

Abstract
Lasso peptides are a new class of ribosomally synthesized and post-translationally modified peptides and thus far are only isolated from proteo- and actinobacterial sources. Typically, lasso peptide biosynthetic gene clusters encode enzymes for biosynthesis and export but not for tailoring. Here, we describe the isolation of the novel lasso peptide paeninodin from the firmicute Paenibacillus dendritiformis C454 and reveal within its biosynthetic cluster a gene encoding a kinase, which we have characterized as a member of a new class of lasso peptide-tailoring kinases. By employing a wide variety of peptide substrates, it was shown that this novel type of kinase specifically phosphorylates the C-terminal serine residue while ignoring those located elsewhere. These experiments also reveal that no other recognition motif is needed for efficient enzymatic phosphorylation of the C-terminal serine. Furthermore, through comparison with homologous HPr kinases and subsequent mutational analysis, we confirmed the essential catalytic residues. Our study reveals how lasso peptides are chemically diversified and sets the foundation for rational engineering of these intriguing natural products.
Keywords: Bacillus, biosynthesis, natural product, phosphorylation, post-translational modification, Firmicutes, Lasso peptide, RiPP, kinase
Introduction
Natural products have long played significant roles in science and medicine. Among them, ribosomally synthesized and post-translationally modified peptides (RiPPs)3 present an important superfamily that has rapidly grown in recent years and encompasses a diverse set of structures and bioactivities (1–3). Lasso peptides are a structurally interesting and pharmacologically relevant class of RiPPs with antimicrobial, antiviral, receptor antagonistic, or enzyme inhibitory activities (3, 4). All lasso peptides are characterized by their unique topology in which the C-terminal tail is threaded through an N-terminally formed macrolactam ring (Fig. 1, a–c) (3–5). To maintain this entropically demanding fold, the tail is trapped by means of steric hindrance, mediated by bulky side chains above and below the ring. Because of this arrangement, lasso peptides exhibit great resistance against chemical and proteolytic degradation as well as, in many cases, thermally induced unthreading (3, 6–12).
FIGURE 1.
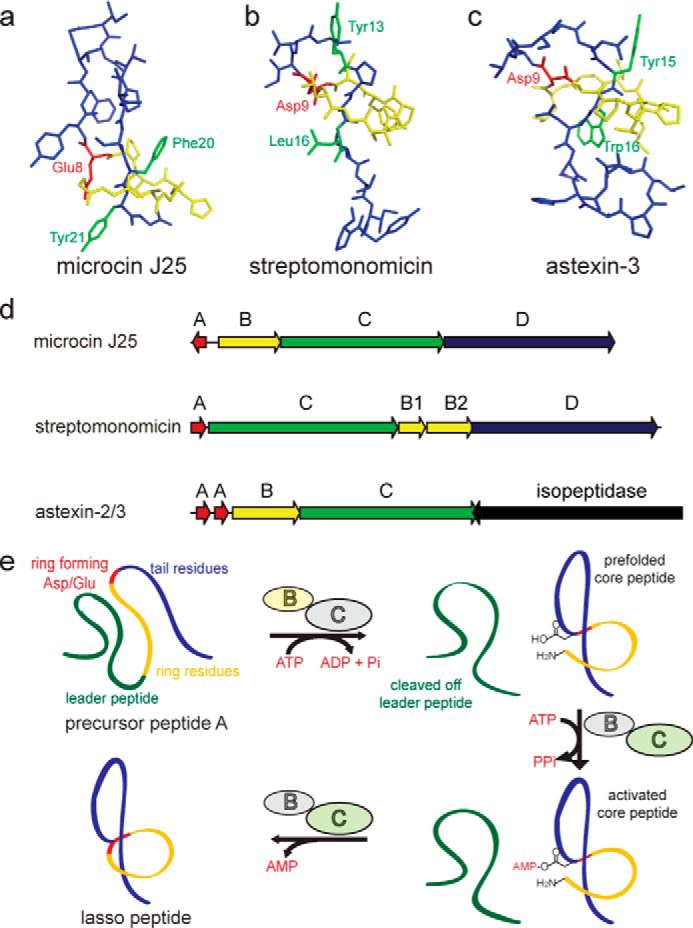
Representative structures of the class II lasso peptides microcin J25 (Protein Data Bank code 1Q71) (27) (a), streptomonomicin (Protein Data Bank code 2MW3) (21) (b), and astexin-3 (Protein Data Bank code 2M8F) (8) (c) are shown. The ring is shown in yellow, the tail in blue, the ring-forming Asp/Glu in red, and the plug side chains in green. d, biosynthetic gene clusters of the three shown lasso peptides (8, 18, 21). e, scheme of the suggested mechanism of lasso peptide biosynthesis (14).
