

Abstract
In this study, diethylnitrosamine-treated male mice were assigned to 3 groups: a 35% high fat ethanol liquid diet (EtOH) with casein as the protein source, the same EtOH liquid diet with soy protein isolate as the sole protein source (EtOH/SPI) and a chow group. EtOH feeding continued for 16 wks. As expected, EtOH increased the incidence and multiplicity of basophilic lesions and adenomas compared to the chow group, p<0.05. Soy protein replacement of casein in the EtOH diet significantly reduced adenoma progression when compared to the EtOH and EtOH/SPI group, p<0.05. Tumor reduction in the EtOH/SPI group corresponded to reduced liver injury associated with decreased hepatic tumor necrosis factor α (Tnfα) and Cd14 antigen (Cd14) expression and decreased nuclear accumulation of NFκB1 protein compared to the EtOH group (p<0.05). Detection of sphingolipids using high resolution MALDI-FTICR Imaging mass spectrometry revealed increased accumulation of long acyl chain ceramide species, and sphingosine-1-phosphate (S1P) in the EtOH group that were significantly reduced in the EtOH/SPI group. Chronic EtOH feeding also increased mRNA expression of β-catenin transcriptional targets, including cyclin D1 (Ccnd1), matrix metallopeptidase 7 (Mmp7) and glutamine synthetase (Glns), which were reduced in the EtOH/SPI group, p<0.05. We conclude that soy prevents tumorigenesis by reducing pro-inflammatory and oxidative environment resulting from EtOH-induced hepatic injury, and by reducing hepatocyte proliferation through inhibition of β-catenin signaling. These mechanisms may involve changes in sphingolipid signaling.
Keywords: carcinogenesis, alcohol, β-catenin, tumor promotion, soy protein isolate, ceramide synthesis
Introduction
Hepatocellular carcinoma (HCC) is the third leading cause of cancer mortality worldwide and is three times more likely to be diagnosed in men (1, 2). Of the known risk factors associated with hepatocellular carcinoma, alcohol consumption is the dominant independent factor linked to increased risk. In China and Japan, epidemiological data suggest that consumption of >80g/d of alcohol over 10 years results in a 2-fold increase in HCC risk, however in Western populations, increased HCC risk is 5-fold (3-5). Even at lower consumption rates, alcohol use is synergistic with other initiating factors, including hepatitis C and B infections or diabetes mellitus, increasing the overall prevalence of HCC in these populations worldwide (6-8). In the U.S., binge and heavy drinking has increased in prevalence, with 25% of adults reporting alcohol misuse or abuse, and is primary contributing risk factor for a third of reported cases (6, 9, 10). Moreover, once initiated, HCC risk does not decrease with abstinence (9) and there are few clinical strategies aimed to reduce risk in alcohol consumers.
Biological mechanisms involved in alcohol-induced liver cancer interact at the level of both initiation and promotion. Tumor initiation results from ethanol (EtOH) metabolism by alcohol dehydrogenase and cytochrome P450 (CYP) 2E1 thus, producing acetaldehyde and reactive oxygen species which interfere with DNA synthesis and repair mechanisms, leading to mutagenicity and tumorigenesis (4, 11). In addition to initiating effects, EtOH also has proliferative and tumor promoting effects in the liver (7, 8, 12). Our laboratory has demonstrated that EtOH stimulates hepatocyte proliferation in rodents in association with liver injury and hepatic vitamin A depletion (13-15). In our study, hepatic proliferation was associated with increased Wnt/β–catenin signaling and tumor multiplicity in male mice receiving EtOH for 16 wks and EtOH-induced tumors were β-catenin positive (13, 16). Likewise, a significant proportion of HCC tumors are β–catenin positive (17), although the extent of β–catenin expression during alcoholic liver disease progression is unknown.
These data suggest that an effective strategy for reducing alcohol-promotion of HCC is to target Wnt signaling. Diets rich in soy protein or soy-derived phytochemicals have been reported to have cancer preventative properties in both epidemiological and experimental animal studies (18-26). Additionally, in certain tissues, these soy rich diets are known to block Wnt signaling pathways. For example, in rat mammary epithelial cells, Su et al. (27) demonstrated that soy' bioactive isoflavone, genistein, inhibits Wnt signaling through increased expression of cadherin 1 and sequestration of β-catenin to the cell membrane. In human colon epithelial cells, Zhang et al. (28) proposed that genistein down regulates Wnt signaling through increased expression of soluble inhibitory factors called secreted frizzled related proteins (sFRP). Therefore, we hypothesized that dietary intervention with soy may prevent EtOH-mediated tumor promoting effects by inhibiting β-catenin signaling and associated proliferation mechanisms. To test this hypothesis, we utilized a two-stage mouse model in which tumor initiation by a genotoxic compound, diethylnitrosamine (DEN), was followed by the promoting agent, an EtOH liquid diet (13,16, 29), containing either casein or soy protein isolate (SPI) as the sole source protein source. Administration of these diets continued for 16 wks, at which livers were analyzed for the presence of adenomas.
Materials and Methods
Animals and experimental design
All experimental procedures involving animals were approved by the Institutional Animal Care and Use Committee at the University of Arkansas for Medical Sciences. Mice were housed in an Association Assessment and Accreditation of Laboratory Animal Care approved animal facility. Male and female C57Bl/6 mice (Jackson Laboratories, Bar Harbor, ME) were used to establish a breeding colony to generate male pups, who received either a single i.p. injection of 10 mg/kg DEN (n=54) or saline (n=25) on postnatal day 14. Mice were weaned to and maintained on rodent chow (Harlan Laboratories, Houston, TX) until postnatal day 60, at which the DEN-injected mice were randomly assigned to three weight-matched diet groups: a chow diet (n=10, chow), an EtOH-containing liquid diet (n=21, EtOH) and an EtOH-containing liquid diet containing soy protein isolate, SPI, (n=23, EtOH/SPI). All groups had access to water ad libitum. Liquid diets were formulated according to the Lieber-De-Carli diet of 35% of energy from fat, 18% from protein, and 47% from carbohydrates (Dyets, Inc., Bethlehem, PA), with SPI (Dupont Nutrition & Health) replacing casein as the sole protein source in the EtOH/SPI liquid diet. EtOH was added to the Lieber-De-Carli liquid diet slowly by substituting EtOH for carbohydrate calories in a stepwise manner until 28% total calories were reached as previously described (13). This dose constitutes a final EtOH concentration of 4.9% (v/v), respectively, and was maintained until sacrifice (4 mo), at which livers were fixed in formalin for pathological evaluation. At postnatal day 60, an additional group of saline-injected mice were randomized into three liquid diet groups, a chow diet (n=5), an EtOH (n=10), an EtOH/SPI (n=10). Diets were maintained for 4 mo. as described above. At sacrifice liver pieces were flash frozen in liquid nitrogen and stored at -70°C. Total serum isoflavone concentrations were extracted and analyzed as previously described (30). Serum alanine aminotransferase (ALT) levels were assessed as a measure of liver damage by using the Infinity ALT liquid stable reagent (Thermo Electron, Waltham, MA) according to manufacturer's protocols.
