

Abstract
A 16-year-old boy with widening of the large joints of the extremities and bilateral genu valgum had been extensively treated with oral vitamin D, with little clinical benefit. A diagnosis of vitamin D-resistant rickets was considered initially but a thorough clinical examination and skeletal survey was suggestive of mucopolysaccharidosis. The diagnosis was confirmed biochemically and subtype classification pointed toward the type I variety of the storage disorder. Absence of mental retardation is very unusual in mucopolysaccharidosis type I, which itself is an uncommon clinical entity. This particular disease can be misdiagnosed as vitamin D-resistant rickets in the absence of thorough systemic examination and an attentive look at the skeletal surveys. Spondyloepiphyseal dysplasia is another close differential of mucopolysaccharidosis and it should be ruled out in all cases of suspected spondyloepiphyseal dysplasia.
Background
Mucopolysaccharidosis type I (MPS I), a lysosomal storage disorder, is characterised by progressive and widespread accumulation of glycosaminoglycans (GAG) within the lysosomes, leading to multiorgan dysfunction and damage.1 It is inherited in an autosomal-recessive manner and is caused by a deficiency of the lysosomal enzyme α-L-iduronidase. Patients appear normal at birth and usually present in early childhood with coarse facial features, corneal clouding, a large tongue and hepatosplenomegaly. A number of skeletal abnormalities such as an over-prominent forehead, widening of wrists and ankles, deformities of the spine, knees and hip joints, and a short stature, are not uncommon. Progressive skeletal dysplasia, also known as dysostosis multiplex, occurs in individuals with severe MPS I in which all the bones of the body are involved. The incidence of MPS I is estimated to be about 1 case per 100 000 live births.2 Historically, three clinical phenotypes have been described in order of decreasing severity, namely, MPS I-H (Hurler syndrome), MPS I-H/S (Hurler-Scheie syndrome) and MPS I-S (Scheie syndrome). In 2009, an International Consensus Panel reclassified MPS I into two major groups: severe MPS I (Hurler syndrome) and attenuated MPS I (Hurler-Scheie and Scheie syndromes).3
Case presentation
A 16-year-old boy presented with bowing of both legs associated with progressive difficulty in walking since early childhood. With a clinical diagnosis of rickets, he had for several years been treated with oral cholecalciferol and calcium by his primary care physician, without much clinical improvement. Born of a consanguineous union, his parents noticed progressive bowing of both the legs starting at around the age of 6 years. His perinatal history was unremarkable and developmental milestones with respect to language, fine and gross motor aspects were normal.
On examination, he had severe short stature (height 110 cm, height standard deviation score (SDS) (−8.59); with an upper:lower segment ratio of 0.67), bilateral genu valgum, pectus carinatum and broadening of bilateral wrists, elbows and ankles, consistent with an initial clinical diagnosis of vitamin D-resistant rickets (figure 1A, B). During evaluation to confirm underlying aetiology, a thorough systemic examination revealed coarse facial features, bilateral corneal clouding (figure 2), bushy eyebrows, increased dental spacing, short neck, kyphoscoliosis, mild hepatosplenomegaly and a left parasternal early diastolic decrescendo murmur suggestive of aortic regurgitation. Interestingly, the wrist, elbow, ankle and knee joints were lax and hypermobile. The boy's intelligence was judged to be normal (full scale IQ score 92 on Wechsler adult intelligence scale IV).
Figure 1.

Bilateral wrist and elbow joint widening, pectus carinatum, prominent costal cartilages and bushy eyebrows (A); genu valgum and widening of ankle joints (B).
Figure 2.
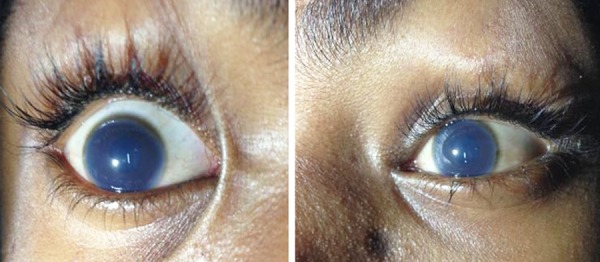
Bilateral corneal clouding.
Investigations
A look through the skeletal survey revealed flattened and dysplastic femoral epiphysis, coxa valga, minimal metaphyseal flaring of distal radius, irregular carpal bones, and pointed proximal metacarpals and phalanges (bullet-shaped). Radiographs of the spine and chest showed platyspondyly, hypoplastic vertebra, anteroinferior vertebral body beaking, dorsal kyphosis and widening of anterior ribs rendering them oar-shaped (figure 3A–C).
Figure 3.

Radiological features: oar-shaped ribs and femoral epiphyseal dysgenesis (A); epiphyseal dysgenesis, minimal metaphyseal widening, bullet-shaped metacarpals and proximal phalanges, irregular carpal bones (B); platyspondyly, hypoplastic vertebra, anteroinferior vertebral body beaking (white arrows) (C).