The biosynthesis of lasso peptides has been studied previously, particularly for the microcin J25 (MccJ25) system (3, 13–19). This biosynthetic cluster contains four genes (Fig. 1d) as follows: mcjA encodes the precursor peptide; mcjB is an ATP-dependent cysteine protease; mcjC is an ATP-dependent asparagine synthetase homolog; and mcjD is an ABC transporter. Although McjD is only needed to export the mature antimicrobial compound (conferring immunity to the producing strain), McjB and McjC directly participate in the maturation of McjA into MccJ25. To accomplish this, McjB cleaves off the leader peptide, and McjC adenylates and then cyclizes the side chain of Glu-8 with the new N-terminal α-amino group of the core peptide, yielding a macrolactam ring with an isopeptide bond (Fig. 1e). The ATP-dependent character of McjB is postulated to mediate prefolding of the precursor peptide prior to macrocyclization. In some lasso peptide operons, such as those responsible for the biosynthesis of lariatin, lassomycin, streptomonomicin, sviceucin, and chaxapeptin as well as a family of putative lasso peptides from proteobacteria, the B protein is split into two separate open reading frames (ORFs) homologous to the N- and C-terminal domains of McjB (3, 7, 20–24). The N-terminal domain (B1) has been shown to bind the leader peptide with nanomolar affinity, although the C-terminal domain (B2) accommodates the cysteine protease catalytic triad (2, 3, 7). A series of recently reported proteobacterial biosynthetic gene clusters lacks the gene encoding an ABC transporter; instead, a neighboring inverted cluster encodes a lasso peptide-specific isopeptidase, among other enzymes (3, 6–8, 10, 11, 25, 26). Fig. 1 shows a selection of lasso peptide gene clusters with known arrangements.
Generally, all known lasso peptides carry a Gly, Ala, Cys, or Ser residue at position 1 and a ring-forming Glu or Asp residue at positions 7–9 (3, 4, 7, 10, 11, 21, 25). Their respective processing enzymes are highly specific for the nature and location of these residues (6, 9–11, 16, 28–30). A conserved Thr at the penultimate position of the leader peptide was also shown to be crucial for effective enzymatic processing of the precursor peptide (6, 9–11, 16, 28, 29).
Until now, all isolated lasso peptides originate from proteobacterial or actinobacterial sources (3, 4, 7, 31), although biosynthetic clusters from other bacterial phyla have been predicted (25). Here, we describe a family of lasso peptide biosynthetic gene clusters that are found primarily in Paenibacillus and Bacillus spp. and feature a novel cluster organization. A cluster from Paenibacillus dendritiformis C454 (32) was chosen for further investigation, and the topology of the predicted lasso peptide product (designated here as paeninodin) was strongly supported by MS2, ion mobility-mass spectrometry (IM-MS), and stability studies. Mutational analysis yielded insights into the biosynthesis of paeninodin. Interestingly, the studied gene cluster was found to contain not only the conserved genes encoding the precursor peptide, processing enzymes, and ABC transporter, but an additional gene coding for a putative kinase. Such a gene is notably absent from all characterized lasso peptide biosynthetic gene clusters (3, 4, 7, 25, 31).
Closer examination of this and homologous kinases in vitro and in vivo demonstrated their ability to selectively phosphorylate the hydroxyl side chain of the C-terminal Ser of the precursor peptide prior to processing by the B1, B2, and C homologs, thus yielding a phosphorylated lasso peptide. The most active kinase was further characterized via bioinformatics, mutational analysis, and kinetic studies, and its specificity was tested for a wide range of substrates. Finally, the position of phosphorylation was unambiguously determined by chemical esterification assays and NMR spectroscopy.
Experimental Procedures
Strains and Materials
P. dendritiformis C454 was generously provided to us by Eshel Ben-Jacob and Alin Finkelshtein (Tel Aviv University, Israel). Thermobacillus composti KWC4 (DSM no. 18247) was purchased from the German Collection of Microorganisms and Cell Cultures (DMSZ). Oligonucleotides of HPLC purity were purchased from Sigma. Precursor peptide derivatives were synthesized by Biomatik.
Bioinformatics Analysis
For genome mining studies, the Position-specific Iterated Basic Local Alignment Search Tool (PSI-BLAST) was employed. The split B2 protein from lariatin was used as a query with the following parameters: maximum target sequences of 500, expected threshold of 10, and PSI-BLAST threshold of 0.005. The sequence of each gene cluster was downloaded from GenBankTM. Alignment of precursor peptides was performed with the CLC Main Workbench 6.5 (CLC bio). Conserved domain motif matching was implemented using the MEME Suite. Kinase sequences were retrieved from the gene clusters and aligned in ClustalX2, with standard settings as implemented in Mega 6. Phylogenetic trees were constructed using the minimum evolution and neighbor-joining method, and the Poisson-correction amino acid model. Phylogeny was tested by bootstrapping with 1000 replications. C protein sequences were downloaded from GenBankTM, and phylogenetic trees were constructed in the same manner.
