

Abstract
Inositol polyphosphates represent a group of differentially phosphorylated inositol metabolites, many of which are implicated to regulate diverse cellular processes such as calcium mobilization, vesicular trafficking, differentiation, apoptosis, etc. The metabolic network of these compounds is complex and tightly regulated by various kinases and phosphatases present predominantly in the cytosol. Multiple inositol polyphosphate phosphatase 1 (Minpp1) is the only known endoplasmic reticulum (ER) luminal enzyme that hydrolyzes various inositol polyphosphates in vitro as well as in vivo conditions. However, access of the Minpp1 to cytosolic substrates has not yet been demonstrated clearly and hence its physiological function. In this study, we examined a potential role for Minpp1 in ER stress-induced apoptosis. We generated a custom antibody and characterized its specificity to study the expression of Minpp1 protein in multiple mammalian cells under experimentally induced cellular stress conditions. Our results demonstrate a significant increase in the expression of Minpp1 in response to a variety of cellular stress conditions. The protein expression was corroborated with the expression of its mRNA and enzymatic activity. Further, in an attempt to link the role of Minpp1 to apoptotic stress, we studied the effect of Minpp1 expression on apoptosis following silencing of the Minpp1 gene by its specific siRNA. Our results suggest an attenuation of apoptotic parameters following knockdown of Minpp1. Thus, in addition to its known role in inositol polyphosphate metabolism, we have identified a novel role for Minpp1 as a stress-responsive protein. In summary, our results provide, for the first time, a probable link between ER stress-induced apoptosis and Minpp1 expression.
Electronic supplementary material
The online version of this article (doi:10.1007/s12192-016-0684-6) contains supplementary material, which is available to authorized users.
Keywords: Inositol phosphates, Apoptosis, Multiple inositol polyphosphate phosphatase, Minpp1, ER stress, Endoplasmic reticulum
Introduction
Inositol polyphosphates (InsPs) are a group of naturally occurring compounds distributed widely in both plant and animal kingdom. A large body of literature has shown that InsPs play signaling roles in the regulation of calcium mobilization, cell growth, vesicular transport, gene expression, export of mRNA, apoptosis, and others (Berridge and Irvine 1984, 1989; Chakraborty et al. 2010; Irvine and Schell 2001; York et al. 1999). Among InsP metabolites, inositol hexakisphosphate (InsP6; phytic acid) is the most abundant in both plant and animal cells (Gimm et al. 2001). It is particularly enriched in plant seeds where it serves as the principal phosphate reservoir. Despite its abundant availability from plant-based food sources, mammalian cells synthesize their own InsP6 from cytosolic Ins (1,4,5)-triphosphate (InsP3) produced as a result of receptor activation and hydrolysis of membrane-bound phosphatidylinositol (4,5)-bisphosphate by phospholipase C (Berridge and Irvine 1984). The role of InsP3 in calcium signaling has been studied the most. Highly energetic inositol pyrophosphates InsP7 and InsP8 have been found to be associated with vesicular trafficking and the inhibition of Akt kinase signaling and induction of apoptosis (Chakraborty et al. 2010; Irvine and Schell 2001). In view of their pertinent role in various physiological processes, the intracellular concentrations of InsPs are found to be tightly regulated in mammalian cells by various mechanisms. The metabolism of InsP6 is regulated by inositol phosphate-metabolizing kinases and phosphatases, which phosphorylate and dephosphorylate them respectively to yield more phosphorylated (InsP7, InsP8) or less phosphorylated (InsP5, InsP4, InsP3) InsPs (Menniti et al. 1993; Shears et al. 1995).
Multiple inositol polyphosphate phosphatase 1 (Minpp1) is the enzyme that dephosphorylates InsP6 and other InsPs by removing specifically phosphate group at position 3 (Ali et al. 1993; Ali et al. 1995; Caffrey et al. 1999; Chi et al. 1999). Ali et al. (1993) were the first to demonstrate that Minpp1 is an endoplasmic reticulum (ER) luminal soluble protein. Subsequently, Minpp1 was cloned and characterized from several animal species, including rat (Craxton et al. 1997). In mammalian cells, Minpp1 is the preferred candidate enzyme that controls the metabolic pools of InsP5 and InsP6. Minpp1 also dephosphorylates inositol pyrophosphates (InsP7, InsP8) rather with higher affinity. It has also been shown to dephosphorylate non-InsP organic phosphates such as p-nitrophenyl phosphate (Chi et al. 1999) and 2,3-bisphosphoglycerate to 2-phosphoglycerate. The later action is implicated to have its role in Rapoport-Luebering shunt pathway (Cho et al. 2008). How an ER resident Minpp1 enzyme access to predominantly cytosolic InsP substrates remains an enigma given cytosolic InsPs are physiological substrates. It was recently postulated that (Windhorst et al. 2013) Minpp1 access to cytosolic InsPs pools via secretions and by transport into lysosomes. Nevertheless, no such Minpp1 isoform in cytosol has been characterized. However, Minpp1 engineered to express a truncated cytosolic form of the ER-confined enzyme, elevated InsP3 levels, and triggered Ca2+ release from intracellular stores via the InsP3 receptor/ Ca2+ channel (Yu et al. 2003). Remarkably, a targeted deletion of Minpp1 gene also led to a significant increase in InsP6 and InsP5 levels. These increases were reversed upon exogenous reintroduction of ER Minpp1 into cytosolic location suggesting that Minpp1 plays a significant physiological role in the maintenance of steady-state levels of InsP5 and InsP6 (Chi et al. 2000). Additionally, Minpp1 has been shown to regulate chondrocyte maturation and differentiation (Hidaka et al. 2003; Romano et al. 1998).
Recent studies from various laboratories including data published from our laboratory (Agarwal et al. 2010; Agarwal et al. 2009) have demonstrated a close association between cytosolic concentrations of InsPs and cell death induction by apoptosis. Further studies from other laboratories have shown that lower concentrations of InsP6 promoted cell proliferation, while higher concentrations led to induction of apoptosis (Cho et al. 2008; Helmis et al. 2013). Thus, a steady-state distribution of InsPs is the manifestation of a highly dynamic program of production and turnover by Minpp1 and other enzymes that regulate inositol polyphosphate inter-conversion which impact cell survival and death.
ER is an essential intracellular organelle with multiple physiological roles. ER stress-related studies in cancer cells have uncovered “unfolded protein response” (UPR), autophagy-apoptosis crosstalk, and other shared cell signaling pathways between mitochondria and ER (Naon and Scorrano 2014). It is now being widely accepted that ER as an alternative site of convergence for Bcl-2 family of proteins in the regulation of stress-induced apoptosis (Jin et al. 2014). Other studies have identified InsP3 receptors (InsP3R) and activation of pro-apoptotic protein BH3-only proteins including Bim and Puma during ER stress as critical factors (Boehning et al. 2004; Greenberg et al. 2014; Ivanova et al. 2014). Under steady-state conditions, ER stress can promote cellular repair and sustained cell survival by reducing the load of unfolded proteins (Holczer et al. 2015; Kaufman 2002). However, severe ER stress can result in apoptotic cell death. Central to the regulation of the mitochondrial checkpoint during apoptosis is complex three-way interplay between members of the BCL-2 family; ER homeostasis and calcium signaling; and the pro-apoptotic BAX, BAK, and BH3-domain-only subgroups. During apoptotic cell death, mitochondrial calcium overload alters the mitochondrial permeability characteristics resulting in depolarization, decreased ATP, and release of cytochrome c (Boehning et al. 2004). Ultimately, constituents of all three pathways converge on the ER, an organelle whose critical contributions to apoptosis are only now becoming apparent. There appears to be an inter-organelle signaling network involving ER, mitochondria, and nucleus for apoptosis induction, autophagy, and cell survival (Boehning et al. 2004). In recent years, ER stress-dependent cell death pathways are being targeted in the treatment of cancer, type 2 diabetes and amyotrophic lateral sclerosis (ALS), and others (Lindholm et al. 2006; Soo et al. 2012; Wali et al. 2014).
Since Minpp1 is the only ER resident enzyme that can hydrolyze multiple InsPs, we hypothesized that it may also play an important role in ER stress, autophagy-apoptosis crosstalk, and finally cell survival. At present, specific mechanisms by which calcium signaling participates in apoptotic cascades at the mitochondria-ER interphase have been elusive. Thus, identifying the role of Minpp1 in ER stress and apoptosis might lead to discovery of novel therapies for cellular disorders including cancer, diabetes, neurodegenerative diseases, etc.
In this work, we investigated the proposition that Minpp1 provides one such molecular link for calcium signaling, cytochrome c release from mitochondria followed by activation of caspases. Towards this end, we studied expression of Minpp1 under various experimental conditions of stress. In order to further correlate the role of Minpp1 in apoptosis, Minpp1 expression was inhibited by silencing with siRNA. These findings constitute primary evidence that Minpp1 may be an intermediary between ER stress and mitochondrial apoptosis.
Materials and methods
Materials
d-myo-inositol hexakisphosphate (InsP6, dodecasodium salt), etoposide, cisplatin, wortmannin, acridine orange, sorbitol, thapsigargin, and brefeldin A were purchased from Sigma-Aldrich (St. Louis, MO). [3H]Ins(1,3,4,5)P4 (specific activity 22 Ci/mmol) was bought from American Radiolabeled Chemicals (St. Louis, MO). TRIzol reagent, cDNA reverse transcription, and flow cytometry apoptosis kit (Vibrant no. 4, V-13243) were obtained from Life technologies (Carlsbad, CA). A monoclonal antibody recognizing KDEL motif in ER luminal protein was obtained from Novus Biological (Littleton, CO). A mouse monoclonal anti-CHOP antibody was obtained from Novus Biologicals (Littleton, CO). All other reagents were purchased from Sigma-Aldrich (St. Louis, MO).