Pathological evaluation
All formalin fixed liver lobes from DEN-treated mice receiving chow, EtOH or EtOH/SPI diets were embedded in paraffin, sectioned (4μm), stained, and examined in a blinded fashion under a light microscope a previously described (13) by two independent pathologists (L.H., K.L.). Preneoplastic foci were counted at 40x magnification. Preneoplastic foci can develop into adenomas, which were defined as a compressive lesion of any size without evidence of invasion or other criteria of malignancy, and may develop into hepatocellular carcinoma, which was defined as a compressive and invasive lesion with criteria of malignancy (31).
Immunohistochemistry
Archival liver tissue sections from de-identified patients diagnosed with alcoholic steatohepatitis (n=5) or alcoholic cirrhosis (n=7) were obtained from the UAMS Department of Pathology. β-catenin expression was assessed in liver sections by immunohistochemistry using standard procedures and a monoclonal β–catenin antibody (1:75) detecting the active, dephosphorylated (Ser37 or Thr41) form (Anti-Active-β-catenin, clone 8E7, EMD Millipore, Billerica, MA), as previously described (13). Hepatic proliferating cell nuclear antigen (PCNA) staining was assessed by immunohistochemistry in non-tumor tissue in chow, EtOH and EtOH/SPI fed mice as described (13). Stained slides were evaluated under a light microscope. Nuclei of S-phase cells stained dark brown; a total of 10 observations (100X field counted) were screened per liver sample. Data are expressed as a percentage of PCNA stained nuclei in S-phase.
Gene Expression
Liver RNA was isolated from DEN- and saline-treated mice receiving chow, EtOH or EtOH/SPI diets as previously described. All RNA was reverse transcribed using IScript cDNA synthesis (Bio-Rad Laboratories, Hercules, CA) according to manufacturer's instructions, and subsequent real-time PCR analysis was carried out using SYBR green and an ABI 7500 sequence detection system (Applied Biosystems, Foster City, CA). Results were quantified using the delta CT method relative to 18s or Gapdh. Primer sequences are presented in Supplementary Table 1.
Protein isolation and Western blotting
Nuclear and cytosolic protein fractions were isolated from TEN control and EtOH-treated mouse livers using NE-PER Nuclear and Cytoplasmic Extraction reagent kit (Fisher Scientific, Pittsburg, PA). Membrane fractions were obtained using a modified protocol previously described (32). Proteins (30 μg) were separated by SDS-polyacrylamide gel electrophoresis using standard methods. Blotted cytosolic proteins were incubated with the following antibodies, pAKT, total AKT, pGSK3β (Phospho-GSK-3α/β (Ser21/9) and total GSK3β, from Cell Signaling Technology (Beverly, MA.) using a 1:1000 dilution. Antibodies toward β-catenin (1:1000, Milipore, Billerica, MA), and pNFκB (Cell Signaling Technology, Beverly, MA) were used on blotted nuclear proteins. Membrane fractions were probed with an anti-SPHK1 antibody (1:1000, Cell Signaling Technology, Beverly, MA). Secondary antibodies were diluted (1:5000 to 1:10000) and incubated at room temperature before chemiluminescense detection. Protein bands were quantified using a densitometer and band densities were corrected for total protein loaded by staining the membrane with 0.1% amido black.
Matrix-assisted laser desorption/ionization (MALDI) imaging mass spectrometry
MALDI-Fourier transform ion cyclotron resonance (FTICR) imaging mass spectrometry was used to qualitatively detect sphingolipid species in liver sections from saline-treated mice receiving an EtOH or EtOH/SPI diet using methods previously described (33). Briefly, the left lateral liver lobe (n=1 per diet group) were rapidly frozen in liquid nitrogen for 2 minutes and stored at -80° C until use. Liver tissues were sectioned at 10μm, thaw mounted on indium tin oxide coated slides (Bruker Daltonics) and desiccated at room temperature for five minutes. A 2,5-dihydroxybenzoic acid matrix was added using an ImagePrep spray station (Bruker Daltonics), at a concentration of 0.2 M in 50% methanol and 0.01% trifluoroacetic acid. MALDI imaging mass spectrometry analysis was performed using a Bruker Solarix 7T FTICR mass spectrometer, equipped with a SmartBeam II laser operating at 1000 Hz, collecting spectra across the entire tissue in positive ion mode between (m/z 200–2000). A laser spot size of 25 μm, and a resolution or raster width of 200 μm was utilized for analysis, collecting 800 shots per pixel. Data was reduced to .98 ICR reduction and loaded into FlexImaging 4.0 software (Bruker Daltonics) for data analysis, and generation of lipid images of interest. Within FlexImaging, all data was normalized using root mean square and intensities were thresholded appropriately. Due to the high mass accuracy and resolution capability of a 7T FTICR mass spectrometer used in this study, lipid species were identified by mass accuracy in reference to an internal ceramide data base (33) and to an external database known as Lipid Maps.
Data and statistical analysis
Data presented as means ± SE. Comparisons between three groups were accomplished by One-Way ANOVA followed by a Student-Newman Keuls post-hoc analysis or by Kruskal-Wallis One-way Analysis of Variance on Ranks followed by Dunn's post-hoc analysis. Comparisons between two groups were accomplished using either Student's T-test or Mann-Whitney U rank-sum test. Number of lesions was compared across groups using negative binomial regression which generalizes Poisson regression to account for over-dispersion of the count data. This model included group membership as a set of indicator (dummy) variables. The proportion of new adenomas (incidence) was compared across groups using Fishers exact test. Statistical analysis was performed using the SigmaPlot software package 11.0 (Systat Software, Inc., San Jose, CA) and Stata statistical software 13.1 (Stata Corporation, College Station, TX). Statistical significance was set at P<0.05.
