

Abstract
Next-generation sequencing technologies have revolutionized gene discovery in patients with intellectual disability (ID) and led to an unprecedented expansion in the number of genes implicated in this disorder. We discuss the strategies that have been used to identify these novel genes for both syndromic and nonsyndromic ID and highlight the phenotypic and genetic heterogeneity that underpin this condition. Finally, we discuss the future of defining the genetic etiology of ID, including the role of whole-genome sequencing, mosaicism, and the importance of diagnostic testing in ID.
Keywords: intellectual disability, next generation sequencing, de novo mutations, copy number variations
Introduction
Next-generation sequencing (NGS) has revolutionized the field of medical genomics. The ability to generate large quantities of DNA sequence data in a high-throughput manner at a fraction of the cost of Sanger sequencing has enabled researchers to study more patients and greater genomic DNA real estate than ever before. Not surprisingly, there has been a concomitant and unprecedented success in novel gene discovery for many Mendelian and sporadic disorders, including intellectual disability (ID).
ID is characterized clinically by below-average abilities in intellectual functioning, mental abilities, and adaptive behavior that limit everyday life skills and learning, and presents before the age of 18 years.1 The presentation of ID is broad and ranges from mild (IQ < 70) to profound (IQ < 30) and may occur either in isolation (nonsyndromic) or in conjunction with other clinical manifestations as part of a recognizable syndrome. Cumulatively, ID occurs in 1 to 3% of the population and has an important impact on health care systems.2
A genetic basis for ID is well established. Cytogenetic alterations including aneuploidies, duplications, deletions, translocations, and inversions visible on standard karyotype account for approximately 5% of cases,3 and smaller copy number variants (CNVs) detected by array comparative genomic hybridization (CGH) explain a further 14% of cases.4 Among the monogenic causes, there are more than 100 genes on the X chromosome and more than 50 autosomal recessive genes in which mutations cause ID.5 6 More recently, and primarily due to the widespread use of next-generation sequencing, the importance of de novo mutations in the pathogenesis of ID has become apparent.
In this review, we will touch on gene discovery for syndromic forms of ID, but will focus primarily on the novel causes of sporadic ID and particularly the role of de novo mutations, as well as describe various study designs. We discuss our increasing appreciation for both the phenotypic and genetic heterogeneity of ID and explore future approaches toward defining the molecular etiology of unexplained ID.
Next-Generation Sequencing Approaches for Gene Discovery
The power of next-generation sequencing lies not only in its high-throughput, rapid and cost-effective nature but also in its versatility. Researchers now have the ability to sequence from one to thousands of genes in an individual, based primarily on the enrichment process used to sequence the target regions of interest (Table 1). Currently, whole-exome sequencing (WES) is the most widely employed, and several strategies for gene discovery have been implemented (Fig. 1). These approaches can be broadly defined into two categories: (1) the phenotype-first approach that groups and investigates individuals based on a particular phenotype and (2) the genotype-first approach that groups patients based on the underlying genetic etiology. The phenotype-first approach has been applied to many ID syndromes, while the genotype-first or unbiased approach has been far more widely implemented for nonsyndromic forms of ID.
Table 1. Tools for next-generation sequencing.
| Whole-exome sequencing: The capture and sequencing of all coding regions in the genome in an individual |
| Targeted resequencing: Used to capture from one to many hundreds of genes, using various probe enrichment strategies. Early on, microarrays with probes complimentary to target sequence were used, but have been since replaced by probes in solution that are more versatile and less laborious |
| Whole-genome sequencing: No enrichment process is used; the complete DNA sequence of an individual is sequenced. The high resolution of this approach allows variants to be called across the entire genome, copy number variants >20 bp can be detected, and structural variants such as insertions or inversions may also be identified |
| Somatic mosaicism: Requires the detection of a small number of cells that carry a mutation; all of the aforementioned technologies can be used to detect mosaics, but are limited by the depth of sequencing. New approaches, such as molecular inversion probes and molecular tagging events, allow the detection of these low-frequency variants50 |
Fig. 1.