Results of initial laboratory tests are summarised in table 1.
Table 1.
Initial laboratory parameters of the patient
| Complete blood count | Within normal limits |
| Liver function tests | Within normal limits |
| Creatinine | 72 µmmol/L (64–104) |
| Albumin corrected serum calcium | 2.35 mmol/L (2.2–2.6) |
| Serum phosphorus | 1.24 mmol/L (0.81–1.4) |
| Alkaline phosphatase | 82 U/L (33–96) |
| 25-OH-vitamin D | 135 nmol/L |
| Intact parathyroid hormone | 28.1 pg/mL (12.0–72.0) |
| Free thyroxine | 14.9 pmol/L (9.0–16.0) |
| Thyroid-stimulating hormone | 1.06 mIU/L (0.34–4.25) |
The reference values are given within brackets.
Arterial blood gas analysis was normal. At this point, MPS and spondyloepiphyseal dysplasia (SED) were taken into consideration. A peripheral blood smear revealed cytoplasmic granules in lymphocytes (Alder Reilly granules) (figure 4). MRI of the cervical spine revealed odontoid hypoplasia and features suggestive of atlantoaxial subluxation with mild cord compression. Echocardiography confirmed the auscultatory findings of aortic regurgitation. With clinical, biochemical and radiological features suggesting MPS, we proceeded for confirmation of our diagnosis and subtype classification. A urine toluidine-blue spot test was ordered to screen for MPS, and tested positive for dermatan sulfate and heparan sulfate. Further enzyme assay from peripheral blood leucocytes revealed deficient α-L-iduronidase enzyme activity (0.90 test value, against control value of 12.0; reference 9.0–26.0).
Figure 4.
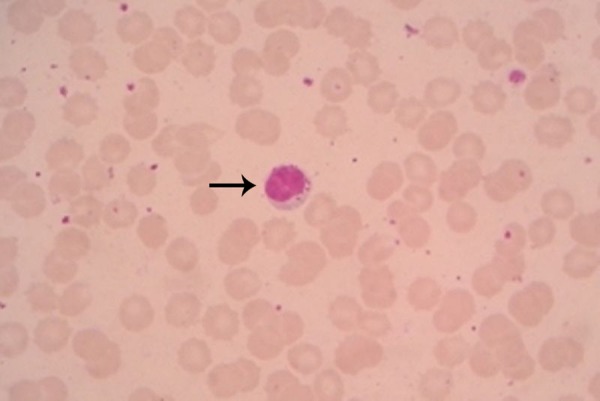
Photomicrograph showing cytoplasmic granules in lymphocytes (Alder Reilly granules) (black arrow).
Differential diagnosis
Spondyloepiphyseal dysplasia
Vitamin D-resistant rickets
Juvenile idiopathic arthritis (destructive polyarticular form)
Outcome and follow-up
A diagnosis of attenuated type I MPS (Hurler/Scheie syndrome) with normal intelligence, a rarity in itself, was made. The patient was offered enzyme replacement therapy but, due to financial constraints, his parents decided against it. He was advised physiotherapy at home and regular follow-up along with periodic pure tone audiometry, echocardiography and sleep studies.
Discussion
Type I MPS is a rare, autosomal recessive lysosomal storage disease due to allelic mutations in the α-L-iduronidase gene located at chromosome 4p16.3, resulting in deficiency of the lysosomal enzyme α-L-iduronidase. This culminates with the inability to degrade dermatan sulfate and heparan sulfate, the GAG normally providing structural support to the extracellular matrix and cartilaginous structures, such as different joints of the body, and heart valves.4 Accumulation of dermatan sulfate and heparan sulfate in various tissues of the body results in variable and progressive organ dysfunction subclassifying type I MPS based on disease severity. MPS I-H (Hurler syndrome) presents as an early onset, progressive disease with severe systemic manifestations including central nervous system (CNS) involvement and death, usually in the first two decades of life; MPS I-S (Scheie syndrome) is a relatively milder variant that is late in onset; it is a slowly progressing disease sparing CNS and having a near normal life expectancy. Finally, there is an intermediate phenotype, MPS I-H/S (Hurler-Scheie syndrome), with variable presentation.1 2
The incidence of MPS I is estimated at 1 case per 100 000 live births, making it a relatively rare metabolic disease. However, in a study undertaken in Australia, from 1980 to 1996, the incidence was estimated to be 1 in 22 500.5 The attenuated form of MPS I comprises almost 20% of all MPS I patients, making it even rarer.6 Short stature due to skeletal dysplasia (dysostosis multiplex) is undoubtedly the major presentation of most forms of MPS.4 7 8 Other associated clinical features include coarse facial features, corneal clouding, joint stiffness, visceromegaly, cardiac involvement (left-sided valve thickening and dysfunction, conduction defects, coronary artery disease and other vascular abnormalities), odontoid hypoplasia and atlantoaxial subluxation, all at variable degrees of severity. The most characteristic finding in bone marrow aspirates or in the peripheral blood smear is the presence of metachromatic granules surrounded by a clear zone within the cytoplasm of the lymphocytes. These