the chip. As can be appreciated in Figure 5b, one of the potential binding sites on MMP8 contains a Lys residue that interacts with Nb14, and thus this interaction might be disturbed in the SPR approach (Figure 5b).
Figure 4.

In silico docking models of the interaction between mMMP8_CD and hMMP8_CD with Nb14. Docking models were obtained for the best Nb14 models (Nb14_m1 and Nb14_m2, in color) in combination with hMMP8_CD (gray) on one hand and the two best models for mMMP8_CD (mMMP8_CD_m1 and mMMP8_CD_m2, gray) on the other hand. These simulations show three possible binding sites (1–3). Two (1 and 3) of which are present outside the active site and one (2) at the active site. Modeling of hMMP8_CD was based on the experimental structure, while homology models (mMMP8m1 and Swissmodel mMMP8) were built for mMMP8_CD. Note the extra linker for mMMP8m1 (dark gray). Multiple templates (PDB: 4LAJ, 3EZJ, 3TPK and 4M3J) were used to construct Nb14, depicted as MMP8_Nbm1 and MMP8_Nbm9. All models were validated by RAMPAGE46 and the best models were used for docking by ClusPro47 to predict binding of the MMP8_Nb to MMP8. Homology models and docking results were analyzed and figures rendered using PyMOL. CD, catalytic domain; h, human; m, mouse; MMP, matrix metalloproteinase; Nb, nanobody.
Figure 5.

Lysine residues present on Nb14 and mMMP8_CD by in silico docking modeling of the interaction between both. (a) mMMP8_CD is depicted in gray and the Lys residues are shown as blue surfaces. The three different binding places where Nb14 could bind mMMP8_CD are shown by the Nb conformation in blue (1), yellow (2), and green (3) of which (2) is located in the catalytic pocket of the enzyme. The sticks in the green Nb14 represent the Lys residue that interacts with the Lys from mMMP8_CD at binding site (3). (b) A detailed view on the Lys-Lys interaction between Nb14 (green) and mMMP8_CD (gray) is shown in stick conformation. In the structure of mMMP8_CD, all Lys residues are shown by the blue sticks. CD, catalytic domain; Lys, lysine; m, mouse; Nb, Nanobody.
Therapeutic potential of MMP8 inhibition in SIRS
Next, we wanted to address the therapeutic potential of MMP8 inhibition in different validated mouse inflammatory models. Therefore, we used an acute sepsis model, in which the endotoxin lipopolysaccharides (LPS) (6 mg/kg) is injected systemically. Kidney I/R, wherein one kidney is removed while the other undergoes ischemia (I) and reperfusion (R), was used as a sterile model for SIRS. In both models, MMP8-deficient mice are protected.20 We explored the therapeutic value of our MMP8-inhibiting Nb and tested whether this Nb could be administered as a cDNA plasmid by in vivo electroporation of the quadriceps. This approach does not require purification of the protein which makes it very cheap and easy. Electroporation is a technique by which electrical shocks are applied to the tissue of interest to achieve an efficient uptake of macromolecules, such as DNA, into the cells. In case of quadriceps electroporation, the Nb is injected intramuscularly as a plasmid (DNA), followed by noninvasive electroporation of the quadriceps by tweezer electrodes. Electroporation parameters were optimized with a luciferase (Luc) expressing plasmid (pCAGGs-Luc). Luc activity was measured in vivo with the IVIS imager after i.p. injection of the mice with the substrate luciferin (Supplementary Figure S4). Both quadricepses were injected intramuscularly with pCAGGs-Luc, but only one side was immediately electroporated. A Luc signal was present 24 hours after electroporation that was stronger than the signal in the nonelectroporated muscle proving the efficiency of plasmid uptake by electroporation. Furthermore, the signal had a long and stable expression for at least 2 months. Both Nb14_NbAlb and Nb14_NbAlb_Nb14 were cloned in a pCAGGS plasmid in which the Nb protein is preceded by a cleavable signal sequence that allows secretion of the Nb from electroporated cells. We confirmed secretion of Nb14_NbAlb_Nb14 in the serum of electroporated mice. Detectable Nb levels were reached 48 hours after electroporation and steady state levels were achieved within 4 days and lasted over 2 weeks (Figure 6a). Administration of Nb14_NbAlb by in vivo electroporation of the quadriceps showed significant protection in both the endotoxemia and kidney I/R model compared to the Nbctrl_NbAlb group (Figure 6b,c). Since binding and inhibition was better with Nb14_NbAlb _Nb14, also this sequence was cloned in the pCAGGs vector and used for in vivo electroporation. Again, mice were electroporated with the Nb14_NbAlb _Nb14 or Nbctrl_NbAlb_Nbctrl construct or PBS, followed by LPS injection or kidney I/R 48h after electroporation (Figure 6d,e). In the LPS model, no significant protection was observed between the different groups (Figure 6d), but in the kidney I/R model, we found a significant survival in the Nb14_NbAlb_Nb14 group of 50%, compared to 0% survival in both control groups (Figure 6e). So, Nb14_NbAlb_Nb14 was more efficacious compared to Nb14_NbAlb in the kidney I/R model. Unfortunately, this was not the case for endotoxemia model. Finally, the effectiveness of the trispecific Nb14 on in vivo biological MMP8 activity was shown by the observed decrease in MMP activity in the serum of Nb14_NbAlb_Nb14 electroporated mice compared to control mice (Figure 6f). Our data show that in vivo quadriceps electroporation can be used as delivery strategy for systemic administration of Nbs.
Figure 6.

Therapeutic potential of in vivo electroporation of an MMP8-inhibiting Nb in systemic inflammation. (a) The OD values (raw data) normalized to baseline (no electroporation) are representative for the serum levels of Nb14_NbAlb_Nb14 after in vivo electroporation of the muscles in mice (n = 8). (b) The survival curve of mice electroporated with a plasmid containing Nb14_NbAlb or Nbctrl_NbAlb (PBS-injected group as control) when challenged with LPS. (n = 17) (c) Survival of mice electroporated with the Nb constructs or PBS after kidney I/R, a sterile model for SIRS. (n = 17) (d) The survival curve of mice electroporated with Nb14_NbAlb_Nb14, Nbctrl_NbAlb_Nbctrl or PBS followed by a challenge with LPS. (n = 6) (e) Survival of mice electroporated with the trispecific Nb constructs or PBS after kidney I/R. (n = 6) (f) Total protease/MMP activity in the serum of mice electroporated with Nb14_NbAlb_Nb14 or PBS. (n = 6) (ctrl, control; I/R, ischemia/reperfusion; LPS, lipopolysaccharide; MMP, matrix metalloproteinase; Nb, Nanobody; OD; optic density; SIRS, systemic inflammatory response syndrome; WT, wild type).
