
Fig. 6.
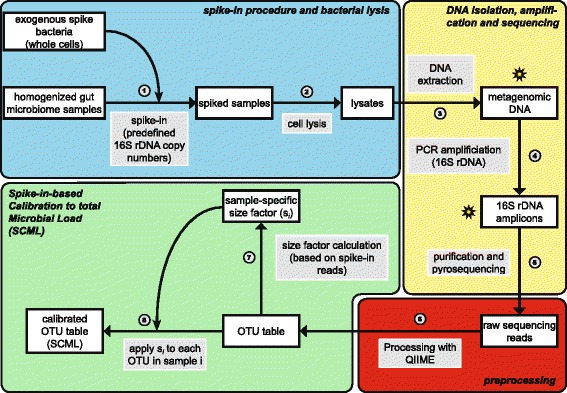
Procedural overview of proposed spike-in procedure and the spike-in-based calibration to total microbial load (SCML). The overview is divided into four sections: spike-in procedure and bacterial lysis (blue), DNA isolation, amplification and sequencing (yellow), pre-processing (red) and the actual spike-in-based calibration to microbial load (green). White-filled boxes depict procedural intermediates, while grey-filled boxes depict the different procedural steps. Each step is numbered. In the first step (1) whole cells of exogenous spike bacteria corresponding to a fixed number of 16S rDNA copies are added to homogenized microbiome samples. Bacterial lysis is performed on the resulting spiked samples (2). Metagenomic DNA is extracted from the lysates (3) and PCR amplified using 16S rDNA specific primers (4), creating 16S rDNA amplicons. These amplicons are purified and pyrosequencing is performed (5). The resulting raw read counts are pre-processed with QIIME (quality filtering, demultiplexing and closed reference OTU picking) to generate OTU read count tables (6). Based on the read counts associated with single or multiple reference spike-in bacteria, a size factor si for each sample i is calculated and applied to each OTU of this particular sample i (8, see methods section). This leads to an OTU read count table calibrated to differences in microbial load. These read counts can be utilized to more accurately assess changes between different samples. All depicted steps are described in detail in the methods section. Stars indicate points in the procedure at which qPCR is performed to identify possible errors in DNA isolation (metagenomic DNA) or PCR amplification (16S rDNA amplicons).