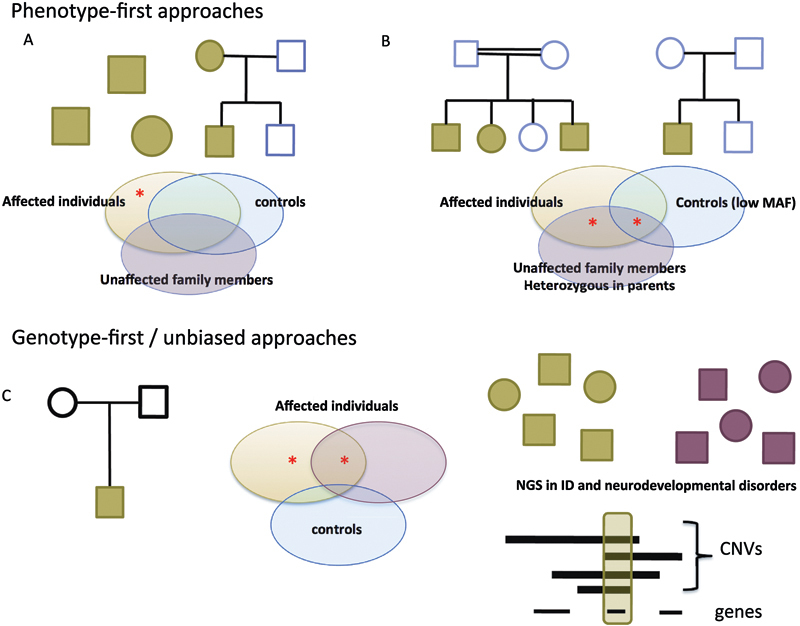
Next-generation sequencing approaches to gene discovery. (A) Autosomal dominant or sporadic “phenotype-first approaches” rely on the identification of mutations (red star) in a common gene in patients with the same syndrome (green) that are absent in controls (blue) and unaffected family members (purple). (B) Similarly, autosomal recessive syndromic forms of ID are caused by either homozygous or compound heterozygous mutations in the same gene in affected individuals. Parents are heterozygous for these mutations and the mutation may be present in control populations at a very low frequency (MAF, minor allele frequency). (C) Genotype-first or unbiased approaches employ WES in trios to identify de novo mutations in an affected individual. Novel ID genes are identified by recurrence, either by targeted or WES studies in individuals with ID (green) or other neurodevelopmental disorders (red) or overlap with known copy number variations (CNVs). ID, intellectual disability; WES, whole-genome sequencing.
Phenotype-First Approaches
The premise of this approach is to identify a commonly mutated gene across individuals with the same recognizable clinical syndrome (Fig. 1A). Kabuki syndrome is a rare autosomal dominant disorder characterized by dysmorphic facial features, multiple congenital abnormalities, and mild–moderate ID, and was one of the first syndromic forms of ID to be solved using WES.7 Ng and colleagues7 sequenced the exomes of ten unrelated probands and identified truncating mutations in seven of the ten probands in MLL2/KMT2D. Follow-up Sanger DNA sequencing identified KMT2D mutations in 2 of the remaining probands and in 26/43 additional cases, showing that KMT2D is the major cause of Kabuki syndrome.
Since then, several syndromic forms of sporadic or milder autosomal dominant forms of ID have been identified. WES in two boys with ID and strikingly similar facial dysmorphisms revealed the same de novo missense mutation in PACS1.8 Four additional patients with this same recurrent PACS1 de novo mutation have since been identified among a large cohort of patients with a variety of childhood developmental disorders, though unfortunately the phenotype was not described.9
Autosomal recessive forms of ID are also amenable to WES using the “common phenotypes approach,” where often individuals with the same phenotype may be in a single family (Fig. 1B). Examples include the discovery of mutations in UBE3B in blepharophimosis–ptosis–intellectual disability10 and WDR62 in polymicrogyria, microcephaly, and ID,11 12 as well as PIGV and PIGO in hyperphosphatasia mental retardation syndrome.13 14
Although effective for gene discovery, phenotype-first approaches are limited by the recognition of multiple individuals with the same clinical features, which can be difficult for rare disorders. Identification of similarly affected individuals within a family is easier, but familial cases, too, are rare, particularly for autosomal dominant conditions. Recessive inheritance is more prevalent in populations where consanguinity is common, and many recessive genes have been identified in these populations, though often only in a single family.15 Moreover, ID more often occurs in the absence of clinical features that can prospectively be recognized as part of a syndrome, necessitating an unbiased, genome-wide approach to gene discovery.
Genotype-First Approaches
The genetic heterogeneity of sporadic, nonsyndromic ID, along with the fact that the causative mutations are usually de novo, has been the significant limiting factor for gene discovery in the past. It is hypothesized that de novo mutations in any one of hundreds of genes may be causative, but until recently, the affordable high-throughput methods needed to identify causative mutations were not available. The trio-WES approach that entails simultaneous sequencing of the proband and parents detects all coding de novo mutations in an individual and has rapidly facilitated gene discovery. This approach was first applied in ten patients with moderate–severe ID, and de novo mutations in six individuals were concluded to be likely pathogenic. Three were mutations in known ID genes, while the remaining mutations were concluded to be likely pathogenic based on gene function and evolutionary genic constraint.16 Subsequent studies in cohorts of 41 to 100 patients identified pathogenic mutations in 16 to 32% in known ID genes, with the majority being de novo mutations.16 17 18 19
The trio approach to identify de novo mutations is very efficient because, on average, each individual carries one to two de novo sequence variants per exome. A major challenge comes with determining which (if any) de novo change is pathogenic, as many of the genes with de novo changes identified in cohort-based WES studies are plausible candidates for this disorder based on gene function and/or evolutionary conservation. A successful approach to tackle this challenge is to identify those genes that are recurrently mutated in patients with the same or similar phenotype. One way to do this is to combine results from different WES studies to identify genes that are recurrently mutated across cohorts (Table 2). The list of genes in Table 2 illustrates the approaches that can be used to establish a candidate genes' role in ID, which include targeted resequencing or WES in cohorts of patients with ID and/or other neurodevelopmental disorders, or identifying overlap with pathogenic CNVs (Fig. 1C).