Discussion
MMPs are involved in the degradation of the extracellular matrix but have a wide range of different substrates, also beyond the extracellular matrix. In normal conditions, MMPs are hardly detectable. However, MMPs are activated during various inflammatory diseases and cancers. Broad-spectrum MMP inhibitors have failed for the treatment of various cancers because broad-range MMP inhibition is associated with several negative side-effects.10,11 These adverse effects are probably caused by inhibition of non-MMP metalloproteinases, such as tumor necrosis factor (TNF)-α converting enzyme (TACE, a disintegrin and metalloproteinase 17, ADAM17).11 As a result, selective or specific targeting of MMPs is a more suitable approach to avoid off-target effects.4 Matrix metalloproteinase 8 (MMP8) is associated with several pathological conditions12 in which some of them, e.g., SIRS, could benefit from MMP8 inhibition.20 However, specific chemical inhibitors and antibodies are lacking. Therefore, single variable domains derived from heavy-chain-only antibodies of Camelidae, called nanobodies (Nbs), are an interesting alternative to target MMP8 because they are specific, small in size, modifiable, and cheap to produce.
Different MMP8-binding Nbs were generated by immunization of Alpacas with mMMP8_CD. These Nbs were produced in large quantities by bacterial (E. coli) and yeast (P. pastoris) expression systems.48 Binding experiments revealed different candidates with a good affinity in the nanomolar range for mMMP8_CD. Inhibitory experiments showed that Nb14 is the most effective inhibitor with an IC50 value in the micromolar range. It is likely that Nbs specific for mouse MMP8 will cross-react with human MMP8 because these proteins show a 74% amino acid identity.49 Indeed, Nb14 was cross-reactive with human MMP8 but not with a long list of other hMMPs or structurally related metalloproteases such as ADAMs. This suggests that possible side-effects, including musculoskeletal pain, are expected to be minimal, making Nb14 suitable for clinical use.
Next, we wanted to explore whether the properties of Nb14 could be improved by conjugating it with an anti-albumin Nb (NbAlb). Binding affinity of the bispecific Nb14_NbAlb and trispecific Nb14_NbAlb_Nb14 for albumin was influenced by these modifications (KD NbAlb, 0.32 nmol/l; KD Nb14_NbAlb, 8.1 nmol/l; KD Nb14_NbAlb_Nb14, 1.5 nmol/l), but it also resulted in an extended half-life (T1/2 Nb14, 2 hours; T1/2 Nb14_NbAlb_Nb14 28 hours), which is in agreement with existing literature.50 The affinity of the different Nb constructs for mMMP8_CD was slightly better compared to the affinity for mMMP8_FL. Since no difference was detected in binding capacity of Nb14 for the nonactivated or activated form of mMMP8_FL (data not shown), the difference in affinity is likely not due to the presence or absence of an intact cysteine switch. Thus, it is more likely that the presence of other protein domains (prodomain and hemopexin domain) can lead to steric hindrance of Nb14 consequently yielding a lower affinity for the full length MMP8. The conjugation of NbAlb to Nb14 did not interfere with binding to MMP8 as shown by ELISA and SPR. Moreover, the addition of a second Nb14 (resulting in Nb14_NbAlb_Nb14) improves the avidity as shown by SPR and to enhanced inhibitory capacity of MMP8. These results prove the ease and usefulness of conjugating different Nbs in order to increase therapeutic efficacy.
A predictive docking model, which was used to visualize the binding of Nb14 with MMP8, disclosed three potential binding sites, i.e., one at the active site of MMP8 and two others at outside the active site. We found no difference in binding capacity of Nb14 for the activated versus nonactivated form of mMMP8_FL (data not shown), indicating that the most probable binding site is not located in the active site of MMP8. This could explain the limited inhibitory capacity of Nb14 despite its good binding capacity. In this view, inhibition is indirectly achieved by steric hindrance of MMP8 by the Nb or by the induction of conformational changes in MMP8 rather than by direct binding of the inhibitor to the catalytic pocket.
Binding of the Nbs was determined by SPR and ELISA, but when comparing KD values, SPR consistently resulted in lower binding affinities. The discrepancy might be explained by the difference in coating, since SPR uses covalent coupling of the substrate (MMP8) via Lys residues to the chip, while ELISA is based on electrostatic forces. The in silico docking model supports this hypothesis because it shows a potential binding site that involves a crucial Lys of MMP8 with Nb14. Several Lys residues are present in the MMP8 molecule which could all be used to covalently couple MMP8 to the chip for SPR. However, only one of them is present at a potential binding site for Nb14, which could prevent the Nb to bind that specific site, leading to a lower total binding affinity measured by SPR. Another explanation might be that SPR has a dynamic character, which is in contrast with the static nature of ELISA.
To explore the therapeutic potential of MMP8 inhibition, the bispecific Nb14_NbAlb and trispecific Nb14_NbAlb_Nb14 sequences were cloned in the pCAGGs vector, containing a cleavable signal sequence to achieve secretion of the Nb into the bloodstream. This plasmid was electroporated in vivo in both quadriceps muscles of the mice. We found that this led to the production of the Nb into the blood stream with detectable levels after 48 hours and maximum steady state levels at 4 days which remained until at least 2 weeks after electroporation. The Nb levels at these time points were high and long enough to protect against lethality during SIRS. Moreover, we found indirect evidence that the production of these Nbs into the circulation has a biological effect shown by the reduction in total MMP activity in the blood. The mice that received the MMP8-inhibiting Nb construct 48 hours before challenge were significantly protected compared to the control groups during systemic inflammation. However, the bispecific Nb14_NbAlb had a lower efficacy compared to Nb14_NbAlb_Nb14 since a smaller group of mice was needed in the latter to achieve significant protection. This is likely due to the fact that the trispecific MMP8-inhibiting construct has better binding and inhibitory capacities. Surprisingly, in contrast with Nb14_NbAlb, we observed no significant protection by Nb14_NbAlb_Nb14 in the endotoxemia model. On one hand, this could be due to the lower number of animals that were used in this experiment. On the other hand, the endotoxemia and kidney I/R models are two very different models for systemic inflammation considering they have different cytokine and MMP profiles. Future research is needed to define the therapeutic niche of Nb14 and whether it is useful to refine this tool, for example, to couple it with specific cell-targeting nanobodies and target, for example, macrophages or neutrophils. Based on the data obtained in MMP8-deficient mice, the MMP8 inhibitor described here may be useful in, e.g., multiple sclerosis (experimental autoimmune encephalomyelitis),25 obliterate bronchiolitis,52 acute hepatitis,32 and bacterial meningitis.53,54
The generation of an MMP8-inhibiting Nb is a logical extension of previous work of our group, in which we showed protection of MMP8-deficient mice in different models of systemic inflammation.20 In those models of SIRS, MMP8-deficient mice were strongly protected, much more pronounced compared to the therapeutic Nb described here. This discrepancy in protection could be explained by the fact that MMP8-deficient mice are completely devoid of MMP8 while the therapeutic tool described here impossibly can target all MMP8, throughout the body.