Cell culture
All cell lines and tissue culture medium used in this study were purchased from ATCC (Manassas, VA). MC3T3-E1 (mouse osteoblastic) and PanC1 (human pancreatic cancer) cells were grown in MEM medium. SW480 (human colon adenocarcinoma) cells were maintained in DMEM medium. All cells were grown at 37 °C in a 5 % CO2 atmosphere in cell culture media supplemented with 10 % fetal bovine serum, penicillin (500 units/ml), and streptomycin (500 ug/ml).
Production and characterization of Minpp1 antibody
A polyclonal anti-peptide antibody against Minpp1 enzyme was custom prepared by FabGennix International (Frisco, TX). Briefly, the peptide sequence corresponding to 26–46 residues of human Minpp1 (SLARCSLLEPRDPVASSLSPY) was used to immunize rabbits following conjugation with Keyhole Limpet Hemocyanin (KLH). This peptide sequence was not only immunogenic but also conserved across three species (rat, mouse, and human). Antibodies were affinity purified using the Minpp1 peptide described above. Subsequently, the antibody was characterized by western blotting. The immunoreactive band was identified and its molecular weight calculated to be 58 kDa from the protein mobility plot. Further characterization of the Minpp1 antibody was accomplished by its ability to immunoprecipitate a 58 kDa protein from mammalian cell lysates which demonstrated to have enzymatic activity typical of Minpp1 against hydrolysis of Ins(1,3,4,5)P4 (InsP4).
Minpp1 enzyme assay
Enzyme activity of Minpp1 was determined as described earlier (Ali et al. 1993). Briefly, enzyme preparations (microsomal subcellular fractions, cell lysates, or immunoprecipitates) were pre-incubated for 15 min at 37 °C in a volume of 50 μl enzyme assay buffer (50 mM bis-Tris, pH 6.1, 100 mM KCl, 1 mM EDTA, 0.5 mM EGTA, 0.05 % w/v bovine serum albumin, and 2 mM CHAPS). To this mixture, [3 H]-Ins(1,3,4,5)P4 (10,000 dpm) was added at a final concentration of 5 μM. After 30 min incubation at 37 °C in a water bath, enzymatic reactions were terminated with 1.0 ml of ice-cold quench medium (0.2 M ammonium formate, 0.1 M formic acid, 1 mg/ml InsP6). Samples were then placed on ice for 30 min and then centrifuged at 10,000×g for 5 min at 4 °C (Microfuge 22 R Centrifuge, Beckman Coulter).
The [3H]-InsPs in the supernatants were analyzed by anion exchange chromatography as described by Agarwal et al. (2010). Briefly, clear supernatants were subjected to gravity fed columns packed with AG-1×8 anion exchange resin (formate form, 100–200 mesh size, Bio-Rad Laboratories, Hercules, CA). Fractions containing inositol mono-, bis-, tris-, and tetrakis-phosphates were separated with increasing concentrations of ammonium formate in 0.1 M formic acid in a batchwise elution. Radioactivity was counted by mixing 1-ml fractions with 9-ml scintillation cocktail in LS6500 Beckman-Coulter beta scintillation counter (Fullerton, CA). The specific enzyme activity was expressed as the rate of InsP4 hydrolyzed (picomoles) per milligram protein per minute at 37 °C, pH 6.1.
Induction of experimental stress
To investigate the role of Minpp1 in cell stress, cells were experimentally subjected to apoptosis, ER stress, osmotic stress, and heat stress. For these experiments, cells were seeded at a density of 5 × 104 cells/well in six-well plates or equivalent densities in 100-mm tissue culture-treated dishes.
Apoptosis in MC3T3-E1, PanC1, and SW480 was induced by treating cells with etoposide (25–200 μM), cisplatin (25–200 μM), and wortmannin (100–1000 nM). Stock solutions were further diluted in PBS at the time of each experiment and incubated with cells at 37 °C for 24 h. Control cells were treated with vehicle alone. At the end of incubation period, cells were harvested, washed free of treatments, and processed for assessment of apoptotic cell death. For ER stress, desired numbers of cells were seeded in 100-mm petri plates and were incubated overnight in the incubator at 37 °C for cells to adhere. Cells were then treated with brefeldin A (0.5–2.0 μM) and thapsigargin (0.5–2.0 μM) or vehicle control. For osmotic stress, cells were treated with d-sorbitol at concentrations ranging from 0 to 200 mM for 15 min as described by Criollo et al. (2007). For heat stress experiments, cells were exposed to different temperatures at 42, 44, and 46 °C for 2 h in an incubator to induce stress as described by Mizzen and Welch (1988). Cell culture medium was replaced after the respective treatments, and culture dishes were returned back to the incubator for the cells to recover from stress. After overnight recovery, cells were harvested and samples were prepared for SDS-PAGE and western blotting.
Subcellular fractionation
Subcellular fractions from MC3T3-E1 cells and freshly harvested rat livers were prepared as described by Ali et al. (1993). Sprague Dawley rat (wild strain) liver samples were kindly provided by Dr. Parimal Chowdhury, Department of Physiology and Biophysics, University of Arkansas for Medical Sciences (UAMS) under the approved UAMS IACUC protocol. Briefly, rat liver tissues were homogenized in an isotonic buffer (10 mM HEPES, pH 7.8, 0.25 M sucrose, 1 mM EGTA, 25 mM KCl) containing a protease inhibitor cocktail [4-(2-aminoethyl) benzenesulfonyl fluoride (AEBSF), pepstatin A, E-64, bestatin, leupeptin, aprotinin] supplemented with 1 mM phenylmethanesulfonylfluoride (PMSF) using a loose-fitting Dounce glass homogenizer by applying 8–10 up and down strokes (Kontes Glass Company, NJ). The homogenate was filtered through three to four layers of cheese cloth to remove any unbroken cells. All steps were performed at 4 °C. Subsequently, different subcellular fractions were separated by differential centrifugation. Nuclear fraction was isolated as a pellet obtained at the bottom of the centrifuge tube by centrifuging the homogenate at 500×g for 10 min (Sorvall RC 6 plus, Thermo Electron Corporation, NC). Mitochondrial fraction was then isolated from the above post-nuclear supernatant by centrifuging at 6000×g for 15 min. In order to isolate the microsomal fraction, the post-mitochondrial supernatant was centrifuged at 100,000×g for 60 min (Optima Ultracentrifuge, Beckman Coulter). The pellet was re-suspended in appropriate volume of isotonic buffer to obtain microsomal subfraction, while the supernatant was used as cytosol.
Preparation of cell lysates, immunoprecipitation, and western blotting
Cells were harvested by trypsinization and washed with cold PBS, pH 7.4. Cell were then suspended in RIPA buffer (25 mM Tris–HCl, pH 7.6, 150 mM NaCl, 1 % NP-40, 1 % sodium deoxycholate, 0.1 % SDS) containing protease inhibitor cocktail along with 0.5 M EDTA and 0.1 M PMSF) and lysed for 15 min on ice. The cell lysates were centrifuged at 13,000×g at 4 °C to remove any cell debris. Clear supernatants were used for follow-up biochemical analysis or stored at −80 °C till further use. Protein concentration was determined by Bradford colorimetric assay using BSA as a standard.
Immunoprecipitation was performed as described by Wyse et al. (2002) with modifications. Briefly, protein samples were solubilized in RIPA buffer and incubated for 4 h with appropriate dilutions of Minpp1 antibody on a rocker at 4 °C. For antibody dose-dependent immunoprecipitation, equal amount of protein samples was incubated with increasing concentrations of Minpp1 antibody. Further incubation was carried out on the rocker for 2 h at 4 °C following the addition of 20 μl of Sepharose beads (50 mg/ml PBS). After three washes with RIPA buffer by centrifugation, immuno-complex was used for the Minpp1 enzyme assay or boiled in 1× SDS sample buffer (50 mM Tris, pH 6.8, 4 % SDS, 200 mM DTT, 2 % glycerol, 0.01 % bromophenol blue) for 5 min to prepare samples for western blotting.
SDS-PAGE and western blotting were performed as described by Agarwal et al. (2009). Briefly, equal amounts of proteins (50 μg) were separated by 10 % SDS-PAGE and the proteins were electrophoretically transferred onto PVDF membranes (Millipore, Billerica, MA). The membrane was blocked with 5 % skim milk prepared in Tris-buffered saline (150 mM NaCl and 20 mM Tris–HCl, pH 7.4) containing 0.05 % Tween-20 (TBST) for 1 h at room temperature. The membranes were then incubated with diluted (2 % skim milk in TBST) primary antibodies overnight at 4 °C with constant shaking. The primary antibody dilution was according to the manufacturer’s recommendations. Hsp40, cytochrome c, and β-actin antibodies were obtained from Cell Signaling Technologies (Danvers, MA). The membranes were washed three times with TBST and incubated with a 1:5000 dilution of an appropriate secondary antibody conjugated to horseradish peroxidase (Sigma-Aldrich, St. Louis, MO) for 2 h in 2 % skim milk in TBST. The signal was detected with a Super Signal West Pico Chemiluminescence kit (Pierce Biotechnology, Rockford, IL) followed by autoradiography.