Table 2. Recurrently mutated genes in ID.
| Gene | Vissers et al16 (n = 10) | de Ligt et al18 (n = 100) | Rauch et al19 (n = 50) | Hamdan et al17 (n = 41) | Total (n = 201) | Additional NGS studies (mutations/cohort size) (study) |
Overlap CNV |
|---|---|---|---|---|---|---|---|
| CHD2 | 1 | 1 | 2 | Targeted: EE (6/500)26
WES: ASD (4/3,486)29 |
Yes 15q26.1del |
||
| COL4A3BP | 1 | 1 | 2 | WES: developmental disorders (3/1,133a)9 | No | ||
| DEAF1 | 1 | 1 | 2 | Targeted: ID (2/2,300)20 | No | ||
| DYNC1H1 | 1 | 1 | 2 | WES and targeted Cortical malformations (9/178)52 |
No | ||
| GATAD2B | 1 | 1 | 2 | Targeted: ID (1/80)53 | Yes 1q21.3del |
||
| KCNH1 | 1 | 1 | 2 | WES: Temple-Baraitser syndrome (6/6)48 | No | ||
| SCN2A | 1 | 3 | 4 | Targeted and WES ASD (6/2,508)25 EE (2/264)54 (5/500)26 Developmental disorder (7/1,133)9 |
No | ||
| SETBP1 | 1 | 1 | 2 | Targeted ID/ DD/ASD (9/4716)33 |
Yes 18q12.3 |
||
| TRIO | 2 | 2 | WES: Developmental disorder (3/1,133a)9 | No |
Abbreviations: ASD, autism spectrum disorder; DD, developmental delay; EE, epileptic encephalopathy; ID, intellectual disability; NGS, next-generation sequencing; WES, whole-exome sequencing.
87% of children in this cohort had ID/DD.
Resequencing in Intellectual Disability
Targeted resequencing is a rapid and cost-effective way to screen many patients in validation studies. For instance, de novo DEAF1 mutations were detected in two individuals using WES.16 19 Subsequent targeted resequencing of more than 2,300 individuals with unexplained ID identified an additional two patients with de novo mutations. All patients had severe ID, profound speech impairment, and behavioral problems, and knockout Deaf1 mice showed learning and memory deficits as well as increased anxiety.20 De novo mutations in KAT6A were recently described in six patients with ID.21 All mutations were detected as part of diagnostic WES from multiple centers, highlighting the importance of WES in diagnostic testing and the power of collaborative studies to identify new genetic etiologies. All patients presented with subtle facial dysmorphisms, microcephaly and/or craniosynostosis, cardiac defects, hypotonia, and poor feeding that may constitute a novel syndrome. In both of these studies, it is noteworthy that patients with pathogenic mutations in the new ID gene shared many clinical features that could be retrospectively classified as new syndromes. Future studies will likely expand the repertoire of new ID syndromes and facilitate molecular diagnosis.
Resequencing in Other Neurodevelopmental Disorders
Patients with ID frequently exhibit comorbid features including epilepsy, autism spectrum disorders (ASDs), and brain malformations. Perhaps not surprisingly this phenotypic overlap is reflected in the shared genetic etiology of these conditions and resequencing studies in these overlapping conditions can be leveraged to validate candidate ID genes. For instance, SCN2A mutations were first described in patients with benign familial neonatal seizures,22 and later in more severe forms of epilepsy including epileptic encephalopathy.23 24 More recently, WES studies in patients with ASD showed a significant enrichment of patients with de novo mutations,25 and SCN2A has the highest recurrence rate of de novo mutations in ID (Table 1). SCN2A is hence associated with a broad spectrum of phenotypes; at the mild end of the spectrum, patients present with benign familial neonatal seizures, outcome is good, and developmental delay is rare, while at the other end of the spectrum patients may have intractable seizures, profound ID, and ASD.
CHD2, GRIN2B, SYNGAP1, SETD5, and TRIO are further examples of genes that have been detected in large cohorts of patients with ID, autism, and epilepsy.9 25 26 27 28 29 These three features—epilepsy, autism, and ID—can co-occur in an individual, and in other cases only one or two of the features may be present. It is likely that the cohort to which each individual is recruited is heavily influenced by the clinicians, health care centers, and researchers with whom these individuals and their families interact. In the future, it will be important to perform careful and comprehensive phenotyping in a genotype-first manner to determine the true phenotypic range and to establish genotype–phenotype correlations, which may have implications for molecular diagnostic testing.