We previously showed that protection of the MMP8-deficient mice to acute inflammation was reflected in the absence of cleavage of collagen type I at the blood–cerebrospinal fluid barrier.20 Here, we show that our Nb could inhibit collagen type I cleavage in vitro, although this is difficult to prove in vivo. Moreover, caution is advisable because some functions/substrates are redundant between MMPs. For example, literature shows that MMP8 deficiency leads to higher MIP1α levels in the lung since it is no longer cleaved by MMP8.21,51 In addition, the absence of MMP8 promotes lung inflammation induced by allergens and during endotoxemia, illustrating that it is not always beneficial to inhibit MMP8 in every organ during systemic inflammation.21,51,55 However, we found no evidence for exacerbation of lung inflammation in MMP8-deficient mice upon systemic inflammation.20,22
In conclusion, we generated an MMP8-specific Nb with a good affinity and inhibition for both mouse and human MMP8. Furthermore, we showed that the multivalent Nbs still maintain good binding and inhibitory abilities for MMP8 and that the addition of NbAlb improves half-life. Our study supports the potential of MMP8-inhibitory Nbs as novel sepsis treatment. Moreover, we demonstrate for the first time the possibility of in vivo electroporation of the muscle for systemic delivery of Nb. Finally, since MMP8 is implicated in various diseases, the tools described here could be interesting to investigate the therapeutic niche of MMP8 inhibition.
Materials and Methods
Production of recombinant mouse MMP8. The catalytic domain of mouse MMP8 (mMMP8_CD) was cloned in a vector containing two different affinity tags: a His6 and maltose-binding protein (HisMBP) at the N-terminal and a Strep2 tag at the C-terminal side. The MBP moiety increases solubility and promotes the proper folding of its fusion partners. The His6 tag and the Strep2 tag facilitate purification. A DEVD sequence was included after the HisMBP tag to allow removal of the tag from the fusion protein by caspase-3 cleavage. The recombinant mMMP8_CD was expressed in E. coli and purification of the mMMP8_CD was done by immobilized metal ion affinity chromatography with nickel sepharose, ion exchange chromatography, and gel filtration. All purification steps were performed in LPS-free conditions.
Nanobody library construction and selection. To generate anti-MMP8 nanobodies, an Alpaca (Vicugna pacos) was immunized by consecutive subcutaneous injections on days 0, 7, 14, 21, 28, and 35 with the recombinant mouse catalytic domain of matrix metalloproteinase 8 (mMMP8_CD) (280 μg/injection). On day 39, blood was collected for the analysis of the immune response and for the preparation of lymphocytes. IgG subclasses were obtained by successive affinity chromatography on protein A and protein G columns. Total plasma and the three purified IgG subclasses (IgG1, IgG2, and IgG3) were tested by ELISA to assess the immune response to mMMP8_CD.
IgG subclasses were obtained and total plasma was tested for IgG subclasses (IgG1, IgG2, and IgG3) to assess the immune response to the antigen. The plasma titer was about 2 × 104-fold and there was an immune response in all IgG subclasses except IgG2. Furthermore, the IgG3 response was lower than the IgG1 response.
A VHH library was constructed and screened for the presence of mMMP8_CD-specific Nanobodies as described before.42,56 In short, total RNA from peripheral blood lymphocytes was used as template for first strand cDNA synthesis with an oligo(dT) primer. Using this cDNA, the VHH encoding sequences were amplified by PCR, digested with PstI and NotI, and cloned into the PstI and NotI sites of the phagemid vector pHEN4. The VHH repertoire was displayed on phages and MMP8-specific phages were enriched by panning on solid-phase coated mMMP8_CD (100 μg/ml, 10 μg/well) for four consecutive rounds. The enrichment for antigen-specific phages after each round of panning was assessed by polyclonal phage ELISA.
About 2 × 108 independent phage transformants were obtained and nearly 90% of these harbored the vector with the right insert size. Four consecutive rounds of panning were performed and enrichment for antigen-specific phages was assessed after each round of panning. A clear enrichment was present after the third and fourth rounds of panning. In total, 190 individual colonies (48, 95, and 47 after second, third, and fourth rounds of panni ng, respectively) were randomly selected and analyzed by ELISA for the presence of antigen-specific VHHs in their periplasmic extracts. Out of 190 colonies, 105 scored positive in this assay (0, 59, and 46 from second, third, and fourth rounds, respectively). The VHHs from 32 positive colonies from the third round were grouped based on restriction fragment length polymorphism (RFLP) analysis using HinfI enzyme.
Cloning of modified nanobodies for protein production. First, MMP8-binding Nbs were recloned from the pHEN4 to pHEN6c vector. This was accomplished by amplification of the Nb gene in the pHEN4 vector by PCR with the following primers; primer A6E (5′ GAT GTG CAG CTG CAG GAG TCT GGA/A GGA GG 3′) and primer 38 (5′ GGA CTA GTG CGG CCG CTG GAG ACG GTG ACC TGG GT 3′), followed by overnight digestion with PstI at 37 °C (all restriction enzymes purchased from Promega, Eupen, Germany). After purification, a second overnight digest was done with BstEII at 50 °C. Both digestion steps were also performed on the pHEN6c vector with an incubation period of 3 hours. Next, the Nb gene was ligated into the pHEN6c vector and transformed into electrocompetent WK6 cells. Bacteria were grown on Luria broth (LB) plates with ampicillin (100 µg/ml) overnight at 37 °C and positive colonies were screened by PCR. Verification of the clones was done by sequencing.
Construction of the bispecific Nb14_NbAlb and trispecific Nb14_NbAlb_Nb14 was done by cloning the desired Nb sequence from the pUC57 vector (GenScript) into the pHEN6c vector, for bacterial transformation, or into the pAOXZalfa vector, for transformation in yeast. Nb14 was linked to an anti-albumin Nb (NbAlb) and, in case of the trispecific construct, to a second Nb14 via flexible [Gly4Ser]3 linkers. Each construct contains a C-terminal His6-tail for purification and detection purposes. Nb14_NbAlb_Nb14 was cloned, together with its equivalent control Nb (Nbctrl), in both the pHEN6c and pAOXZalfa vector. A well-characterized Nb, the anti-β-lactamase Nb, was used as control Nb (Nbctrl) because it only binds the bacterial component β-lactamase.42
To produce recombinant Nbs in the periplasm of E. coli, Nb sequences were cloned from the pUC57 into the pHEN6c vector. Digestion of both vectors was done using NCoI and EcoRI for 3 hours at 37 °C. The digested vectors were run on a 1% agarose gel and the bands of interest were purified using a PCR cleanup gel extraction kit (Machery-Nagel). Ligation was performed at room temperature for 3 hours, followed by another digestion with XbaI for 1 hour at 37 °C to linearize incorrect ligated constructs (counterdigestion). Electrocompetent WK6 cells were then transformed with the purified constructs and transformants were selected using LB agar plates containing ampicillin (100 µg/ml) and 1% glucose. Positive clones were selected by digestion with the same restriction enzymes (NCoI and EcoRI) followed by gel electrophoresis and the identity of these clones was verified by sequencing. The same Nb sequence was also cloned in the pAOXZalfa vector for transformation in P. pastoris using the same protocol but with the restriction enzymes XhoI and XbaI for digestion of the vectors and EcoRV for the counterdigestion.