RT-PCR and quantitative real-time PCR
Total RNA was prepared from MC3T3-E1 cells using TRIzol reagent (Invitrogen, NY). Total cDNA was synthesized using a high capacity cDNA reverse transcription kit (Applied Biosystems, CA) and oligo (dT) primer. For RT-PCR, 12.5 μl PCR master mix was mixed with 1 μM each primer and 100 ng cDNA in a total volume of 25 μl. PCR with initial denaturation at 94 °C for 2 min, 40 cycles of 94 °C for 30 s, 60 °C for 1 min, and 68 °C for 2:30 min, and final extension at 68 °C for 7 min was performed. PCR products were analyzed on 1 % agarose gel followed by visualization under UV light. The primers used were (5'-3') Minpp1 forward GAT CAC CAG CTC CAA GCA CC and reverse TGT AAG CGG TCA GAG GCT CC and GAPDH forward ACC ACA GTC CAT GCC ATC AC and reverse TCC ACC ACC CTG TTG CTG TA. These primers were also used for real-time PCR in a Bio-Rad iCycler optical system using iQ™ SYBR green real-time PCR kit (Bio-Rad Laboratories, Inc., CA) following manufacturer’s protocol. The levels of Minpp1 mRNA expression in MC3T3-E1 cells were analyzed after normalizing cycle thresholds against the housekeeping gene GAPDH. The real-time data represented as a fold change value was determined by 2¯ΔΔC T method (Livak and Schmittgen (2001).
Silencing of Minnp1 expression by siRNA
Cells were seeded at a density of 1 × 105/well in six-well plates 24 h prior to transfection. Before transfection, complete medium was replaced with fresh serum-free medium. The siGENOME smartpool of short interfering RNA (siRNA) for human Minpp1 (M009705) was obtained from Dharmacon (Lafayette, CO). Cells were transfected as per manufacturer’s protocol. Briefly, 55 nM Minpp1 siRNA mixed in 25 μl transfection reagent (Polyplus transfection, France) was added to a total volume of 250 μl medium. After 24 h incubation, the transfection was repeated with same concentration of the siRNA and the incubation continued for further 48 h. Cells were then harvested for biochemical analysis.
Apoptosis assays
Assessment of apoptotic cell death was carried out by one of the four different assay methods.
Acridine orange/ethidium bromide staining (AO/EB)
Apoptotic cells were enumerated under the microscope after staining with AO/EB at a ratio of 1:1 as outlined by Agarwal et al. (2009). For each treatment condition, at least 200 cells were counted from five random fields and the percentage of apoptotic cells was calculated.
Flow cytometric analysis
Apoptosis in MC3T3-E1 cells was assayed by flow cytometry using a commercially available Vybrant apoptosis assay kit (Catalog no. V-13243, Molecular Probes, Carlsbad, CA). This assay detects changes in cell membrane permeability characteristics with YO-PRO-1 dye, a green fluorescent nucleic acid stain that is permeant only to apoptotic cells but not to live cells. Necrotic cells are labeled with red fluorescent propidium iodide. This assay detected three types of cell populations: (1) live cells show a low level of green fluorescence, (2) early apoptotic cells show an incrementally higher level of green fluorescence, (3) and late apoptotic (dead) cells show both red and green fluorescence. Using single color-stained cells, standard compensation was performed and cell debris was gated out (Estaquier et al. 1996; Idziorek et al. 1995). Briefly, 1 × 106 cells/ml were incubated with 1 μl of YO-PRO-I and 1 μl of PI for 30 min incubation at 4 °C and were analyzed in a BD FACSCalibur flow cytometer (UAMS Core Facility) within 1 h. At least 10,000 cells were analyzed for each treatment condition, and the data was analyzed by FlowJo to detect different cell populations. Data in the flow histogram is presented as percentages of apoptotic cells.
Caspase-3 activity
Caspase-3 activity, an index of apoptosis, was carried out as per manufacturer’s recommendations. Briefly, cells were washed once, lysed in ice-cold lysis buffer supplied in the kit, and incubated for 10 min on ice. Debris-free supernatant was prepared by centrifugation at 10,000×g for 1 min. Aliquots of 50 μl of crude cell lysate were incubated with caspase-3 substrate DEVD-pNA (BioVision, Milpitas, CA) at 37 °C for 2 h. The caspase-3 activity was quantified by spectrophotometry at 405 nm.
Cytochrome c release assay
During apoptotic conditions, cytochrome c is released from inner matrix of the mitochondria to the cytosol (Desagher and Martinou 2000; Reed 1997). We assayed cytochrome c release in the subcellular fraction by western blotting. Detailed experimental procedure is outlined under western blotting.
MTT assay for cell death
MC3T3-E1 cells were seeded in 96-well plates at 1 × 104 cells/well. Following induction of apoptosis, cell viability was measured with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide dye (Kannan et al. 2000). The color development was measured in an ELISA plate reader (iMark, Bio-Rad, CA) at 570 nm.
Determination of ROS by DCFH-DA fluorescence
Reactive oxygen species (ROS) production during ER stress-induced apoptosis was detected using 7′-dichlorodihydrofluorescein diacetate (DCFH-DA), a redox-sensitive fluorescent probe from Cell Signaling (Danvers, MA). First, MC3T3 cells were plated at a cell density of 1.5 × 104 cells per well in a black 96-well fluorescence assay plate with clear bottom (Costar, Catalog no. 07-300-562). Triplicate wells were treated with vehicle alone, etoposide (100 μM), brefeldin A (1 μM), and thapsigargin (1 μM) for 24 h. At the end of incubation, cells were incubated with 10 μM DCFH-DA for an additional period of 30 min at 37 °C. Cells were washed to remove any unbound excess fluorescence probe. Fluorescence intensity was determined in Synergy-H4, fluorescence microplate reader (BioTek, Winooski, VT) at 485/535 nm. Relative fluorescence intensity for each treatment condition was calculated after subtracting the background fluorescence. Experiment was repeated twice.
Statistical analysis
All calculations were performed using GraphPad prism v 6.0 software (San Diego, CA). Each experiment was repeated at least three times. All data that show error bars are presented as mean ± SE unless otherwise mentioned. The significance of difference in the mean values was determined using two-tailed Student’s t test or by ANOVA using Prism software. A p < 0.05 was considered as significant.
Results
Characterization of Minpp1 antibody
A rabbit polyclonal antibody against Minpp1 was custom-made and characterized biochemically. The specificity of the custom Minpp1 antibody was confirmed by its ability to immunoprecipitate the expected molecular size of the protein and subsequently the presence of Minpp1 enzymatic activity in the immunoprecipitate. As shown in Fig. 1a, Minpp1 antibody was able to immunoprecipitate a native protein from rat liver extract in an antibody dose-dependent manner. The molecular weight of the immunoprecipitated band was determined to be 58.0 ± 1.7 kDa (n = 3) based on its electrophoretic mobility relative to standard molecular weight protein markers (Fig. 1b). This molecular weight is close to the predicted size (56 kDa) of human Minpp1 calculated based on its amino acid composition. The antibody also immunoprecipitated a 58 kDa protein from mouse osteoblastic cell (MC3T3-E1) extracts (Fig. 1c) further confirming its specificity. In comparison, pre-immune serum (internal control) did not result in any immunoprecipitation (Fig. 1c) suggesting that the Minpp1 antibody we used was specific to the Minpp1.
Fig. 1.

Characterization of Minpp1 antibody by immunoprecipitation and western blotting. a Minpp1 antibody concentration-dependent immunoprecipitation. Dose-dependent immunoprecipitation was done using different concentrations of Minpp1 antibody as illustrated in the figure and then these immunoprecipitates were used for western blot analysis. Densitometry analysis was carried out to quantify antibody dose-dependent Minpp1 immunoprecipitation. Results shown here are a representative data out of two independent experiments with similar results. b Determination of molecular weight of immune-reactive Minpp1 band by electrophoretic mobility of proteins in a SDS-PAGE using standard protein markers with different sizes. From the plot, the relative molecular weight of Minpp1 band was calculated to be 58 kDa. A representative graph is shown out of three independent experiments. c Western blot showing an immunoreactive band in the presence of immune serum. Pre-immune and immune serum were reacted with immunoprecipitated proteins of microsomes isolated from rat liver and MC3T3-E1 cells. Immune serum gave a stronger reaction suggesting the presence of Minpp1-specific antibodies. The blot is a representative data of three independent experiments with similar results. d Graph showing the inhibition of Minpp1 enzyme activity by Minpp1 antibody. Inhibition of enzyme activity was assessed with increasing concentrations of Minpp1 antibody with CHAPS-solubilized liver microsomes. Minpp1-specific enzyme activity was plotted against antibody concentration. Values are means ± S.E. from three independent experiments, each determined in triplicates
To ensure that the immunoprecipitated protein was indeed Minpp1, we also determined a qualitative Minpp1 enzyme activity (hydrolysis of InsP4 to InsP3) in immunoprecipitates obtained from MC3T3-E1 as well as rat liver extracts (Table 1). These results indicate that the antibody indeed recognizes the Minpp1 protein. Furthermore, this antibody inhibited Minpp1 enzyme activity dose-dependently when mixed with rat liver microsomes in an enzyme assay (Fig. 1d) further confirming that the antibody specifically interacts with Minpp1 and this interaction interferes with its activity. We have used this approach earlier to show specificity of the antibody to Minpp1 protein (Craxton et al. 1995). This batch of antibody was then frozen in smaller aliquots for all further studies.
Table 1.
Determination of the specificity of Minpp1 antibody by the presence of Minpp1 enzymatic activity in immunoprecipitates
| Sample | % hydrolysis of [3H]-Ins(1,3,4,5)P4 |
|---|---|
| MC3T3—control | 0.0 |
| MC3T3—IP | 13.3 |
| Rat liver—control | 0.0 |
| Rat liver—IP | 38.3 |
Cell lysates prepared from MC3T3-E1 cells and rat liver tissues were subjected to immunoprecipitation using Minpp1 antibody as described in “Materials and methods” section. Immunoprecipitates (IP) were incubated at 37 °C for 1 h in the Minpp1 enzyme assay buffer using [3H] Ins (1,3,4,5) P4 as substrate. A qualitative hydrolysis of the substrate was determined by analysis of radioactive products using anion exchange chromatography as described before. Qualitative data shown as average values from two experiments indicate that the Minpp1 antibody was specifically able to immunoprecipitate Minpp1 protein
Apoptosis triggers elevated expression of Minpp1 but does not change its localization
Ali et al. (1993) were the first to demonstrate that InsP-hydrolyzing enzyme Minpp1 (InsP 3-phosphatase) is an ER luminal protein. Recent work including the work from our laboratory has demonstrated a close association between apoptosis and cellular levels of InsPs that may also serve as potential substrates for Minpp1. However, the role of Minpp1 in apoptosis is not yet clear. Accumulating literature has shown that ER is emerging as a critical regulator of cell homeostasis, cell survival, and cell death mechanisms (Deegan et al. 2013). Under conditions of prolonged ER stress, cells can switch from protective signaling mode to pro-apoptotic response. Thus, the characterization of this network of signaling molecules would enhance our understanding of ER stress-induced apoptosis (Kim et al. 2008), and any role Minpp1 plays in this process.
