
Figure 1.
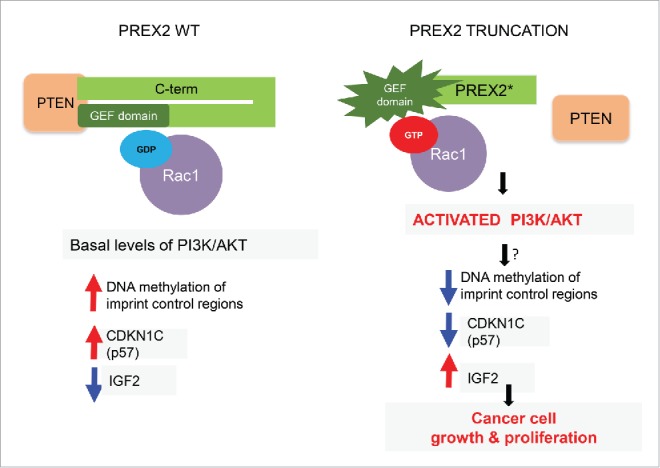
Mechanistic consequences of PREX2 truncating mutations. In wild-type cells PREX2 is bound to PTEN and its GEF domain is kept in an inactive state through association with its C-terminus (C-term), with resultant basal levels of downstream signaling events such as GDP-loaded RAC1. In cells and tumors with truncating PREX2 mutations, PREX2 is unable to bind to PTEN and its intramolecular inhibitory conformation is lost, resulting in activation of its GEF activity. This results in GTP loading of RAC1, which in turn leads to direct activation of the PI3K/AKT pathway. These alterations (through a still unknown link) perturb DNA methylation at a critical genomic imprint control region leading to downregulation of a key cell cycle regulator CDKN1C (also known as p57) and upregulation of IGF2, which favors accelerated cell growth and proliferation. GDP, guanosine diphosphate; GEF, guanine nucleotide exchange factor; GTP, guanosine triphosphate; IGF2, insulin like growth factor 2; PREX2, phosphatidylinositol-3, 4, 5-triphosphate-dependent Rac exchange factor 2; PTEN, phosphatase and tensin homolog; RAC1, ras-related C3 botulinum toxin substrate 1.