Cloning of modified nanobodies for electroporation. For in vivo electroporation, a special construct was made by cloning Nb14_NbAlb (and Nbctrl_NbAlb) from the pUC57 into the pCAGGs vector that contains the AG (chicken-actin β-globin) promotor (Supplementary Figure S5). The pUC57 vector containing the Nb sequence was ordered from GenScript and comprises of Nb14 linked to an anti-albumin Nb (NbAlb) by a flexible [Gly4Ser]3 linker. Furthermore, it contains a C-terminal His6-tail for purification and detection and an N-terminal signal sequence that is cleaved off after secretion out of the cell. Both vectors were digested with BglII and HindIII for 3 hours at 37 °C. The digested vector was run on a 1% agarose gel and the bands of interest were purified with the PCR cleanup gel extraction kit (Machery-Nagel). Ligation was done overnight at 4 °C, followed by a 10-minute heat inactivation step at 65 °C. In this case, a counterdigestion was not possible. WK6 cells were transformed by heat shock with the purified construct and transformants were selected using LB agar plates with ampicillin (100 µg/ml) and 1% glucose. Positive clones were selected by digestion with the same restriction enzymes (BglII and HindIII) followed by gel electrophoresis and sequencing was done for verification of the clones. Correct clones were grown in LB medium containing ampicillin at 37 °C and the cDNA was purified from the bacteria with a high yield (10 µg/µl).
Nanobody production and purification. Nb genes can be translated into proteins by transformation of the cloned vectors in either bacteria (E. coli) or yeast (P. pastoris). Expression of the recombinant monomeric Nbs, bispecific Nb14_NbAlb and trispecific Nb14_NbAlb_Nb14 (and their control counterparts) was accomplished by transformation of the WK6 strain of E. coli with the correct pHEN6c_Nb vector followed by induction with isopropyl β-D-1-thiogalactopyranoside (IPTG), as the Nb genes are under control of the lacUV5 promoter. The expression plasmid is provided with a PelB leader sequence at the N-terminus of the Nb which makes it possible to secrete the Nb into the periplasm of E. coli. The transformed bacteria were grown in LB medium, supplemented with 1% glucose, 1 mmol/l MgCl2 and 100 µg/ml ampicillin overnight at 37 °C. Next, bacteria were inoculated in Terrific Broth (TB) medium, supplemented with 0.1% glucose, 1 mmol/l MgCl2, and 100 µg/ml ampicillin at 28 °C. When an optical density of 1 was reached at 600 nm, expression was induced by addition of 1 mmol/l IPTG for at least 4 hours. Next, the bacterial pellet was resuspended in 50 mmol/l NaH2PO4 pH 8.0, 300 mmol/l NaCl, 1 mmol/l PMSF, and 1 mg/100 ml DnaseI at 3 ml/g and stirred for 1 hour at 4 °C. The periplasmic fraction (supernatant) was isolated by centrifugation at 18,000 × g for 30 minutes at 4 °C.
A higher Nb yield was achieved by expression of the Nb genes in the yeast P. pastoris. The wild-type GS115 P. pastoris strain was used for transformation with the pAOXZalfa_Nb vector encoding the trispecific Nb14_NbAlb_Nb14. The expression vector, which is a derivate of the pPICZα vector (Life Technologies) is supplemented with the AOX1 promoter fused to the α-mating factor pre-pro-signal sequence followed by the gene coding for the Nb construct.57 Again, the Nb sequence included a C-terminal His6-tag. After selection of the appropriate expression clone, a 20-l production was performed in baffled shake flasks. A preculture of the transformed yeast was grown in yeast extract-peptone (YP) medium supplemented with 100 µg/ml zeocyin for 48 hours at 28 °C. Then the medium was switched to YP medium containing glucose (YPD) for 24 hours at 28 °C. Finally, protein expression was induced by addition of 1.25% methanol for at least 24 hours. The medium fraction, containing the Nb, was isolated by centrifugation at 18,000 × g for 30 minutes at 4 °C and diafiltered to a new buffer containing 20 mmol/l NaH2PO4 pH 7.5, 500 mmol/l NaCl, 20 mmol/l imidazole, and 1 mmol/l PMSF.
The periplasmic E. coli fraction and the diafiltered fraction of P. pastoris were applied onto a Ni-Sepharose 6 FF column (GE Healthcare), equilibrated with 20 mmol/l NaH2PO4 pH 7.5, 500 mmol/l NaCl, 20 mmol/l imidazole, and 1 mmol/l PMSF. After loading, the column was washed with the same buffer in presence of 0.1% empigen as detergent, followed by one washing step with the equilibration buffer without detergent. Elution was done with 20 mmol/l NaH2PO4 pH 7.5, 20 mmol/l NaCl, 400 mmol/l imidazole, and 1 mmol/l PMSF. The elution fraction was diluted 1/20 with 25 mmol/l sodium acetate pH 5.5 and loaded on a Source 15S column (GE Healthcare Europe, Upsalla, Sweden) to remove contaminants and LPS. After equilibration, the Nb was eluted by a linear gradient of NaCl from 0 to 1 M in 25 mmol/l sodium acetate pH 5.5. Finally, the recombinant Nb was injected on a Superdex 75 gelfiltration column with PBS, as running solution, for formulation and to remove minor contaminants. The obtained fractions were analyzed by SDS-PAGE and the concentration was determined using the Micro-BCA assay (Pierce, Erembodegem, Belgium).
Docking models. Homology modeling and docking was performed to predict the binding of Nb14 with MMP8 (both mMMP8_CD and hMMP8_CD). A homology model of the monomeric Nb14 was built with Modeller.58 Structural information on hMMP8_CD is available in the Protein DataBank (PDB) (PDB: 2OY4), however, homology models for mMMP8_CD had to be built with Modeller templates 2OY4 and 1FBL (pig MMP1). Homology models of Nb14 were also generated by Modeller using multiple templates from PDB namely 4LAJ, 3EZJ, 3TPK, and 4M3J.59 All models were validated by RAMPAGE46 and the best models were used for docking by ClusPro47 to predict binding of Nb14 to mMMP8_CD and hMMP8_CD. Homology models and docking results were analyzed and figures were rendered using PyMOL.
