

Abstract
Advanced-stage prostate cancer usually metastasizes to bone and is untreatable due to poor biodistribution of intravenously administered anticancer drugs to bone. In this study, we modulated the surface charge/composition of biodegradable nanoparticles (NPs) to sustain their blood circulation time and made them small enough to extravasate through the openings of the bone’s sinusoidal capillaries and thus localize into marrow. NPs with a neutral surface charge, achieved by modulating the NP surface-associated emulsifier composition, were more effective at localizing to bone marrow than NPs with a cationic or anionic surface charge. These small neutral NPs (~150 nm vs. the more usual ~320 nm) were also ~7-fold more effective in localizing in bone marrow than large NPs. We hypothesized that NPs that effectively localize to marrow could improve NP-mediated anticancer drug delivery to sites of bone metastasis, thereby inhibiting cancer progression and preventing bone loss. In a PC-3M-luc cell-induced osteolytic intraosseous model of prostate cancer, these small neutral NPs demonstrated greater accumulation in bone within metastatic sites than in normal contralateral bone as well as co-localization with the tumor mass in marrow. Significantly, a single-dose intravenous administration of these small neutral NPs loaded with paclitaxel (PTX-NPs), but not anionic PTX-NPs, slowed the progression of bone metastasis. In addition, neutral PTX-NPs prevented bone loss, whereas animals treated with the rapid-release drug formulation Cremophor EL (PTX-CrEL) or saline (control) showed >50% bone loss. Neutral PTX-NPs did not cause acute toxicity, whereas animals treated with PTX-CrEL experienced weight loss. These results indicate that NPs with appropriate physical and sustained drug-release characteristics could be explored to treat bone metastasis, a significant clinical issue in prostate and other cancers.
Keywords: Cancer therapy, nanomedicine, drug delivery, biodistribution, bone marrow
Graphical abstract
1. Introduction
Bone is a common site for metastasis in a number of human cancers, in large part because of the relatively slow blood flow in bone marrow and the presence of adhesion receptors on bone marrow capillary endothelial cells that support cancer cell localization in the bone. These characteristics, together with the fact that bone marrow is an environment rich in growth factors and cytokines, all promote progression of bone metastasis [1, 2]. Among cancers that metastasize to bone, prostate cancer presents with a significantly high incidence: ~70–80% of patients develop bone metastases. The 5-year survival rate of patients with bone metastases is very low compared with those in whom the disease is localized (20% vs. 100%). As with prostate cancer, the majority of patients with advanced-stage breast cancer show evidence of skeletal metastases by the time of their death [3]. The consequences of bone metastasis are often devastating; it affects bone remodeling, causes bone pain, fractures and nerve compression, and in prostatic disease is the major cause of prostate cancer-related morbidity and mortality [4].
Since bone is less highly perfused than soft-tissue organs (7% of cardiac output goes to bone vs. 30% to liver) [5], intravenously administered anticancer chemotherapeutics do not achieve enough of a therapeutic dose at bone metastatic sites to suppress tumor growth. A major fraction of the administered drug is either excreted and/or metabolized, often via hepatic processes; or may accumulate in other, more highly perfused body compartments or tissues prior to reaching the marrow in sufficient doses. To overcome this challenge, such approaches as conjugating anticancer drugs [6] or drug-loaded nanoparticles (NPs) to bone-seeking agents (e.g., bisphosphonates [7], tetracycline [8], or E-selectin [overexpressed in bone marrow endothelium] [9]) have been investigated, but these remain inefficient. For example, Wang et al. [10] found that bisphosphonate-conjugated to NPs made with bovine serum albumin did not target bone in vivo, despite in vitro results showing that they had significantly higher affinity than unconjugated NPs to hydroxyapatite, a major component of bone. In a recent study in a bone metastasis model of myeloma, Swami et al. [11] found no significant differences in the efficacy (bone loss, tumor burden, and survival) of alendronate (bisphosphonate)-conjugated NPs loaded with the proteasome inhibitor bortezomib compared with unconjugated NPs or drug alone.
It is now increasingly recognized that for cancer chemotherapy to succeed, effective treatment of metastatic and advanced-stage tumors is critical. Recently, some experimental and clinical studies have shown better outcomes (and reduced toxicity) from the use of drug-loaded nanocarriers than drugs alone [12, 13], mostly for those cancers that progress not to bone but to soft tissue [14, 15]. Hence, specific approaches to improving drug delivery to bone are needed for effective treatment of bone metastasis.
Bone marrow possesses sinusoidal capillaries with intercellular fenestrations or clefts between endothelial cells; some of these clefts are as wide as 170 nm [16, 17]. Therefore, we surmised that designing NPs of a diameter smaller than the opening of these intercellular clefts of sinusoidal capillaries could extravasate from the circulation to the marrow. However, for bone marrow extravasation to be effective, intravenously injected NPs first need to remain in the circulation long enough to pass through the bone’s sinusoidal capillaries into marrow. Therapeutic efficacy also requires sustained retention of the extravasated NPs in the bone marrow at the metastatic tumor site.
Some studies have suggested that NPs with a neutral surface charge have reduced interactions with proteins that promote opsonization [18], are less toxic compared with cationic NPs, and are less prone to aggregation in a biological environment [19, 20]. However, no studies have yet investigated how NP surface charge might influence the localization of NPs to bone marrow and thus the efficacy of these NPs to deliver anticancer drugs to treat bone metastasis. Therefore, in this study, we formulated biodegradable NPs of similar size but varying surface charges (anionic, neutral and cationic) by modulating the emulsifier composition at the NP interface. In addition, we controlled NP size to facilitate preferential extravasation of the intravenously administered NPs through the openings of the bone’s sinusoidal capillaries into marrow. We hypothesized that NPs that effectively localize to marrow could improve NP-mediated anticancer drug delivery to sites of bone metastasis to inhibit cancer progression and prevent bone loss. Our data demonstrated that (a) following intravenous (IV) administration, NPs with a neutral surface charge were more effective in localization to and retention in marrow than anionic or cationic NPs and (b) a single dose of PTX-loaded neutral NPs (PTX-NPs) inhibited progression of bone metastasis and prevented bone loss.
2. Materials and Methods
Materials included the following: Poly (D,L-lactide-co-glycolide) (PLGA; 50:50, inherent viscosity of 0.26–0.54 dL/g) was purchased from LACTEL Absorbable Polymers (Birmingham, AL). Poly (vinyl alcohol) (PVA; 87–90% hydrolyzed, mol wt 30,000–70,000), sucrose, Cremophor EL (CrEL) and cetyltrimethylammonium bromide (CTAB) were purchased from Sigma-Aldrich (St. Louis, MO). Near-infrared (NIR) dye (SDB5700) was obtained from H.W. Sands Corp. (Jupiter, FL). Chloroform was obtained from Fisher Scientific (Pittsburgh, PA). Paclitaxel (PTX) was purchased from LC Laboratories (Woburn, MA).
2.1 Formulation and characterization of NPs of different surface charges
NPs were formulated by a single oil-in-water emulsion solvent-evaporation method, but conditions were optimized to obtain NPs of similar size but different surface charges. Briefly, NPs with either an anionic, neutral or cationic surface charge were formulated by modulating the amount of PVA associated with NPs or using the cationic surfactant CTAB in combination with PVA, as per our previously described procedures [21, 22] (see Supplemental Material). It is important to note that the surface-associated emulsifier is that which remains associated with NPs at the interface despite repeated washing. The emulsifier remains because of the anchoring and integration of the hydrophobic segment of the emulsifier (polyvinyl acetate in the case of PVA or the acyl chain in the case of CTAB) with the polymer matrix at the interface. In several of our previous studies, we have determined the role of the residual PVA on physical and biological properties (cellular uptake and intracellular trafficking) of PLGA-NPs [23–26]. To study the effect of size, PLGA NPs of larger size than those used above were also formulated and tested for their localization in bone (see Supplemental Material).