are called Alder-Reilly granules and are caused by the accumulation of partially degraded mucopolysaccharides within the lysosomes. The granules may occur in clusters, rather than being diffuse, throughout the cytoplasm in all types of mature white cell count, and sometimes in primitive cells, and resemble toxic granulations in neutrophils. Interestingly, the boy in this case presented with lax and hypermobile joints, which, we believe, has not been reported before in the world literature. Moreover, in this particular case of attenuated MPS I, absence of CNS involvement, and presence of normal intelligence and of genu valgum, pectus carinatum with broadening of wrist, elbow and ankle joints mimicking classical rickets, prompted the physicians to overlook the underlying diagnosis. Effective treatment options for MPS I include haematopoietic stem-cell transplantation (HSCT) and enzyme replacement therapy. HSCT is presently considered for severe disease phenotypes (MPS I-H) in patients under 2 years with absent or minimal cognitive impairment, and it has the potential to preserve intellectual development if performed early in the course of the disease.3 The musculoskeletal abnormalities (dysostosis multiplex), however, do not improve with HSCT. Enzyme replacement therapy with recombinant human α-L-iduronidase (laronidase) is proven to be safe and effective in the treatment of MPS I, however, since laronidase cannot cross the blood–brain barrier at recommended doses (100 IU/kg/week), it is unlikely to improve the neurocognitive features of the disease spectrum.5 9 10 Untreated children commonly die of cardiorespiratory compromise.
Learning points.
A thorough clinical examination is mandatory in all patients presenting with clinical features consistent with rickets, vitamin D-resistant rickets in particular.
Mucopolysaccharidosis (MPS) is a very close differential of spondyloepiphyseal dysplasia (SED), and MPS should be ruled out in all children and young adults presenting with clinical and radiological features corroborating SED.
Footnotes
Contributors: PPC, SNB, SR and SKD were involved in patient management. PPC, SNB and SR were involved in the literature search and writing the manuscript. PPC finalised the manuscript.
Competing interests: None declared.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Neufeld EF, Muenzer J. The mucopolysaccharidoses. In: Scriver C, Beaudet A, Sly W et al., eds. The metabolic and molecular bases of inherited disease. New York, NY: McGraw Hill, 2001:3421–52. [Google Scholar]
- 2.Meikle PJ, Hopwood JJ, Clague AE et al. . Prevalence of lysosomal storage disorders. JAMA 1999;281:249–54. 10.1001/jama.281.3.249 [DOI] [PubMed] [Google Scholar]
- 3.Muenzer J, Wraith JE, Clarke LA. Mucopolysaccharidosis I: management and treatment guidelines. Pediatrics 2009;123:19–29. 10.1542/peds.2008-0416 [DOI] [PubMed] [Google Scholar]
- 4.Spranger J. Mucopolysaccharidoses. In: Kliegman RM, Stanton BF, St. Geme JW, Schor NF, Behrman RE, eds. Nelson textbook of pediatrics, 19th edn Philadelphia, PA: Saunders Elsevier, 2012:509–16. [Google Scholar]
- 5.Kakkis ED, Muenzer J, Tiller GE et al. . Enzyme-replacement therapy in mucopolysaccharidosis I. N Engl J Med 2001;344:182–8. 10.1056/NEJM200101183440304 [DOI] [PubMed] [Google Scholar]
- 6.Scott HS, Bunge S, Gal A et al. . Molecular genetics of mucopolysaccharidosis type I: diagnostic, clinical, and biological implications. Hum Mutat 1995;6:288–302. 10.1002/humu.1380060403 [DOI] [PubMed] [Google Scholar]
- 7.Barr DG. Metabolic diseases. In: Hendrickse GR, Harrison J, eds. Pediatrics in the tropics. Oxford: Blackwell, 1991:521–39. [Google Scholar]
- 8.Wit JM, Rekers-Mombarg LT, Cutler GB et al. . Growth hormone (GH) treatment to final height in children with idiopathic short stature: evidence for a dose effect. J Pediatr 2005;146:45–53. 10.1016/j.jpeds.2004.08.055 [DOI] [PubMed] [Google Scholar]
- 9.Clarke LA, Wraith JE, Beck M et al. . Long-term efficacy and safety of laronidase in the treatment of Mucopolysaccharidosis I. Pediatrics 2009;123:229–40. 10.1542/peds.2007-3847 [DOI] [PubMed] [Google Scholar]
- 10.Kakkis ED, McEntee MF, Schmidtchen A et al. . Long-term and high-dose trials of enzyme replacement therapy in the canine model of mucopolysaccharidosis I. Biochem Mol Med 1996;58:156–67. 10.1006/bmme.1996.0044 [DOI] [PubMed] [Google Scholar]