First, we carried out subcellular fractionation in MC3TC-E1, an osteoblast cell line of mouse origin, and demonstrated that Minpp1 is mostly found in the microsomal fraction (Fig. 2a) shown to be enriched in ER (Ali et al. 1993). However, upon the treatment with 100 μM etoposide, the expression of Minpp1 was found increased significantly only in microsomes prepared from etoposide-treated cells; it was absent in cytosolic fraction. Thus, etoposide treatment did not cause any redistribution of Minpp1 to cytosolic fraction; it remained confined to microsomal fraction.
Fig. 2.

Subcellular localization and redistribution of proteins during apoptosis. Apoptosis in MC3T3-E1 cells was induced by incubating with 100 μM etoposide for 24 h followed by subcellular fractionation into microsomal (micro), cytosolic (cyto), and mitochondrial fractions. Equal amounts of proteins (50 μg) of various subcellular fractions separated by SDS-PAGE followed by western blotting as described in the “Materials and methods” section. a Immunoreactive bands were identified using anti-Minpp1 antibodies. Results show that Minpp1 expression was increased in etoposide-treated cells, but it was retained within microsomal fraction suggesting no redistribution to other compartments. Shown is a representative immunoblot out of three independent experiments with similar results. b Immunoreactive bands were identified using anti-cytochrome c antibody. Results show that etoposide treatment led to changes in mitochondrial membrane function and leakage of cytochrome c to the cytosol. Shown is a representative immunoblot out of three independent experiments with similar results. c Immunoreactive bands were identified using anti-KDEL antibody known to detect ER luminal proteins. This antibody detected only two bands (75 and 58 kDa) in MC3T3-E1 cells. Results show that the 75 kDa protein was distributed in both cytosolic and microsomal fractions, but the 58 kDa protein was present only in microsomal fraction, the expression of which was increased by etoposide treatment (C, control; E, etoposide). Shown is a representative immunoblot out of three independent experiments with similar results
Next, we asked whether any other ER resident protein is leaked to cytosol during apoptotic conditions. To elucidate this possibility, we used an antibody that recognizes KDEL sequence containing proteins. Most ER luminal proteins contain a KDEL motif or its variant at C-terminus which acts as a signal for these proteins to be retained inside ER. Using this analytical tool, we examined the subcellular distribution of KDEL motif containing proteins to study the redistribution of ER proteins during apoptosis (Fig. 2c). In our hands, this antibody detected only two major proteins in MC3T3-E1 cells with relative molecular weight of 58 and 75 kDa. While 75 kDa protein was detected both in cytosolic and microsomal fractions, the 58 kDa protein was detected only in the microsomal fraction, the expression of which was also increased upon etoposide treatment. It is plausible that this 58 kDa KDEL motif containing protein is Minpp1 which is overexpressed but remained in ER during apoptosis. At present, the identity of the 75 kDa protein is not clear, although this KDEL antibody is known to recognize a similar protein and some additional proteins in other cell lines.
To better understand the involvement of mitochondria and ER stress-induced apoptosis and ER-mitochondrial interactions, we analyzed the release of cytochrome c (cyto-c) from mitochondria as a loss function of mitochondrial membrane integrity. Our subcellular fractionation results showed a release of cyto-c from mitochondria to cytosol upon etoposide treatment (Fig. 2b) suggesting mitochondrial membrane integrity was compromised during apoptosis. As compared to etoposide-treated cells, control cell preparations had intact mitochondria and no leakage of cyto-c was detected in the cytosol.
Increased expression of Minpp1 correlates with apoptosis
Extending our above observations, we sought whether alteration in Minpp1 expression (Fig. 3a) was associated with changes in enzyme activity. We found that the enzyme activity of Minpp1 increased approximately twofold in 100 μM etoposide-treated cells over vehicle alone-treated controls (specific activity: control 18.0 ± 1.4 vs etoposide 40.0 ± 1.0 pmol/min/mg protein) as shown in Fig. 3b. Furthermore, there was a direct correlation between apoptosis and Minpp1 expression. Treatment with etoposide that resulted in an increased expression of Minpp1 protein and its enzyme activity also accompanied an appropriate increase in apoptotic markers as analyzed by flow cytometry (Fig. 3c) and caspase-3 activation (Fig. 3d) suggesting a strong correlation between apoptosis and Minpp1. These results were further confirmed by enumerating apoptotic cells by acridine orange/ethidium bromide staining (data not shown).
Fig. 3.

Effect of etoposide-induced apoptosis on the expression of Minpp1. a Western blotting of Minpp1 expression during apoptosis. MC3T3-E1 cells were treated with DMSO (vehicle) or 100 μM of etoposide for 24 h followed by cell harvesting and western blotting as detailed in “Materials and methods” section. Results show a marked increase in Minpp1 expression (*p < 0.01). b Enzyme activity of Minpp1 was estimated as outlined in the “Materials and methods” section, and the data is expressed as picomoles of IP4 hydrolyzed per milligram of protein per minute [*p < 0.01]. c Flow cytometry analysis of apoptotic cells using the Vybrant apoptosis assay kit. After treatment with 100 μM etoposide, MC3T3-E1 cells were stained with Yo-Pro-1 (green fluorescence) and propidium iodide (PI; red fluorescence) as described in “Materials and methods” section. Early apoptotic cells were identified by Yo-Pro-1 staining, whereas late apoptotic events were identified by dual staining of Yo-Pro-1 and PI. Results show a marked increase in early and late apoptotic events in etoposide-treated cells. Shown is a representative histogram out of three experiments with similar results. d Apoptosis was confirmed by caspase-3 activity assay as described in “Materials and methods” section. Each data point represents mean ± S. E. from three experiments determined in triplicates *p < 0.01
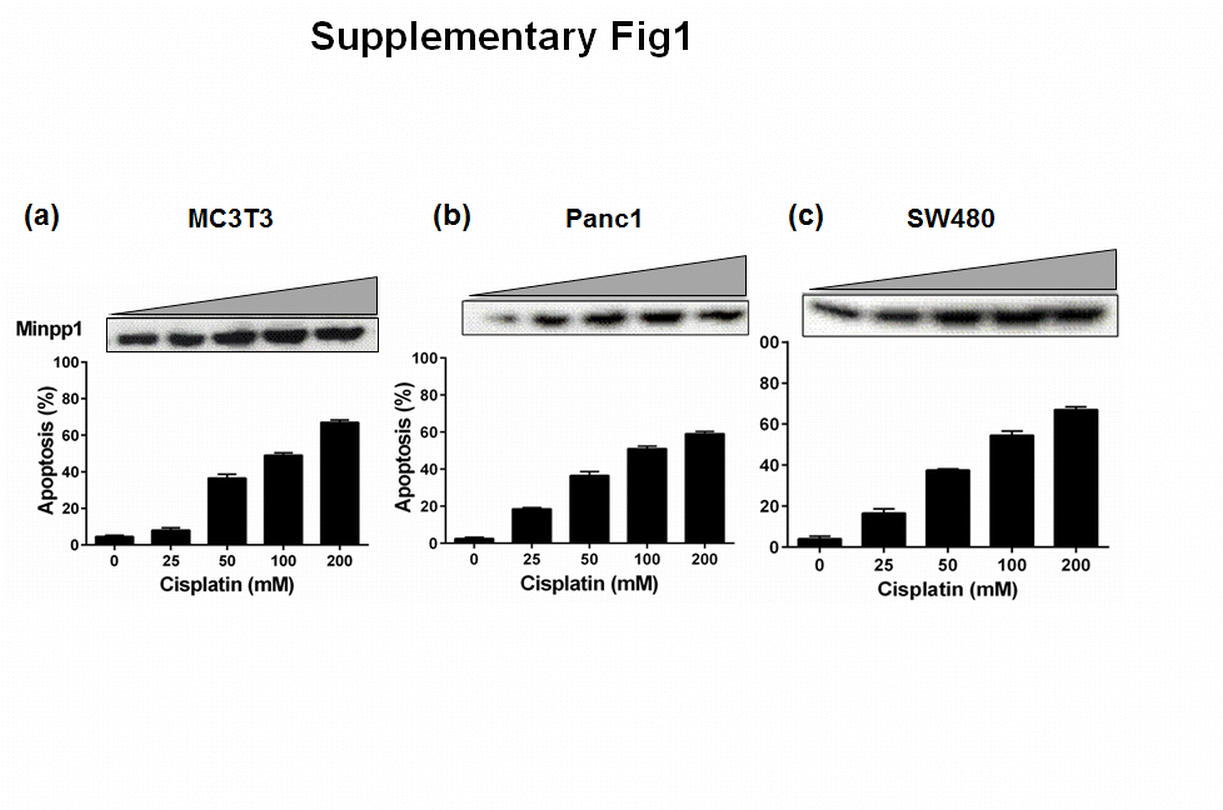
Having established that Minpp1 expression is elevated during etoposide-induced apoptosis, we wondered whether this expression is an isolated event restricted to a particular cell type or cell line. We therefore examined the causal relationship between Minpp1 expression and apoptosis by extending our studies in PanC1 and SW480, a human pancreatic and colon adenocarcinoma cell lines, respectively. In addition, we included other apoptosis-inducing agents, namely wortmannin and cisplatin. As shown in Fig. 4a–f and Supplementary Fig. 1, a similar trend was observed in all three cell lines and with all three apoptosis-inducing agents. Taken together, these data strongly suggest that Minpp1 expression during apoptosis is uniformly upregulated in both mouse osteoblasts and in human tumor cell lines. However, the significance of this positive correlation between Minpp1 expression and apoptosis is not clear at present. Many of the apoptosis-inducing agents also cause significant molecular changes in proteins of ER and its homeostasis resulting in upregulation of ER resident proteins, release of Ca2+ and caspase-12, and other pro-apoptotic molecules (Adams 2003; Orrenius et al. 2003; Schonthal 2013; Shore et al. 2011). It is thus possible that Minpp1, a luminal protein of ER, is also triggered as part of the biochemical cascade during apoptosis. Further research work is necessary to delineate the true nature of the molecular interactions between Minpp1 and UPR pathway for a direct link to ER stress.