To monitor their biodistribution in vivo, NPs were loaded with NIR dye SDB5700. Our laboratory has previously evaluated this dye for in vivo imaging and biodistribution of NPs in breast [27] and prostate [22] xenograft models. The dye offers several advantages, including a stable signal (even after repeated laser exposure), a high-fluorescence yield, no background signal, and only an insignificant amount of dye leaching from NPs because of the hydrophobic nature of the dye and its low loading (0.1% w/w polymer weight) [22]. PTX-loaded NPs were formulated to determine therapeutic efficacy. The hydrodynamic diameter and ζ-potential of NPs were determined in water by dynamic light scattering with a NICOMP 380 ZLS (Particle Sizing Systems, Santa Barbara, CA). In addition, NPs were characterized for surface-associated PVA, surface morphology, and size using atomic force microscopy (AFM) [22]. PTX loading in NPs was determined by extracting the drug from NPs using methanol. PTX release from NPs was carried out in double diffusion chambers under sink conditions as described previously [28]. PTX levels in samples were analyzed by high-performance liquid chromatography (see Supplemental Material for detailed methods used for formulation and characterization of NPs).
2.2 Animal Studies
Cleveland Clinic’s Institutional Animal Care and Use Committee approved all animal procedures, and these were carried out according to federal and internal guidelines. Studies were performed with 5- to 6-week-old male athymic nude mice (Charles River Laboratories, Wilmington, MA).
2.3 Biodistribution of NPs
A single dose of a 100-μL suspension of NIR dye-loaded NPs (30 mg/mL) in saline was injected via tail vein into each mouse, and animals were imaged at different time points post injection with a Maestro EX Imaging system (PerkinElmer, Waltham, MA) using blue and NIR filters set at exposure times of 500 ms and 1200 ms, respectively. To visualize localization and relative quantification of signal intensity (in counts) due to NPs in bone, the skin, muscles, and fat were removed to expose skeletons prior to imaging as above. To quantify relative signal intensity due to NPs localized in tibia, the region of interest (ROI) covering the entire tibia was used. Although we analyzed other bones for NP localization, we focused here on the tibia because this is the most common site used for tumor induction, as described below.
2.4 Induction of bone metastasis and imaging
PC-3M-luc cells (obtained from the NIH) were cultured in RPMI 1640 supplemented with 10% FBS at 37 °C and 5% CO2. Bone metastasis was induced as described by Park et al. [29]. Briefly, mice under ketamine/xylazine anesthesia were injected with 4 × 105 PC-3M-luc cells in 20 μL PBS intraosseously in the lumen of the right tibia, then monitored for induction of bone metastasis and its progression using changes in bioluminescence signal intensity (photons per second per square centimeter steradian; [p/sec/cm2/sr]) and micro-computed tomography (micro-CT) to determine bone loss. For bioluminescence, animals were imaged 15 min following intraperitoneal injection of luciferin (200 mg/kg; VivoGlo™ Luciferin, Promega, Madison, WI) using the IVIS® Lumina II (PerkinElmer). In addition, the IVIS® Lumina II was used to co-register the bioluminescence signal of cancer cells and the fluorescence signal of the dye-loaded NPs in metastasized bone. To determine bone loss, limbs resected at the head of the femur were imaged using an in vivo micro-CT for preclinical procedures (eXplore Locus RS Micro-CT, GE Healthcare, London, ON, Canada). The micro-CT images were acquired using an X-ray tube (80 kV, 490 μA) for an exposure time of 1.8 seconds. The detector bin was set to 1 × 1 spatial resolution, providing a full resolution of 20 μm. Images were acquired for every degree of rotation, creating 360 raw data projections. These projections were corrected, unwarped and reconstructed using GE’s proprietary reconstruction algorithms to create a full three-dimensional reconstruction of the scanned specimen.
2.5 Flow cytometry analysis of bone marrow cells
To determine cellular uptake of NPs in bone marrow, animals without tumor were injected with NIR dye-loaded NPs (3 mg in 100 μL saline) and euthanized 24 hours following injection; their femurs were resected and flushed with saline to recover the bone marrow. The collected marrow was incubated in red blood cell lysis buffer (Sigma-Aldrich) for 10 min, centrifuged to recover nucleated cells, and washed twice with saline. Flow cytometry was performed on nucleated cells in the red channel to determine the percentage of cells with dye-loaded NPs (BD FACSAria II, BD Biosciences, San Jose, CA).
2.6 Treatment to determine efficacy of PTX-loaded NPs
Micro-CT and bioluminescence signals confirmed the induction of tumor in marrow at 7 days post inoculation. At this point, mice with confirmed intraosseous metastases were divided into three groups, receiving: (a) a single-dose IV injection of PTX in Cremophor/ethanol (PTX-CrEL), (b) PTX-NPs (dose of PTX =7 mg/kg or 110 mg/kg PTX-NPs), or (c) saline as a control. The PTX dose was calculated from the standard dose of 175 mg/m2 used in prostate cancer patients. Tumor growth was monitored by weekly bioluminescence imaging of cancer cells using IVIS®. At 5 weeks post treatment, animals were euthanized, and both hind legs were harvested. The difference in the weight of the contralateral leg and the tumor-bearing leg was used to calculate tumor burden [30, 31].
2.7 Statistical analysis
All numerical data were expressed as mean ± standard error of the mean. Statistical significance for both NP uptake by bone marrow cells and their localization in metastatic sites was determined by Student’s t test. One-way analysis of variance was performed to analyze the biodistribution and localization of different NP formulations in tibia and also for therapeutic efficacy studies. Statistical significance was set at P < 0.05
3. Results
3.1 NP formulation and characterization
All small NP formulations showed a similar hydrodynamic diameter and size distribution (polydispersity index) but varied in their surface charge (ζ-potential) (Table 1, Fig. 1). AFM images of the NPs showed their spherical shape, irrespective of surface charge (Fig. 1A). Mean NP diameter as determined by AFM was smaller than the hydrodynamic diameter measured in water using the dynamic light scattering technique (Table 1). The larger NPs had a mean hydrodynamic diameter of 321 nm (range, 248–460 nm; polydispersity index, 0.1) and ζ-potential of −15 mV. As determined, surface-associated PVA was greater for neutral NPs (146 μg/mg NPs) than for cationic (125 μg/mg NP) or anionic (106 μg/mg NP) NPs (Fig. 1B). There was no significant difference in size and ζ-potential of the dye- or drug-loaded NPs compared with the respective NPs formulated without dye or drug (Table 1).
Table 1.
Characteristics of different formulations of nanoparticles
| NP Formulations | Diameter by DLS (nm)/PI* (Range) | Diameter by AFM (nm)** (Range) | ζ-Potential (mV)* |
|---|---|---|---|
| Anionic NPs (without dye) | 164 ± 5/0.055 (139–189 nm) | 112 ± 5 (65–175 nm) | −18 ± 2 |
| Dye-loaded anionic NPs | 168 ± 3/0.035 (159–177 nm) | ND | −17 ± 2 |
| Neutral NPs (without dye) | 152 ± 4/0.020 (136–168 nm) | 112 ± 4 (73–178 nm) | −2 ± 3 |
| Dye-loaded neutral NPs | 148 ± 2/0.022 (144–152 nm) | ND | −3 ± 3 |
| Cationic NPs (without dye) | 162 ± 23/0.088 (139–185 nm) | 91 ± 4 (44–136 nm) | 13 ± 1 |
| Dye-loaded cationic NPs | 169 ± 8/0.063 (161–177 nm) | ND | 12 ± 1 |
| PTX-loaded NPs | 151 ± 6/0.028 (145–157 nm) | ND | −2 ± 3 |
Data are shown as mean ± standard deviation; n = 3.