Results
Study design and observations
Male mice were assigned to an EtOH or EtOH/SPI liquid diet, and a chow fed diet 43 days post DEN injection. Starting weights for DEN-treated mice were 22.8 g ± 0.47, 22.8 g ± 0.28 and 23.0 g ±0.28 for chow, and EtOH and EtOH/SPI, respectively. EtOH feeding continued for 16 wks. Diet intakes between the EtOH and EtOH/SPI groups did not differ statistically, 15.4 ml ±0.12 and 15.2 ml ± 0.09 respectively, which corresponded to an average daily intake of 18.6 g/kg/d of EtOH. In the saline-treated mice starting weights were 23.0±0.46, 24.4±0.44, and 23.7±0.50, for chow, EtOH and EtOH/SPI, respectively. In these animals, diet intakes were 15.8ml±0.23 and 16.0ml±0.14 for EtOH and EtOH/SPI groups, respectively and corresponded to an average daily intake of 19.7 g/kg/d of EtOH, which resulted in a mean (±SE) blood alcohol concentration of 29±7.2 mg/dL and 23±4.3 mg/dL for EtOH and EtOH/SPI groups, respectively. Total circulating isoflavone concentrations (aglycone+conjugates) were measured in serum of DEN- and saline-treated mice receiving the EtOH/SPI diet, as previously described (28). Equol, daidzein, genistein, and o-desmethylangolensin were present at the following concentrations, 1.15 ± 0.14, 0.13 ± 0.05, 0.05 ± 0.02, and 0.08±0.02 μmol. L-1, respectively, and did not differ between the two groups.
SPI decreases adenoma incidence and multiplicity associated with EtOH feeding in DEN-treated mice
Liver lobes taken from DEN-treated chow, EtOH and EtOH/SPI mice were assessed for presence of lesions, which encompassed basophilic foci and adenomas. Basophilic incidence in the EtOH, 0.90 (19/21), and EtOH/SPI, 0.95 (22/23), groups were increased by 2-fold compared to the chow, 0.5 (5/10) fed mice (Fisher's Exact Test, p<0.05). Likewise, basophilic multiplicity in the EtOH and the EtOH/SPI groups were also significantly increased compared to chow, 4.6 (88/21), 3.8 (88/23) and 1.7 (17/10), respectively (One-way ANOVA followed by Mann-Whitney U rank-sum test, p<0.05). We also observed the presence of adenomas in the DEN-treated EtOH, 0.67 (14/21), which were absent in the DEN-treated chow, 0.0 (0/10) group (Fisher's Exact Test, p<0.05). When compared to the EtOH group, SPI feeding reduced adenoma incidence, 0.23 (6/23) in the DEN-treated EtOH/SPI (Fisher's Exact Test, p<0.05). Likewise, we observed a significant decrease in adenoma multiplicity in the ETOH/SPI, 0.43 (10/23) compared to the EtOH, 1.62 (32/21) fed mice (Mann-Whitney U rank sum test, p<0.05).
Reduced tumor progression is associated with decreased liver pathology in EtOH/SPI mice
SPI substitution for casein protein in the EtOH diet had a marked impact on liver pathology in the DEN-treated mice. As seen in Figure 1, chronic consumption of the DEN/EtOH diet resulted in elevated serum ALT concentrations which corresponded to histological evidence of steatosis and inflammation and increased hepatocyte proliferation in non-tumorigenic tissue when compared to the DEN/chow fed group (p<0.05). In contrast, EtOH/SPI feeding resulted in a significant reduction in serum ALT, decreased overall pathology and a reduction in proliferation in comparison to the EtOH-treated mice (Figure 1). Mechanisms associated with decreased liver pathology were assessed in hepatic tissue taken from saline-treated mice receiving chow, EtOH, and EtOH/SPI diets concurrently with the DEN-treated mice. Similar to the DEN-treated mice, the EtOH/SPI diet decreased liver injury as measured by ALT (37.6 ± 3.6 vs. 8.6 ± 1.58 and 8.6 ± 0.89, for the saline-treated EtOH, EtOH/SPI and chow fed groups, respectively (p<0.05). Likewise, in Figure 2, we observed a 47% and 68% decrease in hepatic Tnfα and Interleukin 6 (Il6) mRNA expression in the EtOH/SPI group compared to the EtOH group. Hepatic liver transcripts of Cd14 and chemokine (C-X-C motif) ligand 2 (Cxcl2) were also decreased 1.5 – to 2-fold, respectively, in the EtOH/SPI fed mice, p<0.05 (Figure 2C, D). These findings were associated with reduced protein accumulation of the pro-inflammatory transcription factor, NFκB, in the nucleus in the EtOH/SPI group (Figure 2E, p<0.05). In addition, fibrosis markers were also decreased in the livers of EtOH/SPI-treated mice. Alpha-smooth muscle actin (α-Sma) and platelet-derived growth factor receptor (Pdgfr) were significantly suppressed in comparison to both the EtOH-treated mice and chow controls; 3.48±1.17vs. 0.31±0.07 and 0.27±0.04 for α-Sma in EtOH, EtOH/SPI and chow controls, respectively, and 1.48±0.10 vs. 0.89±0.13 and 0.88±0.1 for Pdgfr, in EtOH, EtOH/SPI and chow controls, respectively (One-way ANOVA, Student Newman Keuls post hoc analysis, p<0.05).
Fig.1.

The effect of EtOH and EtOH/SPI liquid diets on liver pathology in DEN-treated male mice. (A) Representative hematoxylin and eosin stained liver sections in DEN-treated male mice receiving chow (DEN/Chow), n=10, LieberDeCarli EtOH diet (DEN/EtOH), n=21, or Lieber DeCarli EtOH diet with SPI as sole protein source (DEN/EtOH/SPI), n=23; pathology score, (B) liver injury, (C) steatosis, (D) immune cell infiltration and (E) hepatocyte proliferation were determined as described in the Materials and Methods. Statistical analysis was performed by One-way ANOVA followed by Student-Newman Keuls post hoc analysis. Means that do not share a letter are significantly different at p<0.05.
Fig.2.