Overlap with Copy Number Variations
One of the classic approaches to gene discovery has been refining the critical interval of overlapping microdeletions in several individuals to identify a specific causative gene. Known ID genes including MEF2C (5q14.3del),30 EHMT1 (Kleefstra syndrome 9q34.3del),31 and MBD5 (2q23.1del)32 were identified in this manner, and many of the more recently discovered ID genes overlap with known microdeletions (Table 2). Assuming loss-of-function mutations and microdeletions cause disease by haploinsufficiency, WES can now be used to rapidly facilitate single causative gene identification within microdeletions. For instance, mapping of the critical interval for 3p25.3 microdeletions indicated SETD5 was the only overlapping deleted gene, while WES in 301 patients with ID identified two intragenic de novo SETD5 mutations. All six patients had similar phenotypes including characteristic facial dysmorphism, regardless of the nature of the mutation (deletion or point), suggesting SETD5 is the causative gene for 3p25.3 microdeletions.28
In a much larger study, CNV data from 29,000 patients with developmental delay and approximately 20,000 healthy individuals combined with WES de novo mutation calls from approximately 1,900 patients with ASD, ID, congenital heart defects, and schizophrenia were integrated to identify 26 candidate genes for ID. Targeted resequencing in 4,716 patients with ID/DD or ASD identified two novel ID genes, SETBP1 (18q12.3del), and ZMYND11 (10p15.3del). As previously, patients with either microdeletions or intragenic mutations showed characteristic features, suggesting haploinsufficiency of these genes is sufficient to cause ID.33
Interestingly, while SETBP1 loss-of-function mutations cause ID, gain-of-function mutations cause Schinzel–Giedion syndrome, a more severe phenotype characterized by multiple congenital anomalies, severe ID, and death in infancy.34 This observation illustrates the unsuitability of microdeletion mapping to detect hypermorphs, where gain-of-function mutations do not have the same effects on protein function. It is conceivable that single causative genes reside within microduplications and may act in a similar gain-of-function manner. However, many gain-of-function mutations have an activating effect on protein function, and it is not known whether an increase in gene dosage would act in this activating manner. This reciprocal analysis, integrating putative gain-of-function mutations from WES data with critical interval mapping in microduplications, might yield novel ID genes that cause disease, but would require large numbers of patients to be informative.
Genetic Heterogeneity
Each of the recently identified genes for ID accounts for less than 1% of cases, highlighting the extreme genetic heterogeneity of ID (Table 2). As a result, large numbers of patients are required to detect recurrently mutated genes. This was exemplified in a recent WES study of more than 1,000 children with undiagnosed developmental disorders, 87% of who had ID/DD.9 This study led to the identification of 12 new genes, but none had mutations in more than four patients. Intriguingly, in four of these genes—PCGF2, COL4A3BP, PPP2R1A, and PPP2R5D, the exact same recurrent missense mutation was identified. These mutations are likely to act in a gain-of-function manner and highlight the importance of not purely focusing on loss-of-function mutations in neurodevelopmental disorders, which has been the trend in recent large-scale WES studies. Overall, these results suggest that there are many more genes to be discovered for ID.
Autosomal Recessive Forms of Intellectual Disability
Nonsyndromic autosomal recessive ID is primarily limited to consanguineous populations. In Western populations, where consanguinity is uncommon, families with multiple affected siblings are rare, reaching only 6% in one well-characterized population.35 Autosomal recessive inheritance was explored in four large ID WES studies (Table 1), but recessive mutations in known ID genes were detected only in a single patient who inherited a rare LRP2 variant, and had a second de novo LRP2 mutation.18 This apparent lack of recessive inheritance suggests that this form of inheritance is rare in sporadic ID in nonconsanguineous populations.
The Future: Tackling Undiagnosed Intellectual Disability
In a well-studied cohort of patients with severe ID, pathogenic CNVs account for approximately 12% of cases and WES identified the causative genic mutation in a further 27% of individuals.36 Despite these extensive studies, the etiology of the majority of patients with severe ID (61% in this cohort) remains unexplained. It is likely that an appreciable proportion of unexplained ID cases are due to de novo mutations in coding regions of genes that rarely cause ID. Many of these cases will retrospectively be solved once additional mutations in a gene are identified in other patients. In other cases, the mutation may have been missed by WES, and alternative approaches may be required to identify these alterations.