Diameter by AFM was measured using section profile analysis of 40 individual NPs.
ND, Not determined; nm, nanometer.
Fig. 1. Physical characterization of different formulations of NPs.

A) Characterization of anionic, neutral, and cationic NPs for particle size and size distribution by dynamic light scattering (DLS). NP size and shape characterization was determined by atomic force microscopy (AFM). B) Amount of surface-associated PVA with anionic, neutral, and cationic NPs. Data are shown as mean ± s.e.m. n = 3. C) Release of PTX in vitro from drug-loaded neutral NPs under sink conditions. Data are shown as mean ± s.e.m., n = 3.
PTX loading in NPs was 6.4 ± 0.3% w/w and demonstrated 28% cumulative drug release over 7 weeks (Fig. 1C). Also, since only an insignificant amount of the incorporated dye leaches out from NPs (5% over 4 days under sink conditions), due to the hydrophobic nature of the dye, its high solid-state solubility (no phase separation) in PLGA polymer [32], and very low dye loading in NPs (~0.1% w/w), we considered that the signal seen is primarily from the NPs themselves and not from the released dye. All three small NP formulations with different surface charges had similar signal intensity per unit weight of NPs (~14 counts per microgram of NPs), which allowed us to compare their relative biodistribution (including in marrow) based on optical signal intensity measured using Maestro. Signal intensity of large NPs was 13 counts per microgram of NPs.
3.2 Biodistribution of NPs with different surface charges
The initial biodistribution study was carried out in normal mice (without tumor) to determine which formulation of NPs would show better localization and retention in bone marrow. The imaging data showed differences in the biodistribution of NPs, particularly over time, as a function of their charge. Immediately following injection (2 min), all formulations of NPs primarily showed localization in the liver; however, this biodistribution changed as time went on (Fig. 2A).
Fig. 2. Biodistribution of NPs with different surface charges.

A) Whole-body images taken over time indicated prolonged retention of neutral NPs in the body. B) Quantification of the ROIs of the skin over time as measured using Maestro, demonstrating more prolonged body retention of neutral NPs than of anionic or cationic NPs. Data are shown as mean ± s.e.m., *P < 0.05; n = 3.
Quantification of regions of interest (ROIs) for “skin” at the lower left abdominal area, which we considered as the signal attributable to the NPs in the circulation, demonstrated that neutral NPs remain in the circulation longer than anionic and cationic NPs do (Fig. 2B). Our team and others have previously used this method of tracing skin signals to determine the relative circulation time of NPs [21, 33]. While direct measurement of NPs in blood is required for absolute quantification of NPs, the method of skin tracing enables longitudinal monitoring of NPs in the circulation over time and correlates with direct blood measurements.
Quantification of the ROIs of the tibia at 24 hours post NP administration showed 52% and 41% greater accumulation of neutral NPs than cationic or anionic NPs, respectively. At 96 hrs post injection, the accumulation of neutral NPs in tibia was 2.5-fold higher than anionic or cationic NPs (Fig. 3A). In a separate set of experiments, animals were euthanized and dissected at 24 hrs post injection of neutral NPs to visualize their biodistribution in other bones. Besides localization in the tibia and sternum, neutral NPs were seen in all the bones. Based on signal intensity, a greater accumulation of neutral NPs was seen in the pelvis and vertebral column than in the ribs and craniofacial bones (Fig. 3B). In the vertebral column, neutral NPs were seen more in thoracic than in lumbar vertebrae (Fig. 3C). A close-up image of the tibia showed localization of neutral NPs in the marrow rather than in the bone tissue (Fig. 3D), which was further confirmed from flow cytometry analysis of the marrow, which showed 90% of the nucleated cells with NPs (Fig. 3E, F). As seen in the tibia, an excised section of the femur also showed localization of NPs in the marrow (image not shown). The whole-body skeleton imaging showed an approximately 7-fold greater accumulation of small neutral NPs in bones than large NPs (Supplemental Material, Fig. 1S).
Fig. 3. Localization with NPs of different surface charge in bones.

A) Quantification of fluorescence signals due to NP localization in tibia over time as measured using Maestro, demonstrating greater uptake and sustained retention of neutral NPs than of anionic or cationic NPs. B) Ventral view of skeleton of mouse injected with neutral NPs, showing NP localization in all bones, particularly pelvis, long bones, sternum, and vertebrae. C) Close-up view of the vertebral column shows greater NP localization in cervical than lumbar vertebrae. D) Image of the surgically resected tibia, showing localization of neutral NPs in marrow (arrow) but not in calcified bone. E) Flow cytometry analysis showing uptake of NPs by bone marrow cells. F) Quantification of the flow cytometry data shows that >90% of bone marrow cells internalize neutral NPs. Data are shown as mean ± s.e.m., *P < 0.05, ns = not significant; data shown in B to D at 24 hours post NP administration.
3.3 Nanoparticle localization in metastasized bone
Bioluminescence and micro-CT images confirmed induction of intraosseous tumor in the lumen of the tibia within 1 week post inoculation of bone marrow with PC-3M-luc cells (Fig. 4A, B). The imaging data demonstrated that neutral NPs following IV administration show two-fold greater accumulation in the tibia with metastasis than in the normal contralateral tibia (Fig. 4C, D). Ex vivo imaging of the harvested bone from these animals further confirmed greater localization of neutral NPs in the metastasized tibia than in the normal contralateral tibia (Fig. 4E). In addition, co-localization of bioluminescence signal of cancer cells with fluorescence signal of NPs (Fig. 4F) indicated the delivery of neutral NPs to the tumor mass in the bone marrow.
Fig. 4. Localization of neutral NPs at site of tumor metastasis in bone.

A) Bioluminescence signal due to PC-3M-luc prostate cancer cells at 7 days post inoculation as measured using IVIS® and B) micro-CT images of the bone at 7 days post cancer cell inoculation. C) Fluorescence imaging and D) quantification of signal intensity measured using Maestro, demonstrating greater localization of NPs in tibia with tumor than in normal contralateral tibia. E) Ex vivo image (by Maestro) of the bone with intraosseous tumor at 24 hrs following NP administration. Arrows indicate bone with metastatic tumors. F) Bioluminescence signals due to cancer cells and fluorescence of NPs, demonstrating localization of NPs into metastasized tumor mass. For the above colocalization study, both bioluminescence signals due to cancer cells and fluorescence signals due to NPs were captured using IVIS®. Bright spots seen next to the tumor in tibia (Fig. 4F, fluorescence signal) are due to localization of the injected NPs in lymph nodes and other tissues. Data are shown as mean ± s.e.m., *P < 0.05.
3.4 Inhibition of bone metastasis with paclitaxel-loaded NPs
Bioluminescence imaging demonstrated slower progression of bone metastasis in the animals treated with PTX-NPs than with PTX-CrEL or saline control (Fig. 5A). Interestingly, the animals treated with PTX-CrEL showed relatively greater tumor progression than those receiving saline. Based on the bioluminescence signal intensity at 2 weeks post treatment, animals treated with PTX-NPs demonstrated 89% and 96% lower tumor burden than saline controls or those treated with PTX-CrEL, respectively (P < 0.05). At 3 weeks post treatment, animals that had received PTX-NPs still demonstrated a lower tumor burden: 61% and 143% lower than untreated controls or those treated with PTX-CrEL, respectively (Fig. 5A, B). Tumor burden, calculated from the weight difference between the limb with tumor and the contralateral normal limb at the end of the study (5 weeks post treatment), shows significantly lower tumor burden in animals treated with PTX-NP than in untreated saline controls or the animals treated with PTX-CrEL (P < 0.05) (Fig. 5C). Since anionic and cationic NPs have similar bone marrow uptake and retention (Fig. 3), we tested PTX-loaded anionic NPs for inhibition of bone metastasis. The data show insignificant difference in the bioluminescence signal in treated and saline control at the end of 3 weeks (Supplemental Material, Fig. 2S).