Changes in hepatic mRNA expression of pro-inflammatory cytokines (A) Tnfα, (B) Il6, (C) Cxcl2, and (D) Cd14 in saline-treated male mice receiving EtOH (n=10) and EtOH/SPI (n=10) diets compared to chow (n=5) controls. Western blot analysis of (E) nuclear expression of NFκB expression liver tissue from EtOH and EtOH/SPI fed mice. Data are expressed as mean ± St. Err. For real-time RT-PCR, statistical analysis was determined by Kruskal-Wallis One-way Analysis of Variance on Ranks followed by Dunn's post-hoc analysis, and groups with different letters are significantly different from each other. For the Western, statistical significance was determined by Student's T-test. Means that do not share a letter are significantly different at p<0.05
Immunohistochemical analysis of β-catenin expression and localization in livers from chronic alcoholics
Clinically, a significant percentage of liver tumors from alcoholics are positive for β-catenin expression (17). In Figure 3, we looked for the presence of β-catenin in archival liver tissue taken from patients diagnosed with alcoholic steatohepatitis and cirrhosis by immunohistochemistry. In these tissues, we observed a pattern of increased membrane staining of β-catenin and translocation of β-catenin to the nucleus coinciding with disease progression. These data is consistent with our rodent model demonstrating that Wnt/β–catenin signaling during early stages of steatohepatitis (13), and in the clinical setting, increased activation in cirrhotic livers, where HCC generally occurs in humans.
Fig.3.

Representative liver sections showing the expression and localization of β–catenin in archival liver tissue from patients with normal liver pathology (n=3) or from patients diagnosed with alcoholic steatohepatitis (n=5) and cirrhosis (n=7). Immunohistochemistry was performed as described in the Materials and Methods.
SPI inhibits EtOH-mediated Wnt/β-catenin signaling in hepatocytes
Previously we have reported chronic EtOH feeding in rats promotes hepatocyte proliferation through increased β-catenin activation via the Wnt signaling pathway (13). As seen in Table 1, we observed a 3-fold increase in wingless-type MMTV integration site family, member 2 (Wnt2) mRNA expression, corresponding to approximately a 2-fold increase in the ratio of active (phosphorylated) GSK3β to total GSK3β cytosolic protein expression, and up-regulation of mRNA expression of known β-catenin targets, Ccnd1, Glns, and Mmp7 in saline-treated EtOH mice compared to chow controls; SPI blocks these increases in the EtOH/SPI group (p<0.05), including nuclear accumulation of β-catenin, p<0.05. Consistent with these findings, in the DEN-treated mice receiving EtOH, we observed increased mRNA expression of other soluble Wnts, Wnt2b and Wnt7a (Table 1), and SPI reduced transcript expression 16% (p=0.02) and 34% (p=0.08), respectively, in the EtOH/SPI group. Moreover, we observed increased mRNA expression of soluble Wnt inhibitors, Wnt inhibitory factor 1 (Wif1) and dickkopf-3 (Dkk3) in the EtOH/SPI group compared to DEN-treated EtOH mice. These findings are indicative of inhibition of the canonical Wnt signaling pathway. In our model, we did not see any changes in phosphorylation status of the serine/threonine kinase AKT between saline-treated EtOH and chow fed mice that would suggest that the survival pathway PI3Kinase/AKT/GSK3β was responsible for EtOH-mediated activation of Wnt signaling (Table 1).
Table 1. Hepatic Wnt/β-catenin signaling in saline-treated mice receiving EtOH diets.
| Saline-treated mice | |||
|
| |||
| Chow | EtOH | EtOH/SPI | |
|
| |||
| Gene Expression | |||
| Wnt2 | 0.12±0.01a | 0.40±0.09b | 0.22±0.03a,b |
| Glns | 1.40±0.33a | 2.79±0.43b | 1.28±0.24a |
| Mmp7 | 0.83±0.17a | 2.48±0.72b | 0.56±0.11a |
| Ccnd1 | 0.69±0.11a | 1.97±0.28b | 0.92±0.11a |
| Protein Expression | |||
| pAKT:totalAKT | 1.65±0.40a | 0.71±0.12a | 1.16±0.27a |
| pGSK3β:totalGSK3β | 0.65±0.01a | 1.25±0.12b | 0.94±0.05a |
| Nuclear β-catenin | -- | 1.23±0.18 | 0.71±0.05* |
| DEN-treated mice | |||
|
| |||
| Chow | EtOH | EtOH/SPI | |
|
| |||
| Gene Expression | |||
| Wnt2b | 0.21±0.03a | 1.03±0.04b | 0.86±0.04c |
| Wnt5a | 0.24±0.06a | 0.51±0.04b | 0.46±0.02b |
| Wnt7b | 0.65±0.19a | 1.71±0.24b | 1.12±0.16a,b |
| Dkk3 | 0.25±0.09a | 0.48±0.01b | 0.58±0.03c |
| Wif1 | 0.10±0.03a,b | 0.17±0.02b | 0.06±0.01a |
Data is expressed as mean ± St.Err for saline-treated chow (n=5), EtOH (n=10), and EtOH/SPI (n=10), DEN-treated chow (n=5), EtOH (n=21) and EtOH/SPI (n=23) groups. Gene expression was measured by real-time RT-PCR, and values calculated by 2-ΔΔCT method as described in Materials and Methods. Proteins involved in Wnt/β-catenin signaling were assessed by Westerns as described in Materials and Methods; blots were developed using chemiluminscence detection; band densities were corrected for total protein load by staining with 0.1% amido black. Statistical significance was determined by One-way ANOVA, p<0.05, a<b<c, or Student's T-test,
p<0.05, EtOH vs. EtOH/SPI
Effects of SPI on hepatic ceramide accumulation and ER stress during chronic EtOH feeding
In humans and in rodents, alcohol exposure has been shown to increase ceramide accumulation in the liver, thereby enhancing endoplasmic reticulum stress and contributing to liver injury (34). Consistent with these reports, we observed a significant increase in the mRNA expression of serine palmitoyl transfease subunit (Sptlc1), a key enzyme involved in de novo ceramide biosynthesis, in the EtOH-treated mice compared to chow fed mice (Figure 4A). Ceramide synthase (Cers) gene transcripts were also upregulated; of note Cers2 and Cers4 were highly expressed (6-fold) in EtOH-treated mice, p<0.05 (Figure 4B). In addition, chronic EtOH exposure also significantly increased mRNA expression of acid ceramidase (Asah1), and sphingosine kinase 1 (Spkh1), key enzymes in the synthesis of sphingosine-1P (S1P), and sphingosine-1P receptors (Spr) 2 and 3 (Figure 4C-E). In the EtOH/SPI-treated mice, Sptlc1 mRNA expression was increased compared to the EtOH mice, but did not reach significance (p=0.079). However, we did see a significant decrease in Cers1 mRNA expression in response to EtOH/SPI diets, but no other significant decreases with respect to Cers2,4,5, and 6 expression (Figure 4B, p<0.05). In contrast, SPI did shut down sphingosine-1P production, including the mRNA expression and translocation of Sphk1 to the membrane which is necessary for enzymatic activation (Figure 4F, p<0.05). Mechanistically, Cers1 predominately generates C18:0-ceramide, Cers2 produces longer acyl chain ceramides (C20:0—C26:0 ceramides), and Cers5, 6, synthesizes the shorter C14:0, C16:0 ceramide species. In Figure 5, using a novel MALDI-FTICR imaging mass spectrometry workflow used for on-tissue detection and spatial localization of ceramides and other sphingolipids (33), we qualitatively confirmed the presence of short and long acyl chain ceramides and S1P in livers from mice receiving EtOH. We also observed a subsequent loss of ceramide C18 and sphingosine -1P in the EtOH/SPI livers, which is consistent with the mechanistic data presented in Figure 4.
