
Figure 3. Genomic classical and non-classical transciptional activities of E2-stimultated wild-type ER and ligand-independent mutant ER and potential novel therapeutics.
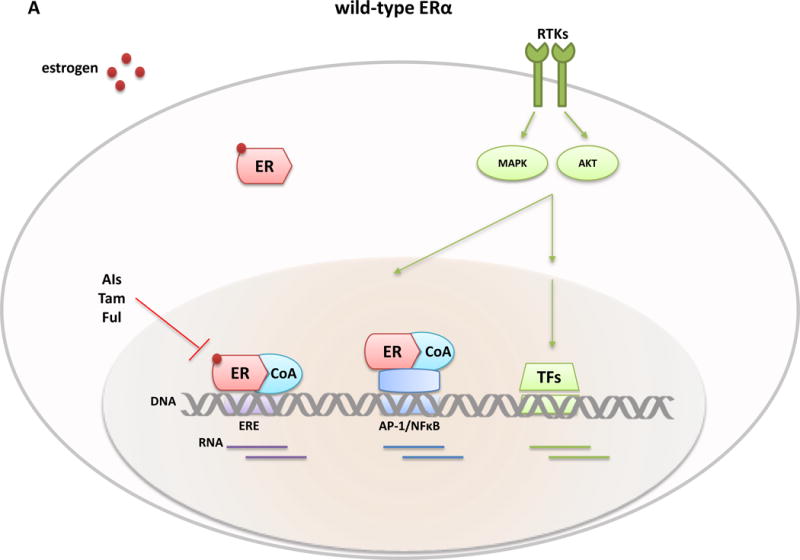
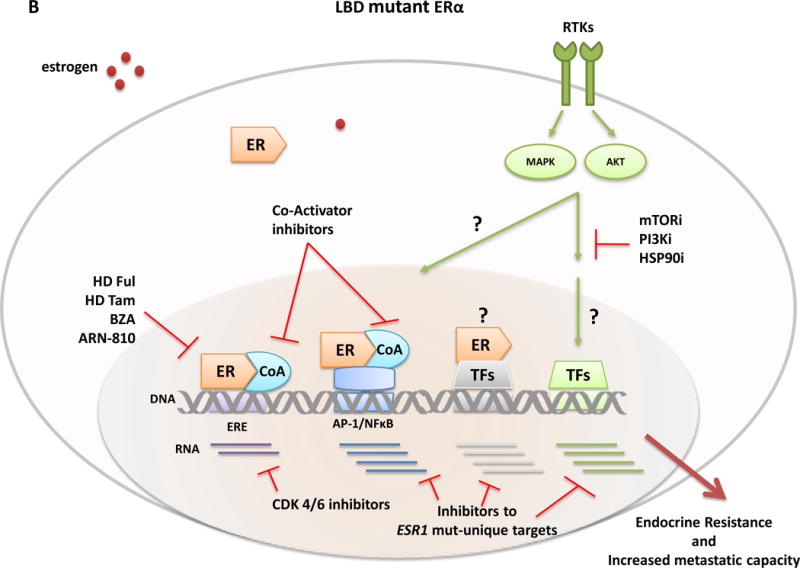
Classical ER transcription activity is mediated by ER binding to DNA at the consensus ERE sites, while in the non-classical ER activity mode, the receptor is tethered to other transcriptional factors such as AP-1 or NFkB and regulates gene expression from their sites. The transcriptional activity of ER and other TFs is further modulated by RTKs and other signaling pathway-induced kinases (e.g., MAPK and AKT) that phosphorylate ER, its coregulatory proteins (e.g., CoA), and other components of the transcriptional machinery to control the overall transcriptional program needed for tumor development and progression. Differential expression profiles between wild-type and mutant ERs suggest an augmented non-classical genomic activity of ER mutants, which may enhance RTK signaling and the metastatic capacity of the tumor cells. Breast cancer cells with wild-type ER (A) are largely sensitive to standard endocrine therapies (aromatase inhibitors, tamoxifen, fulvestrant). In contrast, ESR1 LBD mutant cells (B) display an endocrine resistant phenotype and current findings suggest the need of alternative therapeutic strategies such as higher doses of fulvestrant or tamoxifen, more potent or mutant specific SERMS or SERDs (bazedoxifene and ARN-810, respectively), agents targeting ER co-activators and ER gene products such as, cyclinD1 blockade by CDK4/6 inhibitors, or other signaling pathways and kinase inhibitors (e.g., mTOR, PI3Kinase, and HSP90 inhibitors) alone or in combination with ER inhibitors. Abbreviation: AIs, aromatase inhibitors; BZD, bazedoxifen; CoA, co-activator; ER, estrogen receptor; ERE, estrogen responsive element; Ful, fulvestrant; i, inhibitor; LBD, ligand-binding domain; RTKs, tyrosine kinase receptors; Tam, tamoxifen; TFs, transcription factors.