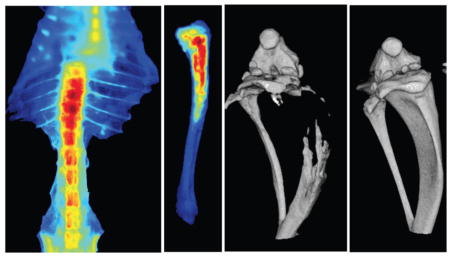
Fig. 5. Efficacy of paclitaxel-loaded neutral NPs in a bone metastasis model.

A) Change in bioluminescence signals due to cancer cells in bone as measured using IVIS® during 3 weeks post treatment. *P < 0.05 at 2 weeks between PTX-NPs and other groups; *P < 0.05 between PTX-NPs and PTX-CrEL at 3 weeks; **no statistical significance between PTX-NPs and saline at 3 weeks days. B) Representative bioluminescence images of the bone with tumor captured using IVIS® at 3 weeks post treatment. C) Tumor burden at 5 weeks post treatment, determined by subtracting the weight of the normal contralateral leg from that of the leg with tumor. D) Representative micro-CT of tibias of mice from different groups at 5 weeks post treatment. Animals treated with PTX-NPs showed no bone loss. E) Changes in body weight of animals post treatment. Data are shown as mean ± s.e.m., *P < 0.05 PTX-NPs and control vs. PTX-CrEL; Not significant, PTX-NPs vs. control, n = 5–6.
Analysis of the harvested tibia at 5 weeks post treatment using micro-CT showed no bone loss in the PTX-NP-treated animals, whereas the PTX-CrEL-treated animals showed >50% bone resorption, similar to the bone loss seen in the saline control animal (Fig. 5D). Animals treated with PTX-CrEL showed weight loss; in contrast, those treated with PTX-NPs gained weight, indicating there was no acute drug toxicity with PTX delivered encased in NPs (Fig. 5E). Animals in all groups showed weight loss at 5 weeks compared to their weights at 3 weeks, but those treated with PTX-CrEL showed greater weight loss than saline control or animals treated with PTX-NPs.
4. Discussion
Progress in the field of cancer nanomedicine is evident from the entry of several NP-based therapies and an increasing number of ongoing clinical studies for treating different types of cancers [34, 35]. In general, the efficacy of NPs has been attributed to either better drug delivery directly to cancerous tissue and/or reduced toxicity compared with drugs alone. However, most of these approaches have been focused on treating primary soft-tissue, highly vascularized, solid tumors. Effective treatment for tumors that arise from advanced-stage cancer metastasis, the major cause of cancer-related mortality, still remains a challenge. In this study, we explored the sinusoidal nature of blood-bone marrow capillaries to deliver NPs to the marrow and found that neutral PTX-NPs are demonstrably effective in slowing the progression of bone metastasis and inhibiting bone loss in an intraosseous model of bone metastasis.
Since neutral NPs have a prolonged circulation time compared with anionic and cationic NPs and since they are also smaller than the opening of the intercellular clefts of bone marrow endothelial cells, neutral NPs are more effective in localizing in the marrow (Fig. 3). Neutral NPs have been reported to have a lower propensity to interact with proteins, which could be the reason for their relatively longer time in the circulation than charged NPs [36]. Furthermore, surface-associated PVA, which is present in greater amounts on neutral NPs than on anionic or cationic NPs, could have played a role in sustaining the retention time of the NPs in the bloodstream. PVA is a copolymer comprising both hydrophobic and hydrophilic portions. A hydrophobic polyvinyl acetate segment integrates within the PLGA-NP matrix, whereas a hydrophilic segment forms an outer corona that could also reduce opsonization [36]. We have previously shown that a significant fraction of PVA remains associated with the NPs over several days when incubated in PBS at 37 °C [37]. This finding means that the NPs’ surface composition would be maintained for a period of time following their IV injection, which is critical, since the surface characteristics of the NPs determine their biodistribution. It would be of further interest to determine the composition of the protein corona formed following systemic administration of NPs with different surfaces to find out what role that may have in influencing circulation time, biodistribution, and localization to bone marrow.
The general strategy used to keep NPs in the circulation is to prevent their recognition by circulating monocytes and their subsequent clearance by the organs of the reticuloendothelial system (RES), particularly the liver and spleen. In our study we found that at 2 minutes post NP administration, the liver signal due to NPs, particularly for neutral NPs, drops over time (Fig. 2A); this drop suggests that the major fraction of these NPs become contained in the vascular compartment of the liver and are not taken up by Kupffer cells or hepatocytes. The liver receives 30% of cardiac output; therefore, the initial high fluorescence signal seen in the liver could be due to the NPs carried with the blood flow to the liver [38]. Generally, NPs taken up by Kupffer cells degrade slowly, whereas those taken up by hepatocytes are excreted through the hepatic biliary duct into the gut; this process is known to occur more rapidly for cationic than anionic NPs [39] but has not yet been investigated for neutral NPs. The relatively more rapid drop in the liver signal seen in our studies in animals injected with neutral NPs than cationic or anionic NPs suggests that neutral NPs are better at escaping uptake by Kupffer cells or hepatocytes than anionic or cationic NPs (Fig. 2A, B).
For delivery to the bone marrow, it is critical that NPs escape sequestering by the liver so that they remain in the circulation long enough to pass through the sinusoidal capillaries in the marrow. Our neutral NPs seem to have avoided sequestering by the liver through their reduced opsonization, thus preventing their uptake by circulating monocytes and subsequent clearance by the organs of the RES, such as liver and spleen. Furthermore, the size of neutral NPs (~150 nm hydrodynamic diameter, 112 nm in the dry state, measured using AFM) is greater than the opening of the fenestrations (~75 nm) in the liver sinusoidal endothelial cell lining [40] but smaller than those in the bone marrow endothelial cell capillaries (170 nm) [16, 17, 41]. In this regard, neutral NPs appear to achieve the above balance in two ways: first, by escaping the liver and remaining in the circulation, but then (following their passage through the bone’s sinusoidal capillaries) becoming sequestered in the marrow because of the marrow’s comparatively sluggish blood flow [42] (Fig. 2B and 3A). Despite a quite uniform size distribution (Table 1), a fraction of the smaller NPs present in the formulation could have passed through the sinusoidal capillaries of the liver. Thus, hepatic uptake of these NPs could not be completely prevented but might be further minimized by controlling particle size within a certain narrow range. Conventionally, NPs are modified with hydrophilic polymers such as polyethylene glycol (PEG; PEGylated NPs) or pluronics to extend their time in the circulation; this is the approach commonly explored, via the enhanced permeation and retention (EPR) effect, for delivery of anticancer therapeutics to vascularized primary tumors [43]. However, PEGylated/pluronic-modified NPs cannot effectively extravasate into the bone marrow [44]. Several studies have reported that uptake of these NPs into bone marrow remains negligible [45]. It is possible that steric hindrance due to the surface-associated PEG/pluronic could have prevented extravasation of these NPs into bone marrow. Considerable data in the recent literature suggest that PEGylation increases the circulation time of NPs yet has the concomitant negative effect of reducing cellular and tissue uptake of NPs; this steric hindrance is thought to be caused by protruding hydrated PEG chains at the NP surface [36, 46]. Similarly, we speculate that rather than extravasating efficiently into bone marrow due to their reduced interactions with the marrow’s cells and tissues, PEGylated NPs remain in the blood circulation. Although it was beyond the scope of these experiments to consider PEGylated NPs, it would be of interest to determine how well they might localize into bone marrow compared with the neutral NPs used in our study. Certainly, a thorough pharmacokinetic and pharmacodynamic study would help us gain insight into how and why neutral NPs (rather than charged NPs) localize to bone marrow.