Fig.4.

Changes in hepatic mRNA expression of de novo ceramide synthesis and sphingosine 1-P production (A) Sptlc1, (B) CerS, (C) Asah1, (D) Spkh1, and (E) S1p receptors 1,2,and 3, in saline-treated male mice receiving EtOH (n=10) and EtOH/SPI (n=10) diets compared to chow (n=5) controls. Western blot analysis of (F) membrane protein expression of Spkh1 in liver tissue from EtOH and EtOH/SPI fed mice. Data are expressed as mean ± St. Err. For real-time RT-PCR, statistical analysis was determined by Kruskal-Wallis One-way Analysis of Variance on Ranks followed by Dunn's post-hoc analysis, and groups with different letters are significantly different from each other. Statistical significance for Western blot analysis was determined by Student's T-test. Means that do not share a letter are significantly different at p<0.05
Fig.5.
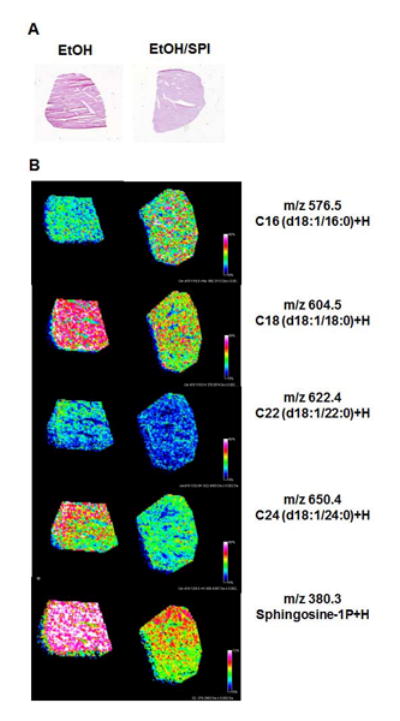
Analysis of ceramide species in a sectioned liver from saline-treated male mice receiving EtOH and EtOH/SPI diets; (A) hematoxylin and eosin stain of serial liver sections, (B) qualitative confirmation of short and long acyl chain ceramides and sphingosine 1P using MALDI-FTICR imaging mass spectrometry previously described for on-tissue detection, spatial localization and structural confirmation of bioactive sphingolipids (33). Representative images, n=1 per diet group, were acquired using a high resolution mass spectrometer, Bruker Solarix 7T FTICR. Images were normalized using root mean squares and thresholded to each other. The intensity of each lipid detected is shown as a reflection of its color intensity with areas of greater distribution of a particular lipid being pink or red, compared to lower signal detection shown as blue or green. All lipid masses were crossed referenced to an internal database (33) and an external database Lipid Maps.
Discussion
Epidemiological data from Asian populations eating traditional soy-based diets have suggested a variety of health benefits which include a reduction in the risk of different cancer types (21, 22, 25, 26, 35). In this study, we examined the possibility that dietary intervention with SPI would also prevent EtOH-mediated hepatic tumor promotion in a rodent model of chemical carcinogenesis (13,16). In response to alcoholic liver injury, most rodent studies have reported a corresponding increase in hepatocyte proliferation (14, 15, 36, 37). This appears to be in response to proliferative signals such as a reduction in retinoid receptor activation and activation of β-catenin transcriptional activity downstream of WNT expression and secretion (8, 13). In the current study, EtOH-mediated hepatocyte proliferation was dependent on Wnt-β-catenin signaling. Moreover, we observed increased β-catenin staining in livers from alcoholics with steatohepatitis and chirrosis, which supports the role of Wnt-β-catenin signaling in tumor growth and progression. Feeding SPI had a highly significant inhibitory effect on EtOH-mediated adenoma tumor progression. Inhibition of β-catenin activation by SPI feeding was accompanied by decreased phosphorylation of GSK3β, decreased expression of Wnt mRNAs and increased expression of mRNA for the Wnt-signaling inhibitor Dkk3. These data suggest that, at least in part, SPI components block EtOH-induced Wnt signaling.
The detailed molecular mechanisms underlying EtOH induction and SPI suppression of hepatic Wnt pathways remain to be elucidated. It has been suggested that Wnt secretion by non-parenchymal cells is involved in regulation of hepatocyte proliferation and in some forms of liver cancer. Studies of hepatocyte proliferation following 2/3 partial hepatectomy have implicated Wnt secretion by Kuppfer cells in this process (38). Recent studies of cholangiocarcinoma have demonstrated characteristic enhancement of Wnt signaling via inflammatory macrophages (39). EtOH promotion of hepatic tumorigenesis has also been demonstrated to be linked to induction of TGFβ signaling and stellate cell activation (7) and enhanced tumorigenesis has also been observed in other experimental models of fibrosis (40). Additional studies are underway to identify if Kupffer cells, other inflammatory cell types, stellate cells or endothelial cells are the cellular source of EtOH-induced Wnts.