Whole-Genome Sequencing
WGS is a completely unbiased approach to genetic diagnosis, and has the power to identify mutations almost anywhere in the genome, including noncoding DNA. In a recent study, WGS was performed in 50 undiagnosed individuals in whom both array CGH and WES failed to identify a pathogenic cause. Overall a positive molecular diagnosis was identified in 21/50 individuals. Of these, 14 were protein-coding mutations in known ID genes—13 de novo and 1 compound heterozygous inheritance—that were not previously detected by WES. Moreover, seven CNVs or structural variants were identified that were not identified by microarray due to size limitations of array CGH.36
These results illustrate the power and utility of WGS in molecular diagnosis for ID over any existing NGS technology. Unlike WES, there is no enrichment step in the preparation of the DNA sequencing library. This prevents sequence bias for regions that are difficult to “capture” and/or amplify (especially GC-rich regions) and more uniform coverage across the genome. The identification of numerous coding mutations that were not previously detected by WES exemplifies this advantage, though it would have been interesting to know explicitly why these variants went undetected by WES, that is, whether there was no coverage, or if variants were not called in the analysis. WGS also offers the most high-resolution map of structural variation in the genome; using the uniform depth of coverage data, changes in copy number for regions >20 bp can be detected, and the “mate pair” data can be used to detect structural variants to a single base–pair resolution. For instance, a partial duplication of TENM3 was detected by CNV analysis by WGS, and further investigation of the discordant mate pairs revealed that this TENM3 sequence was inserted in the known ID gene, IQSEC2. This insertion could be mapped to a single base–pair resolution and RNA studies confirmed the formation of an IQSEC2–TENM3 fusion gene that is likely the cause of ID in this individual.36
The widespread implementation of WGS is, at the moment, limited by cost. However, as sequencing costs continue to decline, the use of this technology in research, and eventually clinically, will become more extensive. While the advantages of WGS are apparent, the ability to scrutinize the genome at such high resolution will present its own unique set of challenges. Per genome, 18 to 82 de novo variants have been reported36 37 and sorting pathogenic from benign will be a challenging task. Our interpretation of variants in coding regions has become increasingly effective. On the contrary, the noncoding regions have been virtually ignored and even the studies that performed WGS in ID and ASD cohorts reported only on the coding regions of the genome.36 38 39 Projects such as the Encyclopedia of DNA Elements (ENCODE) and the Roadmap Epigenomics aim to detect all gene regulatory elements of the genome in various cell and tissue types.40 41 These comprehensive maps of the genome, coupled with further studies of how these elements function, will be important in the future to understand the role of noncoding DNA regions in ID.
Mosaicism
Somatic mosaicism arises when a mutation occurs during prenatal development of the human embryo, resulting in only a subset of cells carrying the mutation. Somatic mutations are a well-known cause of cancer42 and several sporadic overgrowth syndromes.43 44 Somatic mosaic mutations have also been described in neurodevelopmental disorders, primarily in patients with specific brain malformations.45 46 47
The extent to which somatic mosaicism may play a role in nonlesional ID is not known. There are certainly instances of somatic mosaicism in syndromic ID; Cornelia de Lange syndrome is a characteristic ID syndrome caused by mutations in one of five genes, with the majority in NIPBL. In 10 of 13 patients without a detectable genetic cause, mosaic mutations were identified in the buccal cells of individuals that could not be detected in lymphocytes, suggesting that 23% of cases are caused by somatic mutation.48 Moreover, in the aforementioned WGS ID study, three putatively pathogenic changes were likely to be mosaic with a range of 20 to 22%.36 Traditionally, DNA sequencing studies are performed on DNA isolated from the lymphocytes of blood; the aforementioned studies suggest that studies in a different tissue such as skin or buccal swabs may be appropriate in undiagnosed cases. Large-scale studies in undiagnosed patients will be important to determine whether somatic mutations play a significant role in the etiology of ID.
The timing of somatic mosaicism may also pose interesting implications for genotype–phenotype correlations for certain genes. For instance, Temple-Baraitser syndrome is a developmental disorder characterized by ID, epilepsy, and several other characteristic features that is caused by de novo mutations in KCNH1. However, intriguingly two mothers were mosaic carriers (10 and 27%) of pathogenic variants, both had mild epilepsy but were otherwise normal.49 Thus, a somatic mutation may cause only a partial phenotype depending on the proportion and location of cells that carry the mutant allele. Moreover, two KCNH1 mutations were identified in ID patient cohorts; in one case some of the features of Temple-Baraitser were present, while the other patient seems to lack some of the classic features, despite the patients carrying the same mutation.17 19 Whether the phenotypic heterogeneity seen in this condition, and indeed many other diagnosed cases of ID, is due to genetic background, mosaicism, and/or environmental variations will be an intriguing line of investigation in the future.
Next-Generation Sequencing in Diagnostics
Clinical diagnostic testing using NGS is offered by many companies for patients with ID in the form of multigene panels or WES. Multigene panels offer targeted sequencing for mutations in genes known to cause ID. Some panels focus on X-linked ID genes, while others provide sequencing for autosomal causes. The number of genes in each panel varies from dozens to several hundred depending on the specific test. WES is also clinically available and can be considered for patients with ID. Some have argued that early testing by WES will save money and time by ending the “diagnostic odyssey” earlier than if multiple serial tests are performed. In a recent study of children with neurodevelopmental disorders, 34 of 85 (40%) children received a molecular diagnosis via WES (n = 33) or WGS (n = 1); for acutely ill children, the diagnostic rate was a remarkable, 73% by WES (11/15). The cost of prior negative tests was $19,100 per family, and it was estimated that the diagnosis could have been made 6 to 7 years earlier if WES or WGS had been performed at the onset of symptoms.50 While additional studies are needed to confirm these findings, given the genetic heterogeneity of ID, it is clear that gene panels or WES in patients with ID will be the most efficient path to an early diagnosis.