In addition to charge, the size of NPs is known to influence their localization in bone marrow. Our data show that neutral NPs have about a 7-fold greater localization in bone marrow than larger NPs (hydrodynamic diameter ~150 nm vs. ~320 nm), signifying that the openings in the intercellular clefts between endothelial cells lining the bone marrow, which are ~170 nm, regulate the delivery of NPs to the bone marrow [16, 17].
Currently, patients with bone metastasis are treated with bisphosphonates to reduce the risk of deleterious skeleton-related events and to ameliorate bone pain, as bisphosphonates can inhibit bone resorption [47]. Some studies have also reported that bisphosphonates indirectly slow the progression of bone metastasis [48] by inhibiting osteoclast-mediated bone resorption and thereby the release of growth factors necessary to promote cancer cell growth and differentiation and subsequent tumor formation in bone [49]. However, a recent review of data from different clinical studies shows no statistically significant improvement in survival of bisphosphonate-treated patients compared with placebo controls [50]. Furthermore, bisphosphonates show dose-limiting toxicities; with chronic use, they cause osteonecrosis of the jaws, considered to be a consequence of their effect on circulating endothelial progenitor cells, interfering in the normal process of angiogenesis and vasculogenesis required to maintain healthy tissue [51].
We have shown that our neutral NPs localize to and are retained in marrow, where bone metastasis initiates and progresses, rather than to bone itself. Furthermore, the increased accumulation of NPs in bone that has been invaded by cancer compared with normal bone could be due to increased permeability of the blood-bone microvasculature as a result of tumor growth. Since NPs also localize into the metastatic tumor mass, the therapy is effective in suppressing the progression of bone metastasis (Fig. 5).
The most important finding of our study was that a single-dose IV injection of neutral PTX-NPs prevented bone loss (Fig. 5D), suggesting the NPs’ efficacy in delivering drug to marrow to prevent invasion of PC-3 cells in bone matrix. PTX is known to inhibit receptor activator of nuclear factor-kappa B ligand (RANKL)-induced osteoclastogenesis by causing mitotic arrest of osteoclastic precursor cells, thus inhibiting the progression of bone metastasis to pathological osteolysis [52]. Bone loss is a significant clinical issue when prostate cancer metastasizes to bone. However, PTX-NP treatment did not completely cure metastasis, as is evident from the residual bioluminescence signal (Fig. 5A, B). Further optimization of the PTX-NP dosing regimens will be required to effectively suppress bone metastasis.
It is also possible that a sufficient dose of PTX was not released from those NPs that did reach the metastatic tumor site, considering that only 28% of the encapsulated drug was released from NPs during 7 weeks under in vitro conditions (Fig. 1C). PLGA-based NPs typically demonstrate a triphasic release profile: the first release phase is mediated via diffusion of the drug at the interface, followed by a second, steady-release phase during which an insignificant amount of the encapsulated drug is released, and the third phase, in which the polymer matrix degrades, releasing the remaining encapsulated drug. Thus it appears that a considerable fraction of PTX is still entrapped within the NPs. Previously, we have reported a complete release of PTX from a similar formulation of PLGA-based NPs occurring over ~90 days in vitro [53]. Therefore, modulating the drug-release rate from NPs to synchronize it with the retention of NPs in the bone metastatic site, which is ~96 hrs (Fig. 3A), could further improve the outcome. It is not clear why NPs are retained for less than 1 week; it could be due to the dynamic nature of bone marrow with rapid turnover time of marrow cells.
Interestingly, the animals treated with PTX-CrEL demonstrated greater tumor growth than saline controls (Fig. 5A). Most in vivo efficacy studies use repeated dosing of anticancer drugs over a short period of time, which provides a sustained exposure of cancer cells to chemotherapy. In this study, animals received a single dose of PTX-CrEL, and it is possible that only a subtherapeutic level of PTX reached the metastatic sites in bone, stimulating the proliferation of cancer cells, as some teams have reported [54, 55]. Unlike PTX-CrEL (PTX release of 70% in 4 hours and 100% in 12 hours) [56], PTX-NPs provided a continuous localized dose of PTX (~0.7% per day), thus providing the comparison between fast- and sustained-release formulations of PTX on their efficacy. NP albumin-bound (nab) PTX, despite having smaller particle size (~130 nm) than the openings of the bone marrow capillary fenestrations (~170 nm), has in fact been shown to increase the incidence of metastasis, including to bones, in an animal model of breast cancer metastasis [57]. Similar to PTX-CrEL, nab-PTX is a fast-release drug formulation and has a clearance profile similar to that of PTX-CrEL [58]. Our data with PTX-CrEL and the findings with nab-PTX thus signify the importance of sustained drug delivery to inhibit the progression of bone metastasis.
Among the drawbacks of chemotherapy are the severe side effects seen in normal tissues, at times presenting as myelosuppression and weight loss [59, 60]. In our study, we did not see acute toxicity with PTX-NPs, which could be the combined effect of sustained release, of only a fraction of the encapsulated PTX being released from NPs during the experimental time period, and/or of altered biodistribution of PTX with NPs, which other investigators have also reported [61, 62]. A detailed characterization of the pharmacokinetics and pharmacodynamics of drug delivery with NPs and rapid-releasing PTX-CrEL would better explain the efficacy and toxicity results. Further studies may be required to confirm that PTX-NP delivery to bone does not cause myelosuppression. We also need to investigate why weight loss was seen in all groups of mice at 5 weeks post treatment (Fig. 5E). A long-term study would determine if this weight loss is transient or chronic. We did not continue the study beyond 5 weeks because of significant bone loss and tumor burden in the tibias of control animals that affected their normal activity. However, future studies could be aimed at determining the effect of treatment on survival of animals.
We have also shown that, in addition to tibia, neutral NPs localize in the pelvis and vertebrae, which are common sites for metastasis in prostate and breast cancers. At later stages of prostate cancer, there is gross metastasis to the bones involving the ribs, sternum, and long bones [4, 63, 64]. Since neutral NPs localize to these sites (Fig. 3B and C), they could be explored for drug delivery to control late-stage bone metastasis. Although prostate cancer bone metastasis usually is osteoblastic and that of breast cancer is osteolytic, the PC-3M-luc cells used in our study were osteolytic [65]. More importantly, a future study using our model would address the broader issue of effective drug delivery to bone marrow; this aspect could be explored for treating other cancers that metastasize to bone. Prolonged circulation time is considered critical to give the NPs sufficient time to localize into soft-tissue tumors, as NPs often extravasate through the leaky tumor vasculature due to the EPR effect [66]. Hence, neutral NPs could very well be effective in treating both primary tumors and those that have metastasized to bone.
5. Conclusions
We have demonstrated that neutral NPs localize in bone marrow more than anionic or cationic NPs and that PTX-loaded NPs with a neutral surface charge are effective in slowing the progression of bone tumor metastasis, reducing tumor burden, and inhibiting bone loss. Further studies looking at optimal doses and dose regimens for drug-loaded NP delivery could be explored to manage bone metastasis while reducing the side effects associated with conventional IV delivery of drugs. Skeletal complications from bone metastases are associated with many consequences, including a diminished quality of life, increased medical costs, impaired mobility, and a negative impact on survival. Hence, an effective drug-delivery strategy to bone marrow could have significantly broader therapeutic implications in treating bone metastasis, which otherwise is very difficult to treat.
Supplementary Material
Acknowledgments
This study was funded by grants 1R01CA206189 and 1R01EB003975 from the National Institutes of Health to VL. IMA was funded by the “Med into Grad” initiative of the Howard Hughes Medical Institute and predoctoral fellowship 5F31CA150566 from the National Cancer Institute. Flow cytometry was conducted at the Flow Cytometry Core of Cleveland Clinic’s Lerner Research Institute, with assistance from Cathy Shemo and Anne Cotleur. Micro-CT scanning was performed by ImageIQ (Cleveland, OH), with assistance from Rick Rozic and Amit Vasanji, PhD.