SPI feeding also significantly lowered necro-inflammatory injury and Kupffer cell recruitment in the EtOH/SPI group. The hepatoprotective effects of SPI were coincident with reduced mRNA expression of Il6, decreased hepatocyte proliferation and evidence of reduced β-catenin activation. These data support a mechanistic link between soy's inhibitory effects on EtOH-dependent tumor promotion, reductions in Wnt-β-catenin signaling and reduced liver injury. Genistein, the major isoflavone phytoestrogen, is considered the biologically active anti-cancer component in soy foods (27, 28, 41, 42). Recently, using the same model reported here, we have found increased EtOH-mediated hepatic adenomas in male mice receiving genistein in the liquid EtOH diet (43). Our findings suggest that SPI contains additional anti-cancer components. Supporting evidence for this hypothesis comes from a recent report demonstrating feeding of an isoflavone-free SPI diet specifically inhibited NFκB-dependent expression of inflammatory cytokines, including Tnfα in the hyperlipidemic apoE-/- mouse model, thus reducing the presence of atherosclerotic lesions (44). These findings suggest that soy hydrolysates and putative peptides derived from digestion of the major soy storage protein β-conglycinin in the gut may have anti-inflammatory properties.
Recent reports have implied that chronic alcohol consumption influences hepatic ceramide generation (34, 45). In this study, we observed significant elevations of hepatic ceramide synthesis, particularly C18 generation, and S1P production in response to chronic EtOH consumption. These data are consistent with those reported by Longato et al.(34) in chronic alcoholics and from our laboratory using an intra-gastric EtOH treatment in a rat model associated with hepatic insulin resistance (45). In this study, we did not explore the alternative ceramide generation pathways, including complex sphingolipid hydrolysis or through a salvage pathway involving ceramide deacylation (46). However, Longato et al. (34) observed increased sphingomyelin hydrolysis in the hepatic tissue of chronic alcoholics, suggesting that alcohol may effect additional pathways of ceramide synthesis.
Interestingly, EtOH-mediated increases in C18 ceramide and S1P were reversed in the EtOH/SPI group. Different ceramide species have diverse effects on cell death and proliferation (47). Ceramide is converted to S1P, an important pro-inflammatory and pro-fibrotic lipid mediator, via the actions of ceramidases and sphingosine kinases. S1P acts via 5 receptors to stimulate inflammation both up and down-stream of TNFα and to stimulate fibrosis via activation of stellate cells (24, 47,48). Moreover, S1P has anti-apoptotic actions and paracrine proliferative effects on hepatocytes and pancreatic cancer cells mediated via endothelial and stellate cells (47). It is possible that SPI inhibition of ceramide/S1P synthesis is involved in blockade of Wnt/β-catenin signaling, particularly if activated stellate cells are a source of WNTs. Alternatively, SPI may affect other Wnt-independent pathways impacting on tumor promotion such as expansion of tumor initiating stem cell populations or on apoptosis/proliferation balance in transformed cell populations. The role of ceramides and S1P in EtOH-induced hepatic tumor promotion and protection by feeding of SPI are also the subject of ongoing studies.
In conclusion, our data demonstrate that EtOH acts as a tumor promoter in the mouse DEN model as a result of activation of Wnt/ β-catenin signaling. Protection against EtOH-induced tumorigenesis by switching from casein to SPI as the dietary protein source appears to be due to a component other than genistein which acts to block steatosis and progression of liver injury, reverses EtOH activation of β-catenin and prevents EtOH-induced changes in ceramide/sphingosine metabolism.
Supplementary Material
Acknowledgments
This work was funded in part by National Institute of Health R21 CA169389 (M. Ronis) and by the Arkansas Children's Hospital Research Institute and the Marion B. Lyon New Scientist Development Award (K.E. Mercer).
Footnotes
The authors of this manuscript do not have any Conflicts of Interest and therefore have nothing to declare.
References
- 1.Altekruse SF, McGlynn KA, Reichman ME. Hepatocellular carcinoma incidence, mortality, and survival trends in the United States from 1975 to 2005. J Clin Oncol. 2009;27:1485–1491. doi: 10.1200/JCO.2008.20.7753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Giannitrapani L, Soresi M, La SE, Cervello M, D'Alessandro N, Montalto G. Sex hormones and risk of liver tumor. Ann N Y Acad Sci. 2006;1089:228–236. doi: 10.1196/annals.1386.044. [DOI] [PubMed] [Google Scholar]
- 3.Luo RH, Zhao ZX, Zhou XY, Gao ZL, Yao JL. Risk factors for primary liver carcinoma in Chinese population. World J Gastroenterol. 2005;11:4431–4434. doi: 10.3748/wjg.v11.i28.4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morgan TR, Mandayam S, Jamal MM. Alcohol and hepatocellular carcinoma. Gastroenterology. 2004;127:S87–S96. doi: 10.1053/j.gastro.2004.09.020. [DOI] [PubMed] [Google Scholar]
- 5.Shimazu T, Sasazuki S, Wakai K, Tamakoshi A, Tsuji I, Sugawara Y, et al. Alcohol drinking and primary liver cancer: a pooled analysis of four Japanese cohort studies. Int J Cancer. 2012;130:2645–2653. doi: 10.1002/ijc.26255. [DOI] [PubMed] [Google Scholar]
- 6.Yuan JM, Govindarajan S, Arakawa K, Yu MC. Synergism of alcohol, diabetes, and viral hepatitis on the risk of hepatocellular carcinoma in blacks and whites in the U.S. Cancer. 2004;101:1009–1017. doi: 10.1002/cncr.20427. [DOI] [PubMed] [Google Scholar]