Concluding Remarks
In the past 5 years, we have seen unprecedented success in defining the genetic etiology of ID by NGS and hundreds of new genes have been discovered. However, the majority of cases of ID remain unexplained, and many more genes are still likely to be identified. It has been estimated that WES in 1,000 children has only 5 to 10% power to detect a haploinsufficient developmental disorder-associated gene, and the likelihood of identifying gain-of-function recurrently mutated gene is even less.9 This exemplifies the need, and indeed the power, of combining data from many different sources, including WES, targeted resequencing, and array CGH, in both a research and diagnostic setting, and necessitates the standardization and development of large databases to store this data in a collaborative manner. Only by the pooling of the wealth of genomic data will novel ID genes be discovered in a rapid and effective manner in the future. Moreover, many patients will have causative mutations undetectable by traditional WES; the role of mosaicism, structural variants and CNVs detected only with WGS, noncoding variations, and inherited variants will be important and exciting focuses for future research. Finally, at the moment, novel gene discovery is outpacing our understanding of the biological mechanisms that go awry when these genes are mutated. Developing high-throughput and strategic approaches to study pathogenic mechanisms will be important in the coming years, and will ultimately facilitate the development of novel therapeutic options.
References
- 1.American Psychiatric Association . Washington, DC: American Psychiatric Association; 1994. Diagnostic and statistical manual of mental disorders: DSM-IV. Vol. 4th; p. 886. [Google Scholar]
- 2.Leonard H, Wen X. The epidemiology of mental retardation: challenges and opportunities in the new millennium. Ment Retard Dev Disabil Res Rev. 2002;8(3):117–134. doi: 10.1002/mrdd.10031. [DOI] [PubMed] [Google Scholar]
- 3.Stankiewicz P, Beaudet A L. Use of array CGH in the evaluation of dysmorphology, malformations, developmental delay, and idiopathic mental retardation. Curr Opin Genet Dev. 2007;17(3):182–192. doi: 10.1016/j.gde.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 4.Cooper G M, Coe B P, Girirajan S. et al. A copy number variation morbidity map of developmental delay. Nat Genet. 2011;43(9):838–846. doi: 10.1038/ng.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Brouwer A P, Yntema H G, Kleefstra T. et al. Mutation frequencies of X-linked mental retardation genes in families from the EuroMRX consortium. Hum Mutat. 2007;28(2):207–208. doi: 10.1002/humu.9482. [DOI] [PubMed] [Google Scholar]
- 6.Musante L, Ropers H H. Genetics of recessive cognitive disorders. Trends Genet. 2014;30(1):32–39. doi: 10.1016/j.tig.2013.09.008. [DOI] [PubMed] [Google Scholar]
- 7.Ng S B, Bigham A W, Buckingham K J. et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet. 2010;43(9):790–793. doi: 10.1038/ng.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schuurs-Hoeijmakers J H, Oh E C, Vissers L E. et al. Recurrent de novo mutations in PACS1 cause defective cranial-neural-crest migration and define a recognizable intellectual-disability syndrome. Am J Hum Genet. 2012;91(6):1122–1127. doi: 10.1016/j.ajhg.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deciphering Developmental Disorders Study . Large-scale discovery of novel genetic causes of developmental disorders. Nature. 2015;519(7542):223–228. doi: 10.1038/nature14135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Basel-Vanagaite L, Dallapiccola B, Ramirez-Solis R. et al. Deficiency for the ubiquitin ligase UBE3B in a blepharophimosis-ptosis-intellectual-disability syndrome. Am J Hum Genet. 2012;91(6):998–1010. doi: 10.1016/j.ajhg.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murdock D R, Clark G D, Bainbridge M N. et al. Whole-exome sequencing identifies compound heterozygous mutations in WDR62 in siblings with recurrent polymicrogyria. Am J Med Genet A. 2011;155A(9):2071–2077. doi: 10.1002/ajmg.a.34165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bilguvar K, Oztürk A K, Louvi A. et al. Whole-exome sequencing identifies recessive WDR62 mutations in severe brain malformations. Nature. 2010;467(7312):207–210. doi: 10.1038/nature09327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krawitz P M, Schweiger M R, Rödelsperger C. et al. Identity-by-descent filtering of exome sequence data identifies PIGV mutations in hyperphosphatasia mental retardation syndrome. Nat Genet. 2010;42(10):827–829. doi: 10.1038/ng.653. [DOI] [PubMed] [Google Scholar]