Affinity measurements of MMP8-binding nanobodies
ELISA. Microtiter half-area plates (Nunc) were coated overnight at 4 °C with the substrate of interest (50 ng/well) dissolved in Tris-buffered saline (TBS). Unspecific binding was decreased by a blocking step of 1 hour with 5% bovine serum albumin (BSA) in TBS supplemented with 0.05% Tween 20 (TBST) at room temperature. Binding affinity was determined for the following substrates: mMMP8_CD (own production, as described above), full-length mouse MMP8 (mMMP8_FL; R&D systems, 2904-MP-010), full-length human MMP8 (hMMP8_FL; Calbiochem, 444229-5UG), and mouse albumin (Sigma, A3559). When necessary, denaturation of mMMP8_CD was achieved by 15 minutes of boiling. Nbs were diluted in 2.5% BSA in 0.05% TBST starting from 1 µmol/l in a 1/3 dilution and incubated for 1 hour at room temperature. Detection was done by anti-His (1:1,000) (AbD Serotec, MCA1396) or anti-llama (1:5,000) (Bethyl, A160-100) followed by an anti-mouse IgG1-HRP (1:2,000) (GE Healthcare, NA931) or anti-goat IgG-HRP (1:5,000) (Santa Cruz, sc-2020). Visualization was done with the chromogenic substrate 3,3',5,5'-tetramethylbenzidine (TMB) (BD OptEIA, 555214). After a maximal incubation of 30 minutes, stop solution (1 M H2SO4) was added and absorbance (optic density (OD)) was measured at 450 and 595 nm wavelength. The background signal (595 nm) was subtracted from the 450 nm measurement and KD values were determined with GraphPad Prism 6.0 with the nonlinear regression model and the saturation binding equation.
SPR. SPR analysis was performed using the Biacore T200. Mouse MMP8_CD was diluted in acetate pH 5.0 and immobilization was achieved by covalent coupling of mMMP8_CD with 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide and N-hydroxysuccinimide to a Sensor Chip CM5 until an RU (resonance units) of 301.8 was obtained at 25 °C. The Nbs were diluted in a twofold dilution starting from a 500 nmol/l concentration in HEPES-buffered saline buffer (10 mmol/l HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) pH 7.5, 150 mmol/l NaCl, 3.5 mmol/l ethylenediaminetetraacetic acid, and 0.005% Tween 20). Injection of the different Nb concentrations was done at a flow rate of 20 µl/minute. After each cycle, the chip was regenerated with 30 µl of 20 mmol/l NaOH at a flow rate of 30 µl/minute. A blank uncoated channel is used as reference during the injections. The resulting binding sensorgrams were used to calculate the kinetic rate constants kon and koff by the BIA evaluation software to ultimately determine the equilibrium dissociation constant (KD). The sensorgrams were fitted using different diffing models by subtracting the signal of the reference flow. For the monovalent Nb14 and bispecific Nb14_NbAlb, the 1-1 fit model was used, while the two-state binding model was applied for the trispecific Nb14_NbAlb _Nb14.
Nanobody pharmacokinetics. To study the in vivo pharmacokinetic properties of Nbs, blood was sampled via retro-orbital blood collection at different time points (1, 3, 6, 10, 24, 48, and 72 hours) after a single Nb injection (intraperitoneal (i.p.), 100 µg). In case of electroporation with Nb14_NbAlb, blood was sampled 1, 3, 7, and 10 days after electroporation. Serum was prepared from the blood samples. Nbs were detected in the mouse serum by ELISA, as described above using an optimal range of four dilutions of the serum in 0.05% TBST.
Inhibitory capacity of the anti-MMP8 nanobodies. The inhibitory capacity of the different Nbs for MMP8 was investigated using the EnzChek Gelatinase/Collagenase Assay Kit (Molecular Probes, Life Technologies, Ghent, Belgium E12055) according to the manufacturer's instructions. In short, DQ gelatin (1:4) was incubated with recombinant mMMP8_CD after preincubation of 30 minutes with increasing amounts of Nb. Changes in fluorescence over time by cleavage of DQ-gelatin were followed for 2 hours at 37 °C in a fluorescence microplate reader (ex/em = 495/515 nm) (Fluostar Omega). Activity of mMMP8_CD and inhibitory capacity of the Nbs was determined by the changes in fluorescence over time. Plotting the activity in function of the logarithmic Nb concentration gives a sigmoidal shaped curve. A nonlinear regression was used to determine the IC50 value with GraphPad Prism 6.0. The same protocol was used with DQ collagen type I (Invitrogen, Life Technologies, D12060), only the preincubation step was increased (1 hour at 37 °C). The DQ gelatin kit was also used to show general protease activity in the serum of mice. In this case, serum samples (n = 8) were diluted 1/3 without freezing the samples (fresh).
Cross-reactivity for other MMPs and adamlysin family members ADAM10 and ADAM17
Binding affinity for other MMPs. Cross-reactivity of Nb14 for other MMPs was determined by ELISA, as described above. In summary, microtiter half-area plates (Nunc) were coated overnight at 4 °C with the different human MMPs (Enzo Life Sciences, Antwerp, Belgium, BML AK016) (50 ng/well) dissolved in TBS. Nb14 were diluted in 2.5% BSA in 0.05% TBST starting from 1 µmol/l in a 1/3 dilution and incubated for 1 hour at room temperature. Since the MMPs contain a His tag, detection was done by anti-llama (1:5,000) (Bethyl Imtec Diagnostics NV, Antwerp, Belgium, A160-100) followed by anti-goat IgG-HRP (1:5,000) (Santa Cruz Bio-Connect, TE Huissen, The Netherlands, sc-2020).
ADAM10 and ADAM17 recombinant proteins. Cloning, expression, and protein purification was performed as previously described for proADAM10 (ref. 60) and ADAM17.61 Briefly, truncated forms of murine ADAM10/17 containing the pro- and catalytic domains were ligated into pFastBac (Gibco, Thermo Fisher Scientific, Darmstadt, Germany) containing the human meprin β signal peptide followed by a His6-tag. Recombinant protein expression was performed using the Bac-to-Bac expression system (Gibco) following the manufacturer's instructions. All media and supplements were obtained from Gibco.