- 7.Brandon-Warner E, Walling TL, Schrum LW, McKillop IH. Chronic ethanol feeding accelerates hepatocellular carcinoma progression in a sex-dependent manner in a mouse model of hepatocarcinogenesis. Alcohol Clin Exp Res. 2012;36:641–653. doi: 10.1111/j.1530-0277.2011.01660.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chung J, Liu C, Smith DE, Seitz HK, Russell RM, Wang XD. Restoration of retinoic acid concentration suppresses ethanol-enhanced c-Jun expression and hepatocyte proliferation in rat liver. Carcinogenesis. 2001;22:1213–1219. doi: 10.1093/carcin/22.8.1213. [DOI] [PubMed] [Google Scholar]
- 9.Hassan MM, Hwang LY, Hatten CJ, Swaim M, Li D, Abbruzzese JL, et al. Risk factors for hepatocellular carcinoma: synergism of alcohol with viral hepatitis and diabetes mellitus. Hepatology. 2002;36:1206–1213. doi: 10.1053/jhep.2002.36780. [DOI] [PubMed] [Google Scholar]
- 10.Yang JD, Harmsen WS, Slettedahl SW, Chaiteerakij R, Enders FT, Therneau TM, et al. Factors that affect risk for hepatocellular carcinoma and effects of surveillance. Clin Gastroenterol Hepatol. 2011;9:617–623. doi: 10.1016/j.cgh.2011.03.027. [DOI] [PubMed] [Google Scholar]
- 11.Seitz HK, Stickel F. Molecular mechanisms of alcohol-mediated carcinogenesis. Nat Rev Cancer. 2007;7:599–612. doi: 10.1038/nrc2191. [DOI] [PubMed] [Google Scholar]
- 12.Yip-Schneider MT, Doyle CJ, McKillop IH, Wentz SC, Brandon-Warner E, Matos JM, et al. Alcohol induces liver neoplasia in a novel alcohol-preferring rat model. Alcohol Clin Exp Res. 2011;35:2216–2225. doi: 10.1111/j.1530-0277.2011.01568.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mercer KE, Hennings L, Sharma N, Lai K, Cleves MA, Wynne RA, et al. Alcohol consumption promotes diethylnitrosamine-induced hepatocarcinogenesis in male mice through activation of the Wnt/β-catenin signaling pathway. Cancer Prev Res (Phila) 2014;7:675–685. doi: 10.1158/1940-6207.CAPR-13-0444-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ronis MJ, Butura A, Korourian S, Shankar K, Simpson P, Badeaux J, et al. Cytokine and chemokine expression associated with steatohepatitis and hepatocyte proliferation in rats fed ethanol via total enteral nutrition. Exp Biol Med (Maywood) 2008;233:344–355. doi: 10.3181/0707-RM-203. [DOI] [PubMed] [Google Scholar]
- 15.Ronis MJ, Hennings L, Stewart B, Basnakian AG, Apostolov EO, Albano E, et al. Effects of long-term ethanol administration in a rat total enteral nutrition model of alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol. 2011;300:G109–G119. doi: 10.1152/ajpgi.00145.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mercer KE, Hennings L, Ronis MJ. Alcohol consumption, Wnt/β-catenin signaling, and hepatocarcinogenesis. Adv Exp Med Biol. 2015;815:185–195. doi: 10.1007/978-3-319-09614-8_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Edamoto Y, Hara A, Biernat W, Terracciano L, Cathomas G, Riehle HM, et al. Alterations of RB1, p53 and Wnt pathways in hepatocellular carcinomas associated with hepatitis C, hepatitis B and alcoholic liver cirrhosis. Int J Cancer. 2003;106:334–341. doi: 10.1002/ijc.11254. [DOI] [PubMed] [Google Scholar]
- 18.Badger TM, Ronis MJ, Simmen RC, Simmen FA. Soy protein isolate and protection against cancer. J Am Coll Nutr. 2005;24:146S–149S. doi: 10.1080/07315724.2005.10719456. [DOI] [PubMed] [Google Scholar]
- 19.Hakkak R, Korourian S, Shelnutt SR, Lensing S, Ronis MJ, Badger TM. Diets containing whey proteins or soy protein isolate protect against 7,12-dimethylbenz(a)anthracene-induced mammary tumors in female rats. Cancer Epidemiol Biomarkers Prev. 2000;9:113–117. [PubMed] [Google Scholar]
- 20.Illuzzi G, Bernacchioni C, Aureli M, Prioni S, Frera G, Donati C, et al. Sphingosine kinase mediates resistance to the synthetic retinoid N-(4-hydroxyphenyl)retinamide in human ovarian cancer cells. J Biol Chem. 2010;285:18594–18602. doi: 10.1074/jbc.M109.072801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Myung SK, Ju W, Choi HJ, Kim SC. Soy intake and risk of endocrine-related gynaecological cancer: a meta-analysis. BJOG. 2009;116:1697–1705. doi: 10.1111/j.1471-0528.2009.02322.x. [DOI] [PubMed] [Google Scholar]
- 22.Shu XO, Jin F, Dai Q, Wen W, Potter JD, Kushi LH, et al. Soyfood intake during adolescence and subsequent risk of breast cancer among Chinese women. Cancer Epidemiol Biomarkers Prev. 2001;10:483–488. [PubMed] [Google Scholar]
- 23.Simmen RC, Eason RR, Till SR, Chatman L, Jr, Velarde MC, Geng Y, et al. Inhibition of NMU-induced mammary tumorigenesis by dietary soy. Cancer Lett. 2005;224:45–52. doi: 10.1016/j.canlet.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 24.Wang X, Zhang DM, Gu TT, Ding XQ, Fan CY, Zhu Q, et al. Morin reduces hepatic inflammation-associated lipid accumulation in high fructose-fed rats via inhibiting sphingosine kinase 1/sphingosine 1-phosphate signaling pathway. Biochem Pharmacol. 2013;86:1791–1804. doi: 10.1016/j.bcp.2013.10.005. [DOI] [PubMed] [Google Scholar]
- 25.Wu AH, Wan P, Hankin J, Tseng CC, Yu MC, Pike MC. Adolescent and adult soy intake and risk of breast cancer in Asian-Americans. Carcinogenesis. 2002;23:1491–1496. doi: 10.1093/carcin/23.9.1491. [DOI] [PubMed] [Google Scholar]
- 26.Zhang Y, Chen H. Genistein, an epigenome modifier during cancer prevention. Epigenetics. 2011;6:888–891. doi: 10.4161/epi.6.7.16315. [DOI] [PubMed] [Google Scholar]
- 27.Su Y, Simmen RC. Soy isoflavone genistein upregulates epithelial adhesion molecule E-cadherin expression and attenuates β-catenin signaling in mammary epithelial cells. Carcinogenesis. 2009;30:331–339. doi: 10.1093/carcin/bgn279. [DOI] [PubMed] [Google Scholar]