- 14.Krawitz P M, Murakami Y, Hecht J. et al. Mutations in PIGO, a member of the GPI-anchor-synthesis pathway, cause hyperphosphatasia with mental retardation. Am J Hum Genet. 2012;91(1):146–151. doi: 10.1016/j.ajhg.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Najmabadi H, Hu H, Garshasbi M. et al. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature. 2011;478(7367):57–63. doi: 10.1038/nature10423. [DOI] [PubMed] [Google Scholar]
- 16.Vissers L E, de Ligt J, Gilissen C. et al. A de novo paradigm for mental retardation. Nat Genet. 2010;42(12):1109–1112. doi: 10.1038/ng.712. [DOI] [PubMed] [Google Scholar]
- 17.Hamdan F F, Srour M, Capo-Chichi J M. et al. De novo mutations in moderate or severe intellectual disability. PLoS Genet. 2014;10(10):e1004772. doi: 10.1371/journal.pgen.1004772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Ligt J, Willemsen M H, van Bon B W. et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med. 2012;367(20):1921–1929. doi: 10.1056/NEJMoa1206524. [DOI] [PubMed] [Google Scholar]
- 19.Rauch A, Wieczorek D, Graf E. et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet. 2012;380(9854):1674–1682. doi: 10.1016/S0140-6736(12)61480-9. [DOI] [PubMed] [Google Scholar]
- 20.Vulto-van Silfhout A T, Rajamanickam S, Jensik P J. et al. Mutations affecting the SAND domain of DEAF1 cause intellectual disability with severe speech impairment and behavioral problems. Am J Hum Genet. 2014;94(5):649–661. doi: 10.1016/j.ajhg.2014.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tham E, Lindstrand A, Santani A. et al. Dominant mutations in KAT6A cause intellectual disability with recognizable syndromic features. Am J Hum Genet. 2015;96(3):507–513. doi: 10.1016/j.ajhg.2015.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heron S E, Crossland K M, Andermann E. et al. Sodium-channel defects in benign familial neonatal-infantile seizures. Lancet. 2002;360(9336):851–852. doi: 10.1016/S0140-6736(02)09968-3. [DOI] [PubMed] [Google Scholar]
- 23.Kamiya K, Kaneda M, Sugawara T. et al. A nonsense mutation of the sodium channel gene SCN2A in a patient with intractable epilepsy and mental decline. J Neurosci. 2004;24(11):2690–2698. doi: 10.1523/JNEUROSCI.3089-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ogiwara I, Ito K, Sawaishi Y. et al. De novo mutations of voltage-gated sodium channel alphaII gene SCN2A in intractable epilepsies. Neurology. 2009;73(13):1046–1053. doi: 10.1212/WNL.0b013e3181b9cebc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iossifov I, O'Roak B J, Sanders S J. et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515(7526):216–221. doi: 10.1038/nature13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carvill G L, Heavin S B, Yendle S C. et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet. 2013;45(7):825–830. doi: 10.1038/ng.2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lemke J R, Hendrickx R, Geider K. et al. GRIN2B mutations in West syndrome and intellectual disability with focal epilepsy. Ann Neurol. 2014;75(1):147–154. doi: 10.1002/ana.24073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuechler A, Zink A M, Wieland T. et al. Loss-of-function variants of SETD5 cause intellectual disability and the core phenotype of microdeletion 3p25.3 syndrome. Eur J Hum Genet. 2015;23(6):753–760. doi: 10.1038/ejhg.2014.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.O'Roak B J, Stessman H A, Boyle E A. et al. Recurrent de novo mutations implicate novel genes underlying simplex autism risk. Nat Commun. 2014;5:5595. doi: 10.1038/ncomms6595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zweier M, Gregor A, Zweier C. et al. Mutations in MEF2C from the 5q14.3q15 microdeletion syndrome region are a frequent cause of severe mental retardation and diminish MECP2 and CDKL5 expression. Hum Mutat. 2010;31(6):722–733. doi: 10.1002/humu.21253. [DOI] [PubMed] [Google Scholar]
- 31.Kleefstra T, Smidt M, Banning M J. et al. Disruption of the gene Euchromatin Histone Methyl Transferase1 (Eu-HMTase1) is associated with the 9q34 subtelomeric deletion syndrome. J Med Genet. 2005;42(4):299–306. doi: 10.1136/jmg.2004.028464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Talkowski M E, Mullegama S V, Rosenfeld J A. et al. Assessment of 2q23.1 microdeletion syndrome implicates MBD5 as a single causal locus of intellectual disability, epilepsy, and autism spectrum disorder. Am J Hum Genet. 2011;89(4):551–563. doi: 10.1016/j.ajhg.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Coe B P, Witherspoon K, Rosenfeld J A. et al. Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nat Genet. 2014;46(10):1063–1071. doi: 10.1038/ng.3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoischen A, van Bon B W, Gilissen C. et al. De novo mutations of SETBP1 cause Schinzel-Giedion syndrome. Nature genetics. 2010;42(6):483–485. doi: 10.1038/ng.581. [DOI] [PubMed] [Google Scholar]