Fig. 4.

Analysis of Minpp1 expression in different mammalian cell lines during apoptosis. MC3T3-E1, PanC1, and SW480 cells were treated with etoposide (25–200 μM) or wortmannin (100–1000 nM) for 24 h. One set of cells was processed for SDS-PAGE and western blotting for Minpp1 expression as detailed in the “Materials and methods.” The western blots shown are representative experiments repeated independently at least three times on all three cell lines with various treatments. Another set of cells was processed for the assessment of apoptosis following acridine orange/ethidium bromide staining. A minimum of 200 cells were counted from five random fields and percentage of cells undergoing apoptosis was determined as detailed in “Materials and methods” section. The bars indicate the means ± S.E. from three different experiments performed in triplicates. a–c Data on cells treated with etoposide. d–f Data on cells treated with wortmannin
Next, we examined the expression of Minpp1 mRNA by conventional RT-PCR and real-time PCR during apoptosis. Results included in Fig. 5a clearly show a dose-dependent increase in mRNA levels following treatment with etoposide. At 25, 50, and 100 μM etoposide, Minpp1 mRNA levels increased by 1.7-, 1.9-, and 2.1-fold, respectively, over control values. Similar results were obtained (Fig. 5b) when cells were treated with wortmannin (100 nM) and cisplatin (100 μM). To quantify the increased levels of Minpp1 mRNA during apoptosis, we performed real-time PCR. The results included in Fig. 5c show a twofold increase of Minpp1 mRNA in 100 μM etoposide-treated MC3T3-E1 cells. Collectively, these data suggest that both mRNA levels and protein expression of Minpp1 are significantly altered during apoptosis.
Fig. 5.

RT-PCR and real-time PCR showing Minpp1 mRNA expression during apoptosis. a MC3T3 cells were treated with increasing concentrations of etoposide (25–100 μM) for 24 h and changes in Minpp1 mRNA expression during apoptosis were determined by RT-PCR. Shown is a representative experiment out of three with triplicate samples for each treatment. GAPDH was used as an internal control in all these experiments. b Effect of etoposide (ETOP; 100 μM), wortmannin (WMN; 500 nM), and cisplatin (CDDP; 100 μM) were used to induce apoptosis in MC3T3-E1 cells for 24 h. RT-PCR was carried out to determine the changes in expression of Minpp1 mRNA during apoptosis. Shown is a representative experiment out of three with duplicate samples for each treatment. GAPDH was used as an internal control in all these experiments. c Quantitative analysis using real-time PCR was performed on MC3T3 cells to determine changes in expression of Minpp1 mRNA. Data shown is a fold increase calculated from three independent experiments. The bar indicates means ± S.E. *p < 0.01
ER stress-induced apoptosis accompanies Minpp1 expression
After establishing a correlation between apoptosis and expression of Minpp1, we wondered whether ER stress-induced apoptosis has a direct impact on the expression of Minpp1. To test this possibility, we induced ER stress in MC3T3-E1 cells with brefeldin A (BFA) and thapsigargin (TG), the agents known to induce ER stress and investigated apoptosis and Minpp1 expression. The flow histogram in Fig. 6a–c shows ER stress-induced apoptosis by BFA and TG. Results included in Fig. 6d, e demonstrated an increased expression of Minpp1 in response to increasing concentrations of BFA and TG. These results clearly indicate that ER stress-induced apoptosis also accompanies an increased expression of Minpp1. Since CHOP is a well-known downstream marker for UPR-related PERK pathway and is known to be overexpressed in response to ER stress-induced apoptosis, we determined whether CHOP expression is correlated with Minpp1 expression. Our western blot analysis also showed (Fig. 6d, e) that BFA and TG treatment dose-dependently increased CHOP expression under conditions that led to Minpp1 expression and apoptosis. These results suggest a link between Minpp1 expression and UPR-activated PERK pathway that lead to CHOP activation.
Fig. 6.

ER stress-induced apoptosis accompanies elevated expression of Minpp1. a–c Flow cytometry histogram showing apoptotic cell death in control, brefeldin A, and thapsigargin-treated MC3T3-E1 cells. Cells were incubated with ER stress inducers for 24 h followed by flow cytometry analysis of apoptosis by staining with YO-PRO-1 and propidium iodide. Data shows a significant increase in the percent apoptotic cells in brefeldin A and thapsigargin-treated cells. Shown here is a representative experiment repeated three times with similar results. d, e Different concentrations of ER stress inducers brefeldin A and thapsigargin were incubated with MC3T3-E1 cells for 24 h. Minpp1 and CHOP expression was determined in these cells by western blotting. Relative expression of Minpp1/actin was calculated by densitometry. Shown is a representative western blot. Densitometry values for Minpp1/Actin are given as means ± S.E. (error bars) from two separate experiments determined in triplicates for each concentration. When no error bar is shown, the error is smaller than the width of the line
Expression of Minpp1 during heat and osmotic stress
After determining that Minpp1 expression is altered under the apoptotic or ER stress, we wondered whether Minpp1 expression also changes under other stress conditions including heat shock and osmotic stress. For heat stress, MC3T3-E1 cells were subjected to various temperatures (37–46 °C) and protein extracts were analyzed by western blotting. There was a consistent increase in the expression of Minpp1 protein with increasing temperatures up to 46 °C suggesting that Minpp1 is a stable protein even at higher temperatures (Fig. 7a). Hsp40 expression, used as a positive control for heat shock, also increased significantly with increasing temperatures up to 44 °C. At 46 °C, no detectable Hsp40 was observed perhaps due to protein degradation at higher temperature. For osmotic stress, MC3T3-E1 cells were treated with 100 and 200 mM sorbitol for 15 min followed by western blotting of Minpp1 expression. Results included in (Fig. 7b) suggest that osmotic stress induced by sorbitol also results in a significant increase in the expression of Minpp1. Untreated control cells showed only basal levels of Minpp1 in MC3TC-E1 cells. Minpp1 expression was increased even under mild conditions of osmotic stress used in our study, which were perhaps not pro-apoptotic (Yang et al. 2008). Collectively, these data suggest that besides ER and apoptotic stress, other stress conditions including heat and mild osmotic stress can also lead to an increased expression of Minpp1.
Fig. 7.

Western blot analysis of Minpp1 expression under heat and osmotic stress conditions. a To induce heat stress, MC3T3-E1 cells were subjected to increasing temperatures (37 to 46 °C) for 2 h followed by 24 h recovery at 37 °C as detailed in “Materials and methods” section. Minpp1 and Hsp40 expression in these cells was analyzed by western blotting followed by densitometry analysis. Hsp40 used as a positive control for heat stress-associated changes. Results shown are from a representative experiment repeated at least three times. b For osmotic stress, MC3T3-E1 cells were exposed to increasing concentrations of sorbitol for 24 h at 37 °C as detailed in “Materials and methods” section. Minpp1 expression was analyzed by western blotting followed by densitometry analysis. Results shown are from a representative experiment repeated four times
ER stress-induced apoptosis accompanies an increased ROS production
When UPR is prolonged in cells under stress, apoptosis pathway may be triggered. In this study, we wondered whether ROS played any role in ER stress-induced apoptosis. ER stress was experimentally induced by BFA and TG treatment of the MC3T3-E1 cells, and ROS was determined using DCFH-DA fluorescence as a probe for ROS. There was a significant increase in ROS detected in BFA- and TG-treated cells, whereas it was moderate in cells treated with etoposide (Fig. 8). Thus ROS production, Minpp1 expression, cytochrome c release, and apoptosis all point to a link between Minpp1 and cellular stress. Our data on cytochrome c indicate that mitochondria may be the site of ROS production which then may have exerted its effect on ER leading to increased Minpp1 expression as well as triggering apoptosis. Furthermore, this data also point to a causal link between oxidative stress and Minpp1 expression during ER stress.
Fig. 8.

ROS production during ER stress. MC3T3 cells were cultured and treated with etoposide, brefeldin A, and thapsigargin for 24 h as described under “Materials and methods” section. ROS was determined by DCFH-DA fluorescence probe. The signal intensity was detected in a fluorescence plate reader. The data presented is a representative experiment out of two independent experiments performed in triplicate determinations
Silencing of Minpp1 by siRNA attenuates apoptosis
Silencing of Minpp1 was accomplished by transfection of MC3T3-E1 cells using a pool of Minpp1-specific siRNA. Apoptosis was then induced with 100 μM etoposide. Figure 9a showed that the majority of the expression of the Minpp1 was knocked down using the Minpp1 siRNA. Actin used as a loading control remained unchanged. Etoposide, while increased Minpp1 expression in control and knocked down cells, it attenuated apoptosis as evident by the lack of any release of cytochrome c, an apoptotic marker, from mitochondria to cytosolic fractions in Minpp1-knocked down cells; cytochrome c was released in cytosol upon etoposide treatment in control cells (Fig. 9b). We have also measured apoptosis by caspase-3 activation (Fig. 9c) following knockdown of Minpp1 by siRNA. Etoposide induced apoptosis was attenuated in Minpp1 knockdown cells further suggesting a link between Minpp1 and apoptosis.