Peptide-based activity assay. To investigate the inhibitory capacity of Nb14 and the ctrl counterpart on other metalloproteases, ADAM10 and ADAM17 were used in a quenched fluorogenic peptide based activity assay. 500 nmol/l recombinant ADAM10 or ADAM17 were incubated with different concentrations of Nbs for 20 minutes at RT in 20 mmol/l HEPES buffer (pH 7.5). The fluorogenic peptide substrates Mca-KPLGL(Dnp)AR-NH2 (Peptide Institute) and Mca-PLAQAV(Dpa)RSSSR-NH2 (R&D Systems, Wiesbaden-Nordenstadt, Germany), respectively, were used in final concentrations of 10 µmol/l to measure enzyme activity. Enzyme activity was measured with a TECAN infinite F200 pro-reader at 37 °C and proteolytic cleavage was determined every 30 seconds in relation to the emission at 405 nm with excitation at 320 nm for a time interval of 2 hours.
Mice. The wild-type (WT, C57BL/6) mice used in this study were bred in the specific pathogen-free (SPF) facility of the Inflammation Research Center (IRC, Belgium) in a controlled environment (12-hour light–dark cycle; 20 °C) with food and water ad libitum. Endotoxic shock, a model for sepsis, was induced by a single i.p. injection of 6 mg/kg (LD100) LPS (from E. coli, serotype O55/B5, Sigma-Aldrich, Diegem, Belgium) dissolved in sterile PBS. Kidney ischemia reperfusion (I/R) was used as a sterile model for SIRS and was performed as described earlier.20 In short, the mice were sedated with isoflurane and an abdominal incision was made to expose the left kidney. After applying a clamp on the left kidney, the right kidney was exposed and removed. Ischemia was performed for 45 minutes, followed by reperfusion (removal of the clamp). All animal experiments were approved by the local ethical committee and were conform with the European Community Directive (86/609/EEC).
In vivo electroporation of the bispecific Nanobody Nb14_NbAlb. Optimal in vivo electroporation parameters were determined using a luciferase expressing plasmid (pCAGGs-luc) containing the AG (chicken-actin β-globin) promotor and a firefly-luciferase gene. Animals were anesthetized with isoflurane prior to electroporation. The quadriceps was injected intramuscular with 20 µl (0.5 µg/µl) of the DNA plasmid followed by electroporation (200 V, 8 pulses, 20 ms, 100 ms interval) using tweezer electrodes. Luciferase activity was detected and quantified in function of time by injecting the mice intraperitoneal with luciferin (200 µl, 15 µg/µl), followed by imaging with the in vivo imager IVIS (Caliper LifeSciences). The same electroporation parameters (200 V, 8 pulses, 20 ms, 100 ms interval) and plasmid concentrations were used for the electroporation of the Nb plasmids, but electroporation was done bilaterally (both quadricepses). Mice were challenged with LPS (or operated in case of kidney I/R) 48 hours after electroporation with Nb14_NbAlb (Nb14_NbAlb_Nb14), ctrl_NbAlb (Nbctrl_NbAlb_Nbctrl) or PBS. Body temperatures and lethality were followed up for 7 days.
SUPPLEMENTARY MATERIAL Figure S1. Primary and tertiary structure of the different anti-MMP8 Nanobodies. Figure S2. Inhibitory capacity of the different monomeric MMP8-binding Nanobodies for the catalytic domain of mouse MMP8. Figure S3. Analysis of the cross reactivity of Nb14 for human MMPs, mouse ADAM10 and ADAM17. Figure S4. In vivo imaging of luciferase activity after in vivo electroporation of the quadriceps with the luciferase expressing plasmid pCAGGs-Luc. Figure S5. Overview of Nb14_NbAlb plasmid construct for in vivo electroporation.
Acknowledgments
This work was supported by the Agency for Innovation by Science and Technology in Flanders, the Research Council of Ghent University, the Research Foundation Flanders (FWO Vlaanderen), the Interuniversity Attraction Poles Program of the Belgian Science Policy, and by the Deutsche Forschungsgemeinschaft (DFG) SFB877 “Proteolysis as a Regulatory Event in Pathophysiology” (project A9). The authors wish to thank the VIB Nanobody Service Facility for the generation of the Nanobodies and Joke Vanden Berghe for technical assistance related to breeding and generation of the mice. The authors certify that they have no affiliations with or involvement in any organization or entity with any financial interest or non-financial interest in the subject matter or materials discussed in this manuscript.
Supplementary Material
References
- Dejonckheere, E, Vandenbroucke, RE and Libert, C (2011). Matrix metalloproteinases as drug targets in ischemia/reperfusion injury. Drug Discov Today 16: 762–768. [DOI] [PubMed] [Google Scholar]
- Vandenbroucke, RE, Dejonckheere, E and Libert, C (2011). A therapeutic role for matrix metalloproteinase inhibitors in lung diseases? Eur Respir J 38: 1200–1214. [DOI] [PubMed] [Google Scholar]
- Neto-Neves, EM, Kiss, T, Muhl, D and Tanus-Santos, JE (2013). Matrix metalloproteinases as drug targets in acute pulmonary embolism. Curr Drug Targets 14: 344–352. [DOI] [PubMed] [Google Scholar]
- Vandenbroucke, RE and Libert, C (2014). Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat Rev Drug Discov 13: 904–927. [DOI] [PubMed] [Google Scholar]
- Giuffrida, P, Biancheri, P and MacDonald, TT (2014). Proteases and small intestinal barrier function in health and disease. Curr Opin Gastroenterol 30: 147–153. [DOI] [PubMed] [Google Scholar]
- Mirshafiey, A, Asghari, B, Ghalamfarsa, G, Jadidi-Niaragh, F and Azizi, G (2014). The significance of matrix metalloproteinases in the immunopathogenesis and treatment of multiple sclerosis. Sultan Qaboos Univ Med J 14: e13–e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, XX, Tan, MS, Yu, JT and Tan, L (2014). Matrix metalloproteinases and their multiple roles in Alzheimer's disease. Biomed Res Int 2014: 908636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siefert, SA and Sarkar, R (2012). Matrix metalloproteinases in vascular physiology and disease. Vascular 20: 210–216. [DOI] [PubMed] [Google Scholar]
- Lin, J, Kakkar, V and Lu, X (2014). Impact of matrix metalloproteinases on atherosclerosis. Curr Drug Targets 15: 442–453. [DOI] [PubMed] [Google Scholar]