- 35.Schuurs-Hoeijmakers J H, Vulto-van Silfhout A T, Vissers L E. et al. Identification of pathogenic gene variants in small families with intellectually disabled siblings by exome sequencing. J Med Genet. 2013;50(12):802–811. doi: 10.1136/jmedgenet-2013-101644. [DOI] [PubMed] [Google Scholar]
- 36.Gilissen C, Hehir-Kwa J Y, Thung D T. et al. Genome sequencing identifies major causes of severe intellectual disability. Nature. 2014;511(7509):344–347. doi: 10.1038/nature13394. [DOI] [PubMed] [Google Scholar]
- 37.Genome of the Netherlands Consortium . Whole-genome sequence variation, population structure and demographic history of the Dutch population. Nat Genet. 2014;46(8):818–825. doi: 10.1038/ng.3021. [DOI] [PubMed] [Google Scholar]
- 38.Yuen R K, Thiruvahindrapuram B, Merico D. et al. Whole-genome sequencing of quartet families with autism spectrum disorder. Nat Med. 2015;21(2):185–191. doi: 10.1038/nm.3792. [DOI] [PubMed] [Google Scholar]
- 39.Jiang Y H, Yuen R K, Jin X. et al. Detection of clinically relevant genetic variants in autism spectrum disorder by whole-genome sequencing. Am J Hum Genet. 2013;93(2):249–263. doi: 10.1016/j.ajhg.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chadwick L H. The NIH Roadmap Epigenomics Program data resource. Epigenomics. 2012;4(3):317–324. doi: 10.2217/epi.12.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Consortium E P; ENCODE Project Consortium. The ENCODE (ENCyclopedia Of DNA Elements) Project Science 20043065696636–640. [DOI] [PubMed] [Google Scholar]
- 42.Bamford S, Dawson E, Forbes S. et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br J Cancer. 2004;91(2):355–358. doi: 10.1038/sj.bjc.6601894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lindhurst M J, Sapp J C, Teer J K. et al. A mosaic activating mutation in AKT1 associated with the Proteus syndrome. N Engl J Med. 2011;365(7):611–619. doi: 10.1056/NEJMoa1104017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kurek K C, Luks V L, Ayturk U M. et al. Somatic mosaic activating mutations in PIK3CA cause CLOVES syndrome. Am J Hum Genet. 2012;90(6):1108–1115. doi: 10.1016/j.ajhg.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rivière J B, Mirzaa G M, O'Roak B J. et al. De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat Genet. 2012;44(8):934–940. doi: 10.1038/ng.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Poduri A, Evrony G D, Cai X. et al. Somatic activation of AKT3 causes hemispheric developmental brain malformations. Neuron. 2012;74(1):41–48. doi: 10.1016/j.neuron.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gleeson J G, Minnerath S, Kuzniecky R I. et al. Somatic and germline mosaic mutations in the doublecortin gene are associated with variable phenotypes. Am J Hum Genet. 2000;67(3):574–581. doi: 10.1086/303043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huisman S A, Redeker E J, Maas S M, Mannens M M, Hennekam R C. High rate of mosaicism in individuals with Cornelia de Lange syndrome. J Med Genet. 2013;50(5):339–344. doi: 10.1136/jmedgenet-2012-101477. [DOI] [PubMed] [Google Scholar]
- 49.Simons C, Rash L D, Crawford J. et al. Mutations in the voltage-gated potassium channel gene KCNH1 cause Temple-Baraitser syndrome and epilepsy. Nat Genet. 2015;47(1):73–77. doi: 10.1038/ng.3153. [DOI] [PubMed] [Google Scholar]
- 50.Soden S E, Saunders C J, Willig L K. et al. Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci Transl Med. 2014;6(265):265ra168. doi: 10.1126/scitranslmed.3010076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hiatt J B, Pritchard C C, Salipante S J, O'Roak B J, Shendure J. Single molecule molecular inversion probes for targeted, high-accuracy detection of low-frequency variation. Genome Res. 2013;23(5):843–854. doi: 10.1101/gr.147686.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Poirier K, Lebrun N, Broix L. et al. Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly. Nat Genet. 2013;45(6):639–647. doi: 10.1038/ng.2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Willemsen M H, Nijhof B, Fenckova M. et al. GATAD2B loss-of-function mutations cause a recognisable syndrome with intellectual disability and are associated with learning deficits and synaptic undergrowth in Drosophila. J Med Genet. 2013;50(8):507–514. doi: 10.1136/jmedgenet-2012-101490. [DOI] [PubMed] [Google Scholar]
- 54.Allen A S, Berkovic S F, Cossette P. et al. De novo mutations in epileptic encephalopathies. Nature. 2013;501(7466):217–221. doi: 10.1038/nature12439. [DOI] [PMC free article] [PubMed] [Google Scholar]