Footnotes
Competing Interest: Authors declare that they have no competing interests.
Author contributions
Conception and design: I.M. Adjei and V. Labhasetwar
Development of methodology: I.M. Adjei, B. Sharma, C. Peetla and V. Labhasetwar
Acquisition of data: I.M. Adjei, B. Sharma and C. Peetla
Analysis and interpretation of data: I.M. Adjei, B. Sharma, C. Peetla and V. Labhasetwar
Writing, review and/or revision of the manuscript: I.M. Adjei, B. Sharma, C. Peetla and V. Labhasetwar
Study supervision: V. Labhasetwar
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Coleman RE. Metastatic bone disease: clinical features, pathophysiology and treatment strategies. Cancer Treat Rev. 2001;27:165–176. doi: 10.1053/ctrv.2000.0210. [DOI] [PubMed] [Google Scholar]
- 2.Morrissey C, Vessella RL. The role of tumor microenvironment in prostate cancer bone metastasis. J Cell Biochem. 2007;101:873–886. doi: 10.1002/jcb.21214. Review. [DOI] [PubMed] [Google Scholar]
- 3.Kuru B, Camlibel M, Dinc S, Gulcelik MA, Gonullu D, Alagol H. Prognostic factors for survival in breast cancer patients who developed distant metastasis subsequent to definitive surgery. Singapore medical journal. 2008;49:904–911. [PubMed] [Google Scholar]
- 4.Berruti A, Dogliotti L, Bitossi R, Fasolis G, Gorzegno G, Bellina M, Torta M, Porpiglia F, Fontana D, Angeli A. Incidence of skeletal complications in patients with bone metastatic prostate cancer and hormone refractory disease: Predictive role of bone resorption and formation markers evaluated at baseline. J Urol. 2000;164:1248–1253. doi: 10.1016/S0022-5347(05)67149-2. [DOI] [PubMed] [Google Scholar]
- 5.Ramanlal Chaudhari K, Kumar A, Megraj Khandelwal VK, Ukawala M, Manjappa AS, Mishra AK, Monkkonen J, Ramachandra Murthy RS. Bone metastasis targeting: a novel approach to reach bone using Zoledronate anchored PLGA nanoparticle as carrier system loaded with Docetaxel. J Control Release. 2012;158:470–478. doi: 10.1016/j.jconrel.2011.11.020. [DOI] [PubMed] [Google Scholar]
- 6.El-Mabhouh AA, Nation PN, Abele JT, Riauka T, Postema E, McEwan AJB, Mercer JR. A conjugate of gemcitabine with bisphosphonate (Gem/BP) shows potential as a targeted bone-specific therapeutic agent in an animal model of human breast cancer bone metastases. Oncol Res. 2011;19:287–295. doi: 10.3727/096504011x13021877989874. [DOI] [PubMed] [Google Scholar]
- 7.Thamake SI, Raut SL, Gryczynski Z, Ranjan AP, Vishwanatha JK. Alendronate coated poly-lactic-co-glycolic acid (PLGA) nanoparticles for active targeting of metastatic breast cancer. Biomaterials. 2012;33:7164–7173. doi: 10.1016/j.biomaterials.2012.06.026. [DOI] [PubMed] [Google Scholar]
- 8.Hirabayashi H, Fujisaki J. Bone-specific drug delivery systems: approaches via chemical modification of bone-seeking agents. Clin Pharmacokinet. 2003;42:1319–1330. doi: 10.2165/00003088-200342150-00002. [DOI] [PubMed] [Google Scholar]
- 9.Mann AP, Tanaka T, Somasunderam A, Liu X, Gorenstein DG, Ferrari M. E-selectin-targeted porous silicon particle for nanoparticle delivery to the bone marrow. Adv Mater. 2011;23:H278–282. doi: 10.1002/adma.201101541. [DOI] [PubMed] [Google Scholar]
- 10.Wang G, Kucharski C, Lin X, Uludağ H. Bisphosphonate-coated BSA nanoparticles lack bone targeting after systemic administration. J Drug Target. 2010;18:611–626. doi: 10.3109/10611861003622560. [DOI] [PubMed] [Google Scholar]
- 11.Swami A, Reagan MR, Basto P, Mishima Y, Kamaly N, Glavey S, Zhang S, Moschetta M, Seevaratnam D, Zhang Y, Liu J, Memarzadeh M, Wu J, Manier S, Shi J, Bertrand N, Lu ZN, Nagano K, Baron R, Sacco A, Roccaro AM, Farokhzad OC, Ghobrial IM. Engineered nanomedicine for myeloma and bone microenvironment targeting. Proc Natl Acad Sci U S A. 2014;111:10287–10292. doi: 10.1073/pnas.1401337111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brannon-Peppas L, Blanchette JO. Nanoparticle and targeted systems for cancer therapy. Adv Drug Deliv Rev. 2004;56:1649–1659. doi: 10.1016/j.addr.2004.02.014. Review. [DOI] [PubMed] [Google Scholar]
- 13.Davis ME, Chen ZG, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7:771–782. doi: 10.1038/nrd2614. Review. [DOI] [PubMed] [Google Scholar]
- 14.Cabral H, Murakami M, Hojo H, Terada Y, Kano MR, Chung UI, Nishiyama N, Kataoka K. Targeted therapy of spontaneous murine pancreatic tumors by polymeric micelles prolongs survival and prevents peritoneal metastasis. Proc Natl Acad Sci U S A. 2013;110:11397–11402. doi: 10.1073/pnas.1301348110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hrkach J, Von Hoff D, Mukkaram Ali M, Andrianova E, Auer J, Campbell T, De Witt D, Figa M, Figueiredo M, Horhota A, Low S, McDonnell K, Peeke E, Retnarajan B, Sabnis A, Schnipper E, Song JJ, Song YH, Summa J, Tompsett D, Troiano G, Van Geen Hoven T, Wright J, LoRusso P, Kantoff PW, Bander NH, Sweeney C, Farokhzad OC, Langer R, Zale S. Preclinical development and clinical translation of a PSMA-targeted docetaxel nanoparticle with a differentiated pharmacological profile. Sci Transl Med. 2012;4:128ra139. doi: 10.1126/scitranslmed.3003651. [DOI] [PubMed] [Google Scholar]
- 16.Sarin H. Physiologic upper limits of pore size of different blood capillary types and another perspective on the dual pore theory of microvascular permeability. J Angiogenes Res. 2010;2:14. doi: 10.1186/2040-2384-2-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taichman RS. Blood and bone: two tissues whose fates are intertwined to create the hematopoietic stem-cell niche. Blood. 2005;105:2631–2639. doi: 10.1182/blood-2004-06-2480. Review http://dx.doi.org/10.1182/blood-2004-06-2480. [DOI] [PubMed] [Google Scholar]
- 18.Arvizo RR, Miranda OR, Moyano DF, Walden CA, Giri K, Bhattacharya R, Robertson JD, Rotello VM, Reid JM, Mukherjee P. Modulating pharmacokinetics, tumor uptake and biodistribution by engineered nanoparticles. PLoS ONE. 2011;6:e24374. doi: 10.1371/journal.pone.0024374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Asati A, Santra S, Kaittanis C, Perez JM. Surface-charge-dependent cell localization and cytotoxicity of cerium oxide nanoparticles. ACS Nano. 2010;4:5321–5331. doi: 10.1021/nn100816s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hirsch V, Kinnear C, Moniatte M, Rothen-Rutishauser B, Clift MJ, Fink A. Surface charge of polymer coated SPIONs influences the serum protein adsorption, colloidal stability and subsequent cell interaction in vitro. Nanoscale. 2013;5:3723–3732. doi: 10.1039/c2nr33134a. [DOI] [PubMed] [Google Scholar]