- Devy, L and Dransfield, DT (2011). New Strategies for the Next Generation of Matrix-Metalloproteinase Inhibitors: Selectively Targeting Membrane-Anchored MMPs with Therapeutic Antibodies. Biochem Res Int 2011: 191670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond, AH, Beckett, P, Brown, PD, Bone, EA, Davidson, AH, Galloway, WA et al. (1999). Preclinical and clinical studies of MMP inhibitors in cancer. Ann N Y Acad Sci 878: 228–235. [DOI] [PubMed] [Google Scholar]
- Dejonckheere, E, Vandenbroucke, RE and Libert, C (2011). Matrix metalloproteinase8 has a central role in inflammatory disorders and cancer progression. Cytokine Growth Factor Rev 22:73–81. [DOI] [PubMed] [Google Scholar]
- Craig, VJ, Quintero, PA, Fyfe, SE, Patel, AS, Knolle, MD, Kobzik, L et al. (2013). Profibrotic activities for matrix metalloproteinase-8 during bleomycin-mediated lung injury. J Immunol 190: 4283–4296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albaiceta, GM, Gutierrez-Fernández, A, García-Prieto, E, Puente, XS, Parra, D, Astudillo, A et al. (2010). Absence or inhibition of matrix metalloproteinase-8 decreases ventilator-induced lung injury. Am J Respir Cell Mol Biol 43: 555–563. [DOI] [PubMed] [Google Scholar]
- Zhou, X, Lu, J, Chen, D, Wang, W, Cai, Q, Li, T et al. (2014). Matrix metalloproteinase-8 inhibitors mitigate sepsis-induced myocardial injury in rats. Chin Med J (Engl) 127: 1530–1535. [PubMed] [Google Scholar]
- Sivula, M, Hästbacka, J, Kuitunen, A, Lassila, R, Tervahartiala, T, Sorsa, T et al. (2015). Systemic matrix metalloproteinase-8 and tissue inhibitor of metalloproteinases-1 levels in severe sepsis-associated coagulopathy. Acta Anaesthesiol Scand 59: 176–184. [DOI] [PubMed] [Google Scholar]
- Rella, JM, Jilma, B, Fabry, A, Kaynar, AM and Mayr, FB (2014). MMP-8 genotypes influence the inflammatory response in human endotoxemia. Inflammation 37: 451–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hästbacka, J, Linko, R, Tervahartiala, T, Varpula, T, Hovilehto, S, Parviainen, I et al. (2014). Serum MMP-8 and TIMP-1 in critically ill patients with acute respiratory failure: TIMP-1 is associated with increased 90-day mortality. Anesth Analg 118: 790–798. [DOI] [PubMed] [Google Scholar]
- Lauhio, A, Hästbacka, J, Pettilä, V, Tervahartiala, T, Karlsson, S, Varpula, T et al. (2011). Serum MMP-8, -9 and TIMP-1 in sepsis: high serum levels of MMP-8 and TIMP-1 are associated with fatal outcome in a multicentre, prospective cohort study. Hypothetical impact of tetracyclines. Pharmacol Res 64: 590–594. [DOI] [PubMed] [Google Scholar]
- Vandenbroucke, RE, Dejonckheere, E, Van Lint, P, Demeestere, D, Van Wonterghem, E, Vanlaere, I et al. (2012). Matrix metalloprotease 8-dependent extracellular matrix cleavage at the blood-CSF barrier contributes to lethality during systemic inflammatory diseases. J Neurosci 32: 9805–9816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-López, A, Aguirre, A, López-Alonso, I, Amado, L, Astudillo, A, Fernández-García, MS et al. (2012). MMP-8 deficiency increases TLR/RAGE ligands S100A8 and S100A9 and exacerbates lung inflammation during endotoxemia. PLoS One 7: e39940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solan, PD, Dunsmore, KE, Denenberg, AG, Odoms, K, Zingarelli, B and Wong, HR (2012). A novel role for matrix metalloproteinase-8 in sepsis. Crit Care Med 40: 379–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang, C, Wen, G, Zhang, L, Lin, L, Moore, A, Wu, S et al. (2013). An important role of matrix metalloproteinase-8 in angiogenesis in vitro and in vivo. Cardiovasc Res 99: 146–155. [DOI] [PubMed] [Google Scholar]
- Light, M, Minor, KH, DeWitt, P, Jasper, KH and Davies, SJ (2012). Multiplex array proteomics detects increased MMP-8 in CSF after spinal cord injury. J Neuroinflammation 9: 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folgueras, AR, Fueyo, A, García-Suárez, O, Cox, J, Astudillo, A, Tortorella, P et al. (2008). Collagenase-2 deficiency or inhibition impairs experimental autoimmune encephalomyelitis in mice. J Biol Chem 283: 9465–9474. [DOI] [PubMed] [Google Scholar]
- Lee, EJ, Han, JE, Woo, MS, Shin, JA, Park, EM, Kang, JL et al. (2014). Matrix metalloproteinase-8 plays a pivotal role in neuroinflammation by modulating TNF-α activation. J Immunol 193: 2384–2393. [DOI] [PubMed] [Google Scholar]
- Cox, JH, Starr, AE, Kappelhoff, R, Yan, R, Roberts, CR and Overall, CM (2010). Matrix metalloproteinase 8 deficiency in mice exacerbates inflammatory arthritis through delayed neutrophil apoptosis and reduced caspase 11 expression. Arthritis Rheum 62: 3645–3655. [DOI] [PubMed] [Google Scholar]
- García, S, Forteza, J, López-Otin, C, Gómez-Reino, JJ, González, A and Conde, C (2010). Matrix metalloproteinase-8 deficiency increases joint inflammation and bone erosion in the K/BxN serum-transfer arthritis model. Arthritis Res Ther 12: R224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Lint, P and Libert, C (2006). Matrix metalloproteinase-8: cleavage can be decisive. Cytokine Growth Factor Rev 17: 217–223. [DOI] [PubMed] [Google Scholar]
- Väyrynen, JP, Vornanen, J, Tervahartiala, T, Sorsa, T, Bloigu, R, Salo, T et al. (2012). Serum MMP-8 levels increase in colorectal cancer and correlate with disease course and inflammatory properties of primary tumors. Int J Cancer 131: E463–E474. [DOI] [PubMed] [Google Scholar]
- Balbín, M, Fueyo, A, Tester, AM, Pendás, AM, Pitiot, AS, Astudillo, A et al. (2003). Loss of collagenase-2 confers increased skin tumor susceptibility to male mice. Nat Genet 35: 252–257. [DOI] [PubMed] [Google Scholar]
- Van Lint, P, Wielockx, B, Puimège, L, Noël, A, López-Otin, C and Libert, C (2005). Resistance of collagenase-2 (matrix metalloproteinase-8)-deficient mice to TNF-induced lethal hepatitis. J Immunol 175: 7642–7649. [DOI] [PubMed] [Google Scholar]
- Hidalgo, M and Eckhardt, SG (2001). Development of matrix metalloproteinase inhibitors in cancer therapy. J Natl Cancer Inst 93: 178–193. [DOI] [PubMed] [Google Scholar]
- Basu, B, Correa de Sampaio, P, Mohammed, H, Fogarasi, M, Corrie, P, Watkins, NA et al. (2012). Inhibition of MT1-MMP activity using functional antibody fragments selected against its hemopexin domain. Int J Biochem Cell Biol 44: 393–403. [DOI] [PubMed] [Google Scholar]
- Behdani, M, Zeinali, S, Khanahmad, H, Karimipour, M, Asadzadeh, N, Azadmanesh, K et al. (2012). Generation and characterization of a functional Nanobody against the vascular endothelial growth factor receptor-2; angiogenesis cell receptor. Mol Immunol 50: 35–41. [DOI] [PubMed] [Google Scholar]