Fig. 9.

Effect of Minpp1 silencing on apoptosis as determined by release of cytochrome c. MC3T3-E1 cells were transfected with Minpp1-specific siRNA to knockdown the expression of Minpp1 gene as described in “Materials and methods” section. Apoptosis was then induced with 100 μM etoposide for 24 h. Minpp1 expression was determined in total cell lysates following SDS-PAGE and western blotting using Minpp1 antibody. Mitochondrial release of cytochrome c during apoptosis was determined in cytosolic fractions following SDS-PAGE and western blotting using cytochrome c antibody. Results show a significant silencing of Minpp1 expression in the siRNA-treated cells. Control cells show a usual increase in the expression of Minpp1 following etoposide-induced apoptosis. Cytochrome c released in cytosolic fractions was abolished in the siRNA-treated samples. Controls show a significant release of cytochrome c in cytosol upon etoposide treatment. Data shown are representative experiments repeated at least three times with similar results
Discussion
Endoplasmic reticulum plays a central role in the regulation of various cellular stresses. The signals for these stresses may originate from other organelles including mitochondria. Thus, there exists a dynamic crosstalk between ER and mitochondria. Many of these signaling events converge at the mitochondria-ER interphase via an enlarging network of molecules (Chen and Brandizzi 2013; Hetz et al. 2011). In the present study, we examined the role of an ER luminal protein, Minpp1, which we hypothesized as a potential candidate of this crosstalk and as a sensitive indictor of cellular stress. Additionally, we probed its role in ER stress-induced apoptosis under experimental conditions (Hetz 2007; Kim et al. 2008; Ron 2002). We provide evidence, for the first time, that the expression of Minpp1 parallels various cellular stress conditions. A preliminary finding of this observation was reported earlier in the form of an abstract in the experimental biology meeting (Kilaparty and Ali 2013). Here, we have identified a novel physiological attribute to Minpp1 utilizing a paradigm of ER stress-induced apoptosis. The polyclonal antibody developed in our laboratory against Minpp1 served as a valuable tool in the present study. Using this antibody, we demonstrated that there is a direct correlation between the level of expression of Minpp1 and apoptosis. This phenomenon was observed with multiple apoptosis-inducing agents in multiple mammalian cell lines. The increased expression of Minpp1 was not only observed with apoptotic stress but also with other stress conditions including heat shock and osmotic stress. The stress response by Minpp1 was directly proportional to the severity of the stress. Collectively, our data imply a physiological role for Minpp1 during cellular stress response (Holczer et al. 2015; Jin et al. 2014; Puthalakath et al. 2007; Stentz et al. 2014; Szegezdi et al. 2006).
ER plays a central role in calcium homeostasis, protein folding, secretion, and lipid biosynthesis (Ron and Walter 2007). A large body of evidence has now linked aberrations of ER homeostasis in the pathogenesis of several human diseases (Kim et al. 2008). Thus, it is of significant interest to develop a detailed molecular paradigm on ER stress. In a limited way, our data on Minpp1 expression during ER-stress-induced apoptosis (ERSIA) adds another sensitive indictor of cellular stress. Given that Minpp1 participates in InsP metabolism, it is likely that ERSIA-InsPs-Minpp1 axis plays a regulatory role in ER homeostasis.
It is now well established that abnormal cellular stress could trigger cell death via a complex interaction between mitochondria, ER, lysosomes, and the nucleus (Chen and Brandizzi 2013; Hetz et al. 2011; Kim et al. 2008; Szegezdi et al. 2006). The response of Minpp1 to cellular stress conditions could also be viewed as a molecular adaptive mechanism to sustain ER homeostasis under adverse cellular conditions. In metazoans, stress pathways help monitor intracellular systems and deploy a range of regulatory mechanisms in response to various cellular stresses. UPR is one of the best-characterized and evolutionarily conserved pathways (Hetz et al. 2011) in this regard. UPR is an intracellular signaling pathway that monitors ER homeostasis by ER-bound sensors (IRE1, PERK, and ATAF6) located on the outer ER membrane and associated biochemical machinery. In general, the UPR program activates genes for molecular chaperons, transcription factors, and kinases to alleviate ER stress but it can lead to apoptosis if the system fails to restore homoeostasis (Hetz et al. 2011; Verfaillie et al. 2012). Though a molecular paradigm for UPR has been proposed in the literature, there are still gaps on the role of many ER resident proteins including Minpp1 in cell signaling and regulation of stress pathways. The data included in this study with regard to correlation between Minpp1 and CHOP expression and apoptosis (Fig. 6) suggests that Minpp1 could play a sensor or regulatory role during ER stress-induced apoptosis involving UPR signaling pathway.
Several studies have shown that mitochondrial outer membrane as well as ER membrane integrity are compromised during apoptosis and other stress conditions (Boehning et al. 2003; Chipuk and Green 2008; Kanekura et al. 2015; Wang et al. 2011). For example, cytochrome c released from mitochondria interacts with IP3 receptor present on ER membrane rendering ER leaky to calcium (Kanekura et al. 2015). Wang et al. (2011) suggested that ER stress-induced apoptosis alters ER membrane permeability via a Bax- and Bak-dependent mechanism which leads to redistribution of ER luminal proteins including GRP-78 to cytosol. Even disturbances in ionic homeostasis result in reorganization of ER (Varadarajan et al. 2013). These studies prompted us to investigate whether Minpp1, also an ER luminal, redistributes similarly under apoptotic stress conditions. Our subcellular fractionation studies showed that the expression of Minpp1 was increased under apoptotic conditions. However, it remained confined within the ER lumen. This finding was further validated using another antibody that specifically recognizes KDEL motif present in ER luminal proteins. KDEL motif recognizing antibody recognized a protein with a molecular weight similar to that of Minpp1 the expression of which was also increased in response to apoptosis but remained associated with ER-enriched microsomes. This observation is in contrast to other reported studies showing redistribution of luminal proteins under ER stress conditions (Wang et al. 2011). Our data, however, does not preclude leakage of Minpp1 during other pathological or ER stress conditions including heat shock and osmotic stress.
Assigning a physiological function to Minpp1 in InsP metabolism remains a challenge because of its confined location within ER lumen, away from its potential cytosolic substrates (Chi et al. 2000; Cho et al. 2008; Craxton et al. 1997; Hidaka et al. 2003; Nogimori et al. 1991; Romano et al. 1998; Windhorst et al. 2013; Yu et al. 2003). However, when expressed experimentally in cytosolic location, Minpp1 dephosphorylates InsPs and alters their cellular levels (Yu et al. 2003). A cytosolic isoform of Minpp1 with its physiological function in InsP dephosphorylation has not been unequivocally demonstrated. However, a recent finding that Minpp1 associated with lysosomal fraction dephosphorylates InsPs in lysosomes and also secreted extracellularly in response to excess InsPs (Windhorst et al. 2013) suggests a potential role for Minpp1 in InsP metabolism outside ER lumen. Our computational analysis of available databases to find heterogeneity in Minpp1 structure and function revealed an existence of four human isoforms including two that lack ER retention signal (Kilaparty et al. 2014). It is likely that such truncated isoforms of Minpp1 has a role in InsP metabolism outside ER lumen. Furthermore, it remains to be determined whether Minpp1 is directly involved in other physiological functions that are relevant to ER function and its homeostasis.
Our data also provide a molecular link between Minpp1 expression and ER oxidative stress by correlating the expression with ROS production implying a wider role for Minpp1 in ER stress regulation (Nakajima and Kitamura 2013). The molecular link between Minpp1 function and cellular stress was further strengthened by knocking down the expression of Minpp1 using siRNA-mediated gene silencing. Our data showing resistance to apoptosis in Minpp1 knocked down cells point to an involvement of Minpp1 in ER stress-mediated apoptosis. Studies on Minpp1 knockdown cells relied upon longer incubations of up to 72 h with siRNA transfection reagents. This has caused an overall high background apoptosis in siRNA only transfected control cells perhaps due to toxicity induced by multiple pools of Minpp1 siRNA. Therefore, the interpretation of these results requires caution. Nevertheless, it can be safely stated that there exists a molecular link between Minpp1 expression and cellular levels of stress.
In conclusion, Minpp1 exhibits characteristics of a stress-responsive molecule during ER stress-induced apoptosis, irrespective of the underlying mechanisms involved. Further research is necessary to delineate the role for Minpp1 in stress-adaptive pathways, which can be exploited in therapeutic applications to treat cellular disorders.
Electronic supplementary material
Below is the link to the electronic supplementary material.
{kind=link}
Analysis of Minpp1 expression in different mammalian cell lines during apoptosis. MC3T3-E1, PanC1, and SW480 cells were treated with cisplatin (25–200 mM) for 24 h. One set of cells was processed for SDS-PAGE and western blotting for Minpp1 expression as detailed in the “Materials and methods.” The western blots shown are representative experiments repeated independently at least three times on all three cell lines with various treatments. Another set of cells was processed for the assessment of apoptosis following acridine orange/ethidium bromide staining. A minimum of 200 cells were counted from five random fields and percentage of cells undergoing apoptosis was determined as detailed in “Materials and methods” section. The bars indicate the means ± S.E. from three different experiments performed in triplicates. (GIF 217 kb)
Acknowledgments
This work was supported in part by funds from NASA-EPSCoR, NSF-EPSCoR/P3 Asset II, Arkansas Space Grant Consortium, and Kathleen Thomsen-Hall Charitable Foundation. We also acknowledge partial support from the Center for Advanced Surface Engineering, under the National Science Foundation Grant No. IIA-1457888 and the Arkansas EPSCoR Program, ASSET III. Dr. Kilaparty thanks UALR Tech Launch for a graduate assistantship. The authors thank Dr. Waqar Majeed, UALR Nanotechnology Center, for assistance with siRNA technique and Ms. Andrea Harris and Ms. Aarthi Kannan of the University of Arkansas for Medical Sciences for assistance with flow cytometry and preparation of the manuscript, respectively.