- 21.Sharma B, Peetla C, Adjei IM, Labhasetwar V. Selective biophysical interactions of surface modified nanoparticles with cancer cell lipids improve tumor targeting and gene therapy. Cancer Lett. 2013;334:228–236. doi: 10.1016/j.canlet.2013.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adjei IM, Peetla C, Labhasetwar V. Heterogeneity in nanoparticles influences biodistribution and targeting. Nanomedicine (Lond) 2014;9:267–278. doi: 10.2217/nnm.13.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Panyam J, Labhasetwar V. Dynamics of endocytosis and exocytosis of poly(D,L-lactide-co-glycolide) nanoparticles in vascular smooth muscle cells. Pharm Res. 2003;20:212–220. doi: 10.1023/a:1022219003551. [DOI] [PubMed] [Google Scholar]
- 24.Panyam J, Labhasetwar V. Sustained cytoplasmic delivery of drugs with intracellular receptors using biodegradable nanoparticles. Mol Pharmaceutics. 2004;1:77–84. doi: 10.1021/mp034002c. http://0-dx.doi.org.library.ccf.org/10.1021/mp034002c. [DOI] [PubMed] [Google Scholar]
- 25.Panyam J, Zhou WZ, Prabha S, Sahoo SK, Labhasetwar V. Rapid endo-lysosomal escape of poly(DL-lactide-co-glycolide) nanoparticles: implications for drug and gene delivery. FASEB J. 2002;16:1217–1226. doi: 10.1096/fj.02-0088com. [DOI] [PubMed] [Google Scholar]
- 26.Sahoo SK, Panyam J, Prabha S, Labhasetwar V. Residual polyvinyl alcohol associated with poly (D,L-lactide-co-glycolide) nanoparticles affects their physical properties and cellular uptake. J Control Rel. 2002;82:105–114. doi: 10.1016/s0168-3659(02)00127-x. [DOI] [PubMed] [Google Scholar]
- 27.Foy SP, Manthe RL, Foy ST, Dimitrijevic S, Krishnamurthy N, Labhasetwar V. Optical imaging and magnetic field targeting of magnetic nanoparticles in tumors. ACS Nano. 2010;4:5217–5224. doi: 10.1021/nn101427t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–964. doi: 10.1038/nri1733. Review. [DOI] [PubMed] [Google Scholar]
- 29.Park SI, Kim SJ, McCauley LK, Gallick GE. Pre-clinical mouse models of human prostate cancer and their utility in drug discovery. Curr Protoc Pharmacol. 2001;Ch 14(Unit 14.15) doi: 10.1002/0471141755.ph1415s51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim SJ, Uehara H, Yazici S, He J, Langley RR, Mathew P, Fan D, Fidler IJ. Modulation of bone microenvironment with zoledronate enhances the therapeutic effects of STI571 and paclitaxel against experimental bone metastasis of human prostate cancer. Cancer Res. 2005;65:3707–3715. doi: 10.1158/0008-5472.CAN-04-3601. [DOI] [PubMed] [Google Scholar]
- 31.Kim SJ, Uehara H, Yazici S, Busby JE, Nakamura T, He J, Maya M, Logothetis C, Mathew P, Wang X, Do KA, Fan D, Fidler IJ. Targeting platelet-derived growth factor receptor on endothelial cells of multidrug-resistant prostate cancer. J Natl Cancer Inst. 2006;98:783–793. doi: 10.1093/jnci/djj211. [DOI] [PubMed] [Google Scholar]
- 32.Panyam J, Williams D, Dash A, Leslie-Pelecky D, Labhasetwar V. Solid-state solubility influences encapsulation and release of hydrophobic drugs from PLGA/PLA nanoparticles. J Pharm Sci. 2004;93:1804–1814. doi: 10.1002/jps.20094. [DOI] [PubMed] [Google Scholar]
- 33.Ballou B, Lagerholm BC, Ernst LA, Bruchez MP, Waggoner AS. Noninvasive imaging of quantum dots in mice. Bioconjug Chem. 2004;15:79–86. doi: 10.1021/bc034153y. http://0-dx.doi.org.library.ccf.org/10.1021/bc034153y. [DOI] [PubMed] [Google Scholar]
- 34.Damascelli B, Cantù G, Mattavelli F, Tamplenizza P, Bidoli P, Leo E, Dosio F, Cerrotta AM, Di Tolla G, Frigerio LF, Garbagnati F, Lanocita R, Marchianò A, Patelli G, Spreafico C, Tichà V, Vespro V, Zunino F. Intraarterial chemotherapy with polyoxyethylated castor oil free paclitaxel, incorporated in albumin nanoparticles (ABI-007): Phase I study of patients with squamous cell carcinoma of the head and neck and anal canal: preliminary evidence of clinical activity. Cancer. 2001;92:2592–2602. doi: 10.1002/1097-0142(20011115)92:10<2592::AID-CNCR1612>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 35.Harries M, Ellis P, Harper P. Nanoparticle albumin-bound paclitaxel for metastatic breast cancer. J Clin Oncol. 2005;23:7768–7771. doi: 10.1200/JCO.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 36.Owens DE, 3rd, Peppas NA. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int J Pharm. 2006;307:93–102. doi: 10.1016/j.ijpharm.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 37.Panyam J, Dali MM, Sahoo SK, Ma W, Chakravarthi SS, Amidon GL, Levy RJ, Labhasetwar V. Polymer degradation and in vitro release of a model protein from poly(D,L-lactide-co-glycolide) nano- and microparticles. J Control Release. 2003;92:173–187. doi: 10.1016/S0168-3659(03)00328-6. [DOI] [PubMed] [Google Scholar]
- 38.Wynne HA, Cope LH, Mutch E, Rawlins MD, Woodhouse KW, James OFW. The effect of age upon liver volume and apparent liver blood flow in healthy man. Hepatology. 1989;9:297–301. doi: 10.1002/hep.1840090222. [DOI] [PubMed] [Google Scholar]
- 39.Souris JS, Lee CH, Cheng SH, Chen CT, Yang CS, Ho JA, Mou CY, Lo LW. Surface charge-mediated rapid hepatobiliary excretion of mesoporous silica nanoparticles. Biomaterials. 2010;31:5564–5574. doi: 10.1016/j.biomaterials.2010.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Warren A, Cogger VC, Arias IM, McCuskey RS, Le Couteur DG. Liver sinusoidal endothelial fenestrations in caveolin-1 knockout mice. Microcirculation. 2010;17:32–38. doi: 10.1111/j.1549-8719.2009.00004.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moghimi SM. Exploiting bone marrow microvascular structure for drug delivery and future therapies. Adv Drug Deliv Rev. 1995;17:61–73. Review. [Google Scholar]
- 42.Kahn D, Weiner GJ, Ben-Haim S, Ponto LL, Madsen MT, Bushnell DL, Watkins GL, Argenyi EA, Hichwa RD. Positron emission tomographic measurement of bone marrow blood flow to the pelvis and lumbar vertebrae in young normal adults. Blood. 1994;83:958–963. Erratum in Blood 1994 Nov 15;84(10):3602. [PubMed] [Google Scholar]
- 43.Cabral H, Kataoka K. Progress of drug-loaded polymeric micelles into clinical studies. J Control Release. 2014;190:465–76. doi: 10.1016/j.jconrel.2014.06.042. Review. [DOI] [PubMed] [Google Scholar]
- 44.Kwon IK, Lee SC, Han B, Park K. Analysis on the current status of targeted drug delivery to tumors. J Control Release. 2012;164:108–114. doi: 10.1016/j.jconrel.2012.07.010. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vandorpe J, Schacht E, Dunn S, Hawley A, Stolnik S, Davis SS, Garnett MC, Davies MC, Illum L. Long circulating biodegradable poly(phosphazene) nanoparticles surface modified with poly(phosphazene)-poly(ethylene oxide) copolymer. Biomaterials. 1997;18:1147–1152. doi: 10.1016/s0142-9612(97)00052-5. [DOI] [PubMed] [Google Scholar]