Cloning of the Paeninodin Gene Cluster
Whole genomic DNA of P. dendritiformis C454 was prepared using a standard protocol (FastDNA SPIN Kit, MP Biomedicals). The padeCAKB1B2D gene cluster (GenBankTM accession no. AHKH01000064.1, 23026 … 28726) was amplified by PCR from the genomic DNA (see supplemental Table S1 for primers) using Phusion High Fidelity DNA polymerase. Purified products and pET-41a(+) were digested with NdeI and XhoI and ligated with T4 DNA ligase to yield an expression plasmid containing the padeCAKB1B2D gene cluster. After transforming into Escherichia coli TOP10 cells, plasmids were prepared from individual transformants and confirmed by DNA sequencing. E. coli BL21(DE3) cells were retransformed with the expression construct and cultured in lysogeny broth (LB) medium containing kanamycin (50 μg/ml) at 37 °C with shaking.
Heterologous Production of Paeninodin and Paeninodin-OPO32− in E. coli BL21(DE3)
For heterologous expression of the lasso peptide biosynthetic gene cluster, M9 minimal medium (17.1 g/liter Na2HPO4·12 H2O, 3 g/liter KH2PO4, 0.5 g/liter NaCl, 1 g/liter NH4Cl, 1 ml/liter MgSO4 solution (2 m), 0.2 ml/liter CaCl2 solution (0.5 m), pH 7.0, 10 ml/liter glucose solution (40% w/v) and 2 ml/liter vitamin mix (see supplemental Table S2) were added after autoclaving) was used to screen for the best fermentation conditions. Different temperatures and durations (1–3 days at 37 °C or 3 days at 20 °C) were tested for lasso peptide production. Initially, 60 ml of LB medium supplemented with 50 μg/ml kanamycin was inoculated with E. coli BL21 cells carrying pET41a-padeCAKB1B2D and grown overnight at 37 °C with shaking. From these cultures, 2-liter baffled culture flasks, each containing 600 ml of M9 fermentation media, were inoculated to A600 ≈0.01. For fermentation at 37 °C, IPTG (final concentration of 0.05 mm) was used to induce the expression at A600 ≈0.5–0.6. For fermentation at 20 °C, cells were at first grown at 37 °C to A600 ≈0.4–0.5 in about 4.5 h and then slowly cooled to 20 °C for another 1 h. At A600 ≈0.5–0.6, the cells were induced with the same amount of IPTG and cultivated at 20 °C for 3 days. Cells were harvested by centrifugation (8000 rpm for 15 min at 4 °C) and resuspended by vortexing. Soluble components were extracted by shaking the resuspended pellet of each 600-ml culture in 60 ml of MeOH overnight. After centrifugation (6000 rpm for 20 min at 4 °C), the pellet extract was filtered, and the solvent was evaporated at 40 °C and reduced pressure. Dried extracts were resuspended in a total of 900 μl of 50% MeOH, clarified by centrifugation, and analyzed via LC-MS. Additionally, culture supernatants were extracted by use of XAD16 resin (Sigma, ∼7 g/liter supernatant). The XAD16-containing supernatant was stirred at room temperature for 1 h, and the resin was then removed by filtration. The XAD16 on the filter was washed several times with water and subsequently eluted with a total of 60 ml of MeOH per 600 ml of supernatant. Solvent was removed by evaporation at 40 °C and reduced pressure, and dried extracts were resuspended in a total of 900 μl of 50% MeOH and then clarified by centrifugation and analyzed via LC-MS.
Mass Spectrometric Analysis
MS analysis of each extract (100 μl per measurement) was performed with a high resolution LTQ FT Ultra (Thermo Fisher Scientific) connected to an Agilent 1100 HPLC system equipped with a Nucleosil 300-8 C18 column (125 × 2 mm, Macherey-Nagel). UV absorption was monitored at 215 nm. Analysis was performed by applying the following gradient of water, 0.1% trifluoroacetic acid (solvent A) and MeCN, 0.1% trifluoroacetic acid (solvent B) at a column temperature of 40 °C and flow rate of 0.2 ml/min: isocratic 2% B for 2 min, followed by linear increase from 2 to 30% B for 18 min, subsequent linear increase from 30 to 95% B for 15 min, and then isocratic 95% B for 2 min.
To fragment the target masses, collision-induced dissociation fragmentation studies within the linear ion trap were performed using on-line LC-MS. Different charged ions were selected for fragmentation based on their predominance. For the lasso peptides, [M + 2H]2+ ions were selected; for the precursor peptide and variants thereof, [M + 4H]4+ or [M + 5H]5+ ions were selected. The energy for fragmentation was set to 35% for