
Figure 2.
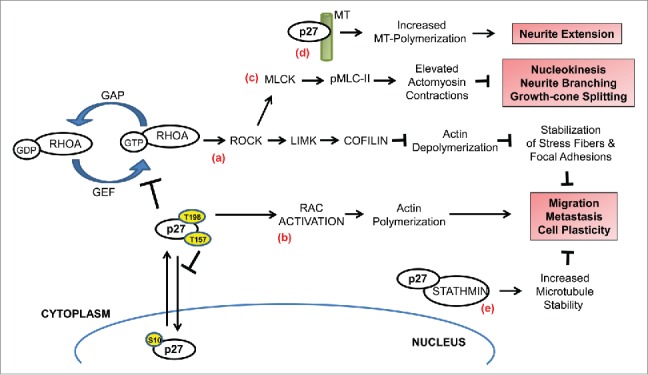
p27 regulates cell migration. Depending on the cell-specific context, p27 can either promote or inhibit cell migration in a CDK-independent manner. (a) RhoA functions as a molecular switch that cycles between inactive GDP-bound and active GTP-bound states that are promoted by GTPase-activating proteins (GAPs) and guanine–nucleotide exchange factors (GEFs), respectively. Upon stimulation, GEFs activate RhoA which in turn activates ROCKs to phosphorylate LIMKs. Thus activated, LIMK then phosphorylates cofilin to inhibit its actin-depolymerizing function to stabilize actin stress fibers and focal adhesions. Cytoplasmic p27 inhibits this pathway by directly interacting with RhoA and preventing its activation by the Rho-GEFs, resulting in increased cell motility. Mitogenic signaling pathways and oncogenic kinases target p27 for cytoplasmic relocalization through phosphorylation of specific residues (indicated by yellow circles)—phosphorylation of S10 promotes nuclear export while phosphorylation of T157 and/or T198 impairs nuclear import. (b) In certain cell types, cytoplasmic p27 can promote Rac activation to stimulate filopodium formation and cell migration (c) In migrating cortical interneurons, sustained activation of RhoA in the absence of p27 results in elevated actomyosin contractions due to increased phosphorylation of myosin-II light chain (pMLC-II) by activated MLC kinase (MLCK). This deregulation contributes to defective nucleokinesis, neurite branching, and tangential migration. Cytoplasmic p27 fine-tunes the actomyosin contractions by inhibiting RhoA activation to correct this defect. (d) p27 is a microtubule (MT)-associated protein and it promotes MT-polymerization, thereby contributing to neurite extension during interneuron migration. (e) p27 controls MT dynamics in a stathmin-dependent manner. Sequestration of stathmin, a MT-destabilizing protein, by p27 blocks the tubulin-sequestration activity of the former resulting in increased MT polymerization and inhibition of migration.