- 46.Oh E, Delehanty JB, Sapsford KE, Susumu K, Goswami R, Blanco-Canosa JB, Dawson PE, Granek J, Shoff M, Zhang Q, Goering PL, Huston A, Medintz IL. Cellular uptake and fate of PEGylated gold nanoparticles is dependent on both cell-penetration peptides and particle size. ACS Nano. 2011;5:6434–6448. doi: 10.1021/nn201624c. [DOI] [PubMed] [Google Scholar]
- 47.Talreja DB. Importance of antiresorptive therapies for patients with bone metastases from solid tumors. Cancer Manag Res. 2012;4:287–297. doi: 10.2147/CMAR.S33983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boissier S, Ferreras M, Peyruchaud O, Magnetto S, Ebetino FH, Colombel M, Delmas P, Delaissé JM, Clézardin P. Bisphosphonates inhibit breast and prostate carcinoma cell invasion, an early event in the formation of bone metastases. Cancer Res. 2000;60:2949–2954. [PubMed] [Google Scholar]
- 49.Green JR, Clézardin P. Mechanisms of bisphosphonate effects on osteoclasts, tumor cell growth, and metastasis. Am J Clin Oncol. 2002;25(6 Suppl 1):S3–9. doi: 10.1097/00000421-200212001-00002. [DOI] [PubMed] [Google Scholar]
- 50.Lopez-Olivo MA, Shah NA, Pratt G, Risser JM, Symanski E, Suarez-Almazor ME. Bisphosphonates in the treatment of patients with lung cancer and metastatic bone disease: a systematic review and meta-analysis. Support Care Cancer. 2012;20:2985–2998. doi: 10.1007/s00520-012-1563-z. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Allegra A, Alonci A, Penna G, Granata A, Nastro Siniscalchi E, Oteri G, Loddo S, Teti D, Cicciù D, De Ponte FS, Musolino C. Bisphosphonates induce apoptosis of circulating endothelial cells in multiple myeloma patients and in subjects with bisphosphonate-induced osteonecrosis of the jaws. Acta Haematol. 2010;124:79–85. doi: 10.1159/000313787. [DOI] [PubMed] [Google Scholar]
- 52.Ang ES, Pavlos NJ, Chim SM, Feng HT, Scaife RM, Steer JH, Zheng MH, Xu J. Paclitaxel inhibits osteoclast formation and bone resorption via influencing mitotic cell cycle arrest and RANKL-induced activation of NF-κB and ERK. J Cell Biochem. 2012;113:946–955. doi: 10.1002/jcb.23423. [DOI] [PubMed] [Google Scholar]
- 53.Sahoo SK, Ma W, Labhasetwar V. Efficacy of transferrin-conjugated paclitaxel-loaded nanoparticles in a murine model of prostate cancer. Int J Cancer. 2004;112:335–340. doi: 10.1002/ijc.20405. [DOI] [PubMed] [Google Scholar]
- 54.Vassileva V, Allen CJ, Piquette-Miller M. Effects of sustained and intermittent paclitaxel therapy on tumor repopulation in ovarian cancer. Mol Cancer Ther. 2008;7:630–637. doi: 10.1158/1535-7163.MCT-07-2117. [DOI] [PubMed] [Google Scholar]
- 55.Vassileva V, Moriyama EH, De Souza R, Grant J, Allen CJ, Wilson BC, Piquette-Miller M. Efficacy assessment of sustained intraperitoneal paclitaxel therapy in a murine model of ovarian cancer using bioluminescent imaging. Br J Cancer. 2008;99:2037–2043. doi: 10.1038/sj.bjc.6604803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nie S, Hsiao WW, Pan W, Yang Z. Thermoreversible Pluronic® F127-based hydrogel containing liposomes for the controlled delivery of paclitaxel: in vitro drug release, cell cytotoxicity, and uptake studies. Int J Nanomedicine. 2011;6:151–166. doi: 10.2147/IJN.S15057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ernsting MJ, Murakami M, Undzys E, Aman A, Press B, Li SD. A docetaxel-carboxymethylcellulose nanoparticle outperforms the approved taxane nanoformulation, Abraxane, in mouse tumor models with significant control of metastases. J Control Release. 2012;162:575–581. doi: 10.1016/j.jconrel.2012.07.043. [DOI] [PubMed] [Google Scholar]
- 58.Desai N, Trieu V, Yao Z, Louie L, Ci S, Yang A, Tao C, De T, Beals B, Dykes D, Noker P, Yao R, Labao E, Hawkins M, Soon-Shiong P. Increased antitumor activity, intratumor paclitaxel concentrations, and endothelial cell transport of cremophor-free, albumin-bound paclitaxel, ABI-007, compared with cremophor-based paclitaxel. ClinCancer Res. 2006;12:1317–1324. doi: 10.1158/1078-0432.CCR-05-1634. [DOI] [PubMed] [Google Scholar]
- 59.Karapanagiotou EM, Roulstone V, Twigger K, Ball M, Tanay M, Nutting C, Newbold K, Gore ME, Larkin J, Syrigos KN, Coffey M, Thompson B, Mettinger K, Vile RG, Pandha HS, Hall GD, Melcher AA, Chester J, Harrington KJ. Phase I/II trial of carboplatin and paclitaxel chemotherapy in combination with intravenous oncolytic reovirus in patients with advanced malignancies. Clin Cancer Res. 2012;18:2080–2089. doi: 10.1158/1078-0432.CCR-11-2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gréen H, Khan MS, Jakobsen-Falk I, Åvall-Lundqvist E, Peterson C. Impact of CYP3A5*3 and CYP2C8-HapC on paclitaxel/carboplatin-induced myelosuppression in patients with ovarian cancer. J Pharm Sci. 2011;100:4205–4209. doi: 10.1002/jps.22680. [DOI] [PubMed] [Google Scholar]
- 61.Farokhzad OC, Cheng J, Teply BA, Sherifi I, Jon S, Kantoff PW, Richie JP, Langer R. Targeted nanoparticle-aptamer bioconjugates for cancer chemotherapy in vivo. Proc Natl Acad Sci U S A. 2006;103:6315–6320. doi: 10.1073/pnas.0601755103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kalaria DR, Sharma G, Beniwal V, Ravi Kumar MN. Design of biodegradable nanoparticles for oral delivery of doxorubicin: in vivo pharmacokinetics and toxicity studies in rats. Pharm Res. 2009;26:492–501. doi: 10.1007/s11095-008-9763-4. [DOI] [PubMed] [Google Scholar]
- 63.Suzuki T, Shimizu T, Kurokawa K, Jimbo H, Sato J, Yamanaka H. Pattern of prostate cancer metastasis to the vertebral column. Prostate. 1994;25:141–146. doi: 10.1002/pros.2990250305. [DOI] [PubMed] [Google Scholar]
- 64.Mundy GR. Metastasis to bone: causes, consequences and therapeutic opportunities. Nat Rev Cancer. 2002;2:584–593. doi: 10.1038/nrc867. Review. [DOI] [PubMed] [Google Scholar]
- 65.Olechnowicz SW, Edwards CM. Contributions of the host microenvironment to cancer-induced bone disease. Cancer Res. 2014;74:1625–1631. doi: 10.1158/0008-5472.CAN-13-2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Maeda H. The enhanced permeability and retention (EPR) effect in tumor vasculature: the key role of tumor-selective macromolecular drug targeting. Adv Enzyme Regul. 2001;41:189–207. doi: 10.1016/s0065-2571(00)00013-3. Review. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.