Compliance with ethical standards
Conflict of Interest
The authors declare that they have no conflict of interest.
References
- Adams JM. Ways of dying: multiple pathways to apoptosis. Genes Dev. 2003;17:2481–95. doi: 10.1101/gad.1126903. [DOI] [PubMed] [Google Scholar]
- Agarwal R, Mumtaz H, Ali N. Role of inositol polyphosphates in programmed cell death. Mol Cell Biochem. 2009;328:155–65. doi: 10.1007/s11010-009-0085-6. [DOI] [PubMed] [Google Scholar]
- Agarwal R, Hassen S, Ali N. Changes in cellular levels of inositol polyphosphates during apoptosis. Mol Cell Biochem. 2010;345:61–8. doi: 10.1007/s11010-010-0560-0. [DOI] [PubMed] [Google Scholar]
- Ali N, Craxton A, Shears SB. Hepatic Ins(1,3,4,5)P4 3-phosphatase is compartmentalized inside endoplasmic reticulum. J Biol Chem. 1993;268:6161–7. [PubMed] [Google Scholar]
- Ali N, Craxton A, Sumner M, Shears SB. Effects of aluminium on the hepatic inositol polyphosphate phosphatase. Biochem J. 1995;305(Pt 2):557–61. doi: 10.1042/bj3050557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ, Irvine RF. Inositol trisphosphate, a novel second messenger in cellular signal transduction. Nature. 1984;312:315–21. doi: 10.1038/312315a0. [DOI] [PubMed] [Google Scholar]
- Berridge MJ, Irvine RF. Inositol phosphates and cell signalling. Nature. 1989;341:197–205. doi: 10.1038/341197a0. [DOI] [PubMed] [Google Scholar]
- Boehning D, Patterson RL, Sedaghat L, Glebova NO, Kurosaki T, Snyder SH. Cytochrome c binds to inositol (1,4,5) trisphosphate receptors, amplifying calcium-dependent apoptosis. Nat Cell Biol. 2003;5:1051–61. doi: 10.1038/ncb1063. [DOI] [PubMed] [Google Scholar]
- Boehning D, Patterson RL, Snyder SH. Apoptosis and calcium: new roles for cytochrome c and inositol 1,4,5-trisphosphate. Cell Cycle. 2004;3:252–4. doi: 10.4161/cc.3.3.705. [DOI] [PubMed] [Google Scholar]
- Caffrey JJ, Hidaka K, Matsuda M, Hirata M, Shears SB. The human and rat forms of multiple inositol polyphosphate phosphatase: functional homology with a histidine acid phosphatase up-regulated during endochondral ossification. FEBS Lett. 1999;442:99–104. doi: 10.1016/S0014-5793(98)01636-6. [DOI] [PubMed] [Google Scholar]
- Chakraborty A, Koldobskiy MA, Bello NT, Maxwell M, Potter JJ, Juluri KR, Maag D, Kim S, Huang AS, Dailey MJ, Saleh M, Snowman AM, Moran TH, Mezey E, Snyder SH. Inositol pyrophosphates inhibit Akt signaling, thereby regulating insulin sensitivity and weight gain. Cell. 2010;143:897–910. doi: 10.1016/j.cell.2010.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Brandizzi F. IRE1: ER stress sensor and cell fate executor. Trends Cell Biol. 2013;23:547–55. doi: 10.1016/j.tcb.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi H, Tiller GE, Dasouki MJ, Romano PR, Wang J, O’Keefe RJ, Puzas JE, Rosier RN, Reynolds PR. Multiple inositol polyphosphate phosphatase: evolution as a distinct group within the histidine phosphatase family and chromosomal localization of the human and mouse genes to chromosomes 10q23 and 19. Genomics. 1999;56:324–36. doi: 10.1006/geno.1998.5736. [DOI] [PubMed] [Google Scholar]
- Chi H, Yang X, Kingsley PD, O’Keefe RJ, Puzas JE, Rosier RN, Shears SB, Reynolds PR. Targeted deletion of Minpp1 provides new insight into the activity of multiple inositol polyphosphate phosphatase in vivo. Mol Cell Biol. 2000;20:6496–507. doi: 10.1128/MCB.20.17.6496-6507.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chipuk JE, Green DR. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 2008;18:157–64. doi: 10.1016/j.tcb.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho J, King JS, Qian X, Harwood AJ, Shears SB. Dephosphorylation of 2,3-bisphosphoglycerate by MIPP expands the regulatory capacity of the Rapoport-Luebering glycolytic shunt. Proc Natl Acad Sci U S A. 2008;105:5998–6003. doi: 10.1073/pnas.0710980105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craxton A, Ali N, Shears SB. Comparison of the activities of a multiple inositol polyphosphate phosphatase obtained from several sources: a search for heterogeneity in this enzyme. Biochem J. 1995;305(Pt 2):491–8. doi: 10.1042/bj3050491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craxton A, Caffrey JJ, Burkhart W, Safrany ST, Shears SB. Molecular cloning and expression of a rat hepatic multiple inositol polyphosphate phosphatase. Biochem J. 1997;328(Pt 1):75–81. doi: 10.1042/bj3280075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criollo A, Galluzzi L, Maiuri MC, Tasdemir E, Lavandero S, Kroemer G. Mitochondrial control of cell death induced by hyperosmotic stress. Apoptosis. 2007;12:3–18. doi: 10.1007/s10495-006-0328-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deegan S, Saveljeva S, Gorman AM, Samali A. Stress-induced self-cannibalism: on the regulation of autophagy by endoplasmic reticulum stress. Cell Mol Life Sci. 2013;70:2425–41. doi: 10.1007/s00018-012-1173-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desagher S, Martinou JC. Mitochondria as the central control point of apoptosis. Trends Cell Biol. 2000;10:369–77. doi: 10.1016/S0962-8924(00)01803-1. [DOI] [PubMed] [Google Scholar]
- Estaquier J, Tanaka M, Suda T, Nagata S, Golstein P, Ameisen JC. Fas-mediated apoptosis of CD4+ and CD8+ T cells from human immunodeficiency virus-infected persons: differential in vitro preventive effect of cytokines and protease antagonists. Blood. 1996;87:4959–66. [PubMed] [Google Scholar]
- Gimm O, Chi H, Dahia PL, Perren A, Hinze R, Komminoth P, Dralle H, Reynolds PR, Eng C. Somatic mutation and germline variants of MINPP1, a phosphatase gene located in proximity to PTEN on 10q23.3, in follicular thyroid carcinomas. J Clin Endocrinol Metab. 2001;86:1801–5. doi: 10.1210/jcem.86.4.7419. [DOI] [PubMed] [Google Scholar]
- Greenberg EF, Lavik AR, Distelhorst CW. Bcl-2 regulation of the inositol 1,4,5-trisphosphate receptor and calcium signaling in normal and malignant lymphocytes: potential new target for cancer treatment. Biochim Biophys Acta. 2014;1843:2205–10. doi: 10.1016/j.bbamcr.2014.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmis C, Blechner C, Lin H, Schweizer M, Mayr GW, Nielsen P, Windhorst S. Malignant H1299 tumour cells preferentially internalize iron-bound inositol hexakisphosphate. Biosci Rep. 2013;33(5):815–22. doi: 10.1042/BSR20130079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz CA. ER stress signaling and the BCL-2 family of proteins: from adaptation to irreversible cellular damage. Antioxid Redox Signal. 2007;9:2345–55. doi: 10.1089/ars.2007.1793. [DOI] [PubMed] [Google Scholar]
- Hetz C, Martinon F, Rodriguez D, Glimcher LH. The unfolded protein response: integrating stress signals through the stress sensor IRE1alpha. Physiol Rev. 2011;91:1219–43. doi: 10.1152/physrev.00001.2011. [DOI] [PubMed] [Google Scholar]
- Hidaka K, Kanematsu T, Caffrey JJ, Takeuchi H, Shears SB, Hirata M. The importance to chondrocyte differentiation of changes in expression of the multiple inositol polyphosphate phosphatase. Exp Cell Res. 2003;290:254–64. doi: 10.1016/S0014-4827(03)00337-9. [DOI] [PubMed] [Google Scholar]
- Holczer M, Marton M, Kurucz A, Banhegyi G, Kapuy O. A comprehensive systems biological study of autophagy-apoptosis crosstalk during endoplasmic reticulum stress. Biomed Res Int. 2015;2015:319589. doi: 10.1155/2015/319589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idziorek T, Estaquier J, De Bels F, Ameisen JC. YOPRO-1 permits cytofluorometric analysis of programmed cell death (apoptosis) without interfering with cell viability. J Immunol Methods. 1995;185:249–58. doi: 10.1016/0022-1759(95)00172-7. [DOI] [PubMed] [Google Scholar]
- Irvine RF, Schell MJ. Back in the water: the return of the inositol phosphates. Nat Rev Mol Cell Biol. 2001;2:327–38. doi: 10.1038/35073015. [DOI] [PubMed] [Google Scholar]
- Ivanova H, Vervliet T, Missiaen L, Parys JB, De Smedt H, Bultynck G. Inositol 1,4,5-trisphosphate receptor-isoform diversity in cell death and survival. Biochim Biophys Acta. 2014;1843:2164–83. doi: 10.1016/j.bbamcr.2014.03.007. [DOI] [PubMed] [Google Scholar]
- Jin HR, Liao Y, Li X, Zhang Z, Zhao J, Wang CZ, Huang WH, Li SP, Yuan CS, Du W. Anticancer compound Oplopantriol A kills cancer cells through inducing ER stress and BH3 proteins Bim and Noxa. Cell Death Dis. 2014;5 doi: 10.1038/cddis.2014.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanekura K, Ma X, Murphy JT, Zhu LJ, Diwan A. ‘IRE1 prevents endoplasmic reticulum membrane permeabilization and cell death under pathological conditions’. Sci Signal. 2015;8:ra62. doi: 10.1126/scisignal.aaa0341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan K, Holcombe RF, Jain SK, Alvarez-Hernandez X, Chervenak R, Wolf RE, Glass J. Evidence for the induction of apoptosis by endosulfan in a human T-cell leukemic line. Mol Cell Biochem. 2000;205:53–66. doi: 10.1023/A:1007080910396. [DOI] [PubMed] [Google Scholar]