- 28.Zhang Y, Chen H. Genistein attenuates WNT signaling by up-regulating sFRP2 in a human colon cancer cell line. Exp Biol Med (Maywood) 2011;236:714–722. doi: 10.1258/ebm.2011.010347. [DOI] [PubMed] [Google Scholar]
- 29.Brandon-Warner E, Walling T, Schrum L, McKilop I. Chronic ethanol feeding accelerates hepatocellular carcinoma progression in a sex-dependent manner in a mouse model of hepatocarcinogenesis. Alcohol Clin Exp Res. 2012;36:641–653. doi: 10.1111/j.1530-0277.2011.01660.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gu L, House SE, Prior RL, Fang N, Ronis MJ, Clarkson TB, et al. Metabolic phenotype of isoflavones differ among female rats, pigs, monkeys, and women. J Nutr. 2006;136:1215–1221. doi: 10.1093/jn/136.5.1215. [DOI] [PubMed] [Google Scholar]
- 31.Cardiff RD, Anver MR, Boivin GP, Bosenberg MW, Maronpot RR, Molinolo AA, et al. Precancer in mice: animal models used to understand, prevent, and treat human precancers. Toxicol Pathol. 2006;34:699–707. doi: 10.1080/01926230600930129. [DOI] [PubMed] [Google Scholar]
- 32.Gyamfi MA, Kocsis MG, He L, Dai G, Mendy AJ, Wan YJ. The role of retinoid X receptor alpha in regulating alcohol metabolism. J Pharmacol Exp Ther. 2006;319:360–368. doi: 10.1124/jpet.106.108175. [DOI] [PubMed] [Google Scholar]
- 33.Jones EE, Dworski S, Canals D, Casas J, Fabrias G, Schoenling D, et al. On-tissue localization of ceramides and other sphingolipids by MALDI mass spectrometry imaging. Anal Chem. 2014;86:8303–8311. doi: 10.1021/ac501937d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Longato L, Ripp K, Setshedi M, Dostalek M, Akhlaghi F, Branda M, et al. Insulin resistance, ceramide accumulation, and endoplasmic reticulum stress in human chronic alcohol-related liver disease. Oxid Med Cell Longev. 2012;2012:479348. doi: 10.1155/2012/479348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sharp GB, Lagarde F, Mizuno T, Sauvaget C, Fukuhara T, Allen N, et al. Relationship of hepatocellular carcinoma to soya food consumption: a cohort-based, case-control study in Japan. Int J Cancer. 2005;115:290–295. doi: 10.1002/ijc.20897. [DOI] [PubMed] [Google Scholar]
- 36.Bojes HK, Germolec DR, Simeonova P, Bruccoleri A, Schoonhoven R, Luster MI, et al. Antibodies to tumor necrosis factor alpha prevent increases in cell replication in liver due to the potent peroxisome proliferator, WY-14,643. Carcinogenesis. 1997;18:669–674. doi: 10.1093/carcin/18.4.669. [DOI] [PubMed] [Google Scholar]
- 37.Diehl AM. Recent events in alcoholic liver disease V. effects of ethanol on liver regeneration. Am J Physiol Gastrointest Liver Physiol. 2005;288:G1–G6. doi: 10.1152/ajpgi.00376.2004. [DOI] [PubMed] [Google Scholar]
- 38.Yang J, Mowry LE, Nejak-Bowen KN, Okabe H, Diegel CR, Lang RA, et al. βeta-catenin signaling in murine liver zonation and regeneration: a Wnt-Wnt situation! Hepatology. 2014;60:964–976. doi: 10.1002/hep.27082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boulter L, Guest RV, Kendall TJ, Wilson DH, Wojtacha D, Robson AJ, et al. WNT signaling drives cholangiocarcinoma growth and can be pharmacologically inhibited. J Clin Invest. 2015;125:1269–1285. doi: 10.1172/JCI76452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Uehara T, Ainslie GR, Kutanzi K, Pogribny IP, Muskhelishvili L, Izawa T, et al. Molecular mechanisms of fibrosis-associated promotion of liver carcinogenesis. Toxicol Sci. 2013;132:53–63. doi: 10.1093/toxsci/kfs342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Taylor CK, Levy RM, Elliott JC, Burnett BP. The effect of genistein aglycone on cancer and cancer risk: a review of in vitro, preclinical, and clinical studies. Nutr Rev. 2009;67:398–415. doi: 10.1111/j.1753-4887.2009.00213.x. [DOI] [PubMed] [Google Scholar]
- 42.Zhang Y, Li Q, Chen H. DNA methylation and histone modifications of Wnt genes by genistein during colon cancer development. Carcinogenesis. 2013;34:1756–1763. doi: 10.1093/carcin/bgt129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mercer KE, Pulliam CF, Hennings L, Lai K, Cleves MA, Jones EE, et al. Diet supplementation with soy protein isolate, but not the isoflavone genistein, protects against alcohol-induced tumor progression in DEN-treated male mice. Adv Exp Med Biol Forthcoming in. 2016 doi: 10.1007/978-3-319-98788-0_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Burris RL, Ng HP, Nagarajan S. Soy protein inhibits inflammation-induced VCAM-1 and inflammatory cytokine induction by inhibiting the NF-kappaB and AKT signaling pathway in apolipoprotein E-deficient mice. Eur J Nutr. 2014;53:135–148. doi: 10.1007/s00394-013-0509-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ronis MJ, Wands JR, Badger TM, de la Monte SM, Lang CH, Calissendorff J. Alcohol-induced disruption of endocrine signaling. Alcohol Clin Exp Res. 2007;31:1269–1285. doi: 10.1111/j.1530-0277.2007.00436.x. [DOI] [PubMed] [Google Scholar]
- 46.Cowart LA. Sphingolipids: players in the pathology of metabolic disease. Trends Endocrinol Metab. 2009;20:34–42. doi: 10.1016/j.tem.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 47.Saddoughi SA, Ogretmen B. Diverse functions of ceramide in cancer cell death and proliferation. Adv Cancer Res. 2013;117:37–58. doi: 10.1016/B978-0-12-394274-6.00002-9. [DOI] [PubMed] [Google Scholar]
- 48.Kunkel GT, Maceyka M, Milstien S, Spiegel S. Targeting the sphingosine-1-phosphate axis in cancer, inflammation and beyond. Nat Rev Drug Discov. 2013;12:688–702. doi: 10.1038/nrd4099. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.