- Harmsen, MM and De Haard, HJ (2007). Properties, production, and applications of camelid single-domain antibody fragments. Appl Microbiol Biotechnol 77: 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumoulin, M, Conrath, K, Van Meirhaeghe, A, Meersman, F, Heremans, K, Frenken, LG et al. (2002). Single-domain antibody fragments with high conformational stability. Protein Sci 11: 500–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold, K, Bordoli, L, Kopp, J and Schwede, T (2006). The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22: 195–201. [DOI] [PubMed] [Google Scholar]
- Korotkov, KV, Pardon, E, Steyaert, J and Hol, WG (2009). Crystal structure of the N-terminal domain of the secretin GspD from ETEC determined with the assistance of a nanobody. Structure 17: 255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muyldermans, S, Baral, TN, Retamozzo, VC, De Baetselier, P, De Genst, E, Kinne, J et al. (2009). Camelid immunoglobulins and nanobody technology. Vet Immunol Immunopathol 128: 178–183. [DOI] [PubMed] [Google Scholar]
- Desmyter, A, Transue, TR, Ghahroudi, MA, Thi, MH, Poortmans, F, Hamers, R et al. (1996). Crystal structure of a camel single-domain VH antibody fragment in complex with lysozyme. Nat Struct Biol 3: 803–811. [DOI] [PubMed] [Google Scholar]
- Conrath, KE, Lauwereys, M, Galleni, M, Matagne, A, Frère, JM, Kinne, J et al. (2001). Beta-lactamase inhibitors derived from single-domain antibody fragments elicited in the camelidae. Antimicrob Agents Chemother 45: 2807–2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss, ML, Jin, SL, Milla, ME, Bickett, DM, Burkhart, W, Carter, HL et al. (1997). Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha. Nature 385: 733–736. [DOI] [PubMed] [Google Scholar]
- Coppieters, K, Dreier, T, Silence, K, de Haard, H, Lauwereys, M, Casteels, P et al. (2006). Formatted anti-tumor necrosis factor alpha VHH proteins derived from camelids show superior potency and targeting to inflamed joints in a murine model of collagen-induced arthritis. Arthritis Rheum 54: 1856–1866. [DOI] [PubMed] [Google Scholar]
- Neumann, E, Frei, E, Funk, D, Becker, MD, Schrenk, HH, Müller-Ladner, U et al. (2010). Native albumin for targeted drug delivery. Expert Opin Drug Deliv 7: 915–925. [DOI] [PubMed] [Google Scholar]
- Lovell, SC, Davis, IW, Arendall, WB 3rd, de Bakker, PI, Word, JM, Prisant, MG et al. (2003). Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins 50: 437–450. [DOI] [PubMed] [Google Scholar]
- Kozakov, D, Beglov, D, Bohnuud, T, Mottarella, SE, Xia, B, Hall, DR et al. (2013). How good is automated protein docking? Proteins 81: 2159–2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muyldermans, S (2013). Nanobodies: natural single-domain antibodies. Annu Rev Biochem 82: 775–797. [DOI] [PubMed] [Google Scholar]
- Balbín, M, Fueyo, A, Knäuper, V, Pendás, AM, López, JM, Jiménez, MG et al. (1998). Collagenase 2 (MMP-8) expression in murine tissue-remodeling processes. Analysis of its potential role in postpartum involution of the uterus. J Biol Chem 273: 23959–23968. [DOI] [PubMed] [Google Scholar]
- Dennis, MS, Zhang, M, Meng, YG, Kadkhodayan, M, Kirchhofer, D, Combs, D et al. (2002). Albumin binding as a general strategy for improving the pharmacokinetics of proteins. J Biol Chem 277: 35035–35043. [DOI] [PubMed] [Google Scholar]
- Quintero, PA, Knolle, MD, Cala, LF, Zhuang, Y and Owen, CA (2010). Matrix metalloproteinase-8 inactivates macrophage inflammatory protein-1 alpha to reduce acute lung inflammation and injury in mice. J Immunol 184: 1575–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatwa, UA, Kleibrink, BE, Shapiro, SD and Subramaniam, M (2010). MMP-8 promotes polymorphonuclear cell migration through collagen barriers in obliterative bronchiolitis. J Leukoc Biol 87: 69–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leppert, D, Leib, SL, Grygar, C, Miller, KM, Schaad, UB and Holländer, GA (2000). Matrix metalloproteinase (MMP)-8 and MMP-9 in cerebrospinal fluid during bacterial meningitis: association with blood-brain barrier damage and neurological sequelae. Clin Infect Dis 31: 80–84. [DOI] [PubMed] [Google Scholar]
- Leib, SL, Leppert, D, Clements, J and Täuber, MG (2000). Matrix metalloproteinases contribute to brain damage in experimental pneumococcal meningitis. Infect Immun 68: 615–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gueders, MM, Balbin, M, Rocks, N, Foidart, JM, Gosset, P, Louis, R et al. (2005). Matrix metalloproteinase-8 deficiency promotes granulocytic allergen-induced airway inflammation. J Immunol 175: 2589–2597. [DOI] [PubMed] [Google Scholar]
- Saerens, D, Kinne, J, Bosmans, E, Wernery, U, Muyldermans, S and Conrath, K (2004). Single domain antibodies derived from dromedary lymph node and peripheral blood lymphocytes sensing conformational variants of prostate-specific antigen. J Biol Chem 279: 51965–51972. [DOI] [PubMed] [Google Scholar]
- Schoonooghe, S, Leoen, J and Haustraete, J (2012). Production of antibody derivatives in the methylotrophic yeast Pichia pastoris. Methods Mol Biol 907: 325–340. [DOI] [PubMed] [Google Scholar]
- Sali, A and Overington, JP (1994). Derivation of rules for comparative protein modeling from a database of protein structure alignments. Protein Sci 3: 1582–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson, P, Wallner, B, Lindahl, E and Elofsson, A (2008). Using multiple templates to improve quality of homology models in automated homology modeling. Protein Sci 17: 990–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jefferson, T, Auf dem Keller, U, Bellac, C, Metz, VV, Broder, C, Hedrich, J et al. (2013). The substrate degradome of meprin metalloproteases reveals an unexpected proteolytic link between meprin β and ADAM10. Cell Mol Life Sci 70: 309–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucher, J, Linke, D, Koudelka, T, Cassidy, L, Tredup, C, Wichert, R et al. (2014). LC-MS based cleavage site profiling of the proteases ADAM10 and ADAM17 using proteome-derived peptide libraries. J Proteome Res 13: 2205–2214. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.