- Kaufman RJ (2002) Orchestrating the unfolded protein response in health and disease. J Clin Invest 110:1389–98 [DOI] [PMC free article] [PubMed]
- Kilaparty SP, Ali N (2013) Changes in expression of multiple inositol polyphosphate phosphatase1 (Minpp1) under apoptotic and cellular stress conditions. In Experimental Biology. FASEB J 27:834.14
- Kilaparty SP, Singh A, Baltosser WH, Ali N. Computational analysis reveals a successive adaptation of multiple inositol polyphosphate phosphatase 1 in higher organisms through evolution. Evol Bioinformatics Online. 2014;10:239–50. doi: 10.4137/EBO.S18948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2008;7:1013–30. doi: 10.1038/nrd2755. [DOI] [PubMed] [Google Scholar]
- Lindholm D, Wootz H, Korhonen L. ER stress and neurodegenerative diseases. Cell Death Differ. 2006;13:385–92. doi: 10.1038/sj.cdd.4401778. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−delta delta C(T)) method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Menniti FS, Oliver KG, Putney JW, Jr, Shears SB. Inositol phosphates and cell signaling: new views of InsP5 and InsP6. Trends Biochem Sci. 1993;18:53–6. doi: 10.1016/0968-0004(93)90053-P. [DOI] [PubMed] [Google Scholar]
- Mizzen LA, Welch WJ. Characterization of the thermotolerant cell. I. Effects on protein synthesis activity and the regulation of heat-shock protein 70 expression. J Cell Biol. 1988;106:1105–16. doi: 10.1083/jcb.106.4.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima S, Kitamura M. Bidirectional regulation of NF-kappaB by reactive oxygen species: a role of unfolded protein response. Free Radic Biol Med. 2013;65:162–74. doi: 10.1016/j.freeradbiomed.2013.06.020. [DOI] [PubMed] [Google Scholar]
- Naon D, Scorrano L. At the right distance: ER-mitochondria juxtaposition in cell life and death. Biochim Biophys Acta. 2014;1843:2184–94. doi: 10.1016/j.bbamcr.2014.05.011. [DOI] [PubMed] [Google Scholar]
- Nogimori K, Hughes PJ, Glennon MC, Hodgson ME, Putney JW, Jr, Shears SB. Purification of an inositol (1,3,4,5)-tetrakisphosphate 3-phosphatase activity from rat liver and the evaluation of its substrate specificity. J Biol Chem. 1991;266:16499–506. [PubMed] [Google Scholar]
- Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol. 2003;4:552–65. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- Puthalakath H, O’Reilly LA, Gunn P, Lee L, Kelly PN, Huntington ND, Hughes PD, Michalak EM, McKimm-Breschkin J, Motoyama N, Gotoh T, Akira S, Bouillet P, Strasser A. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell. 2007;129:1337–49. doi: 10.1016/j.cell.2007.04.027. [DOI] [PubMed] [Google Scholar]
- Reed JC. Cytochrome c: can’t live with it—can’t live without it. Cell. 1997;91:559–62. doi: 10.1016/S0092-8674(00)80442-0. [DOI] [PubMed] [Google Scholar]
- Romano PR, Wang J, O’Keefe RJ, Puzas JE, Rosier RN, Reynolds PR. Hipper, a phosphatase of the endoplasmic reticulum with a role in chondrocyte maturation. J Cell Sci. 1998;111(Pt 6):803–13. doi: 10.1242/jcs.111.6.803. [DOI] [PubMed] [Google Scholar]
- Ron D. Translational control in the endoplasmic reticulum stress response. J Clin Invest. 2002;110:1383–8. doi: 10.1172/JCI0216784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–29. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Schonthal AH. Pharmacological targeting of endoplasmic reticulum stress signaling in cancer. Biochem Pharmacol. 2013;85:653–66. doi: 10.1016/j.bcp.2012.09.012. [DOI] [PubMed] [Google Scholar]
- Shears SB, Ali N, Craxton A, Bembenek ME. Synthesis and metabolism of bis-diphosphoinositol tetrakisphosphate in vitro and in vivo. J Biol Chem. 1995;270:10489–97. doi: 10.1074/jbc.270.18.10489. [DOI] [PubMed] [Google Scholar]
- Shore GC, Papa FR, Oakes SA. Signaling cell death from the endoplasmic reticulum stress response. Curr Opin Cell Biol. 2011;23:143–9. doi: 10.1016/j.ceb.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soo KY, Atkin JD, Farg M, Walker AK, Horne MK, Nagley P. Bim links ER stress and apoptosis in cells expressing mutant SOD1 associated with amyotrophic lateral sclerosis. PLoS One. 2012;7 doi: 10.1371/journal.pone.0035413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stentz R, Osborne S, Horn N, Li AW, Hautefort I, Bongaerts R, Rouyer M, Bailey P, Shears SB, Hemmings AM, Brearley CA, Carding SR. A bacterial homolog of a eukaryotic inositol phosphate signaling enzyme mediates cross-kingdom dialog in the mammalian gut. Cell Rep. 2014;6:646–56. doi: 10.1016/j.celrep.2014.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006;7:880–5. doi: 10.1038/sj.embor.7400779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadarajan S, Tanaka K, Smalley JL, Bampton ET, Pellecchia M, Dinsdale D, Willars GB, Cohen GM. Endoplasmic reticulum membrane reorganization is regulated by ionic homeostasis. PLoS One. 2013;8 doi: 10.1371/journal.pone.0056603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verfaillie T, Rubio N, Garg AD, Bultynck G, Rizzuto R, Decuypere JP, Piette J, Linehan C, Gupta S, Samali A, Agostinis P. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 2012;19:1880–91. doi: 10.1038/cdd.2012.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wali JA, Rondas D, McKenzie MD, Zhao Y, Elkerbout L, Fynch S, Gurzov EN, Akira S, Mathieu C, Kay TW, Overbergh L, Strasser A, Thomas HE. The proapoptotic BH3-only proteins Bim and Puma are downstream of endoplasmic reticulum and mitochondrial oxidative stress in pancreatic islets in response to glucotoxicity. Cell Death Dis. 2014;5 doi: 10.1038/cddis.2014.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Olberding KE, White C, Li C. Bcl-2 proteins regulate ER membrane permeability to luminal proteins during ER stress-induced apoptosis. Cell Death Differ. 2011;18:38–47. doi: 10.1038/cdd.2010.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windhorst S, Lin H, Blechner C, Fanick W, Brandt L, Brehm MA, Mayr GW. Tumour cells can employ extracellular Ins(1,2,3,4,5,6)P(6) and multiple inositol-polyphosphate phosphatase 1 (MINPP1) dephosphorylation to improve their proliferation. Biochem J. 2013;450:115–25. doi: 10.1042/BJ20121524. [DOI] [PubMed] [Google Scholar]
- Wyse B, Ali N, Ellison DH. Interaction with grp58 increases activity of the thiazide-sensitive Na-Cl cotransporter. Am J Physiol Ren Physiol. 2002;282:F424–30. doi: 10.1152/ajprenal.0028.2001. [DOI] [PubMed] [Google Scholar]
- Yang L, Reece JM, Cho J, Bortner CD, Shears SB. The nucleolus exhibits an osmotically regulated gatekeeping activity that controls the spatial dynamics and functions of nucleolin. J Biol Chem. 2008;283:11823–31. doi: 10.1074/jbc.M800308200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- York JD, Odom AR, Murphy R, Ives EB, Wente SR. A phospholipase C-dependent inositol polyphosphate kinase pathway required for efficient messenger RNA export. Science. 1999;285:96–100. doi: 10.1126/science.285.5424.96. [DOI] [PubMed] [Google Scholar]
- Yu J, Leibiger B, Yang SN, Caffery JJ, Shears SB, Leibiger IB, Barker CJ, Berggren PO. Cytosolic multiple inositol polyphosphate phosphatase in the regulation of cytoplasmic free Ca2+ concentration. J Biol Chem. 2003;278:46210–8. doi: 10.1074/jbc.M303743200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Analysis of Minpp1 expression in different mammalian cell lines during apoptosis. MC3T3-E1, PanC1, and SW480 cells were treated with cisplatin (25–200 mM) for 24 h. One set of cells was processed for SDS-PAGE and western blotting for Minpp1 expression as detailed in the “Materials and methods.” The western blots shown are representative experiments repeated independently at least three times on all three cell lines with various treatments. Another set of cells was processed for the assessment of apoptosis following acridine orange/ethidium bromide staining. A minimum of 200 cells were counted from five random fields and percentage of cells undergoing apoptosis was determined as detailed in “Materials and methods” section. The bars indicate the means ± S.E. from three different experiments performed in triplicates. (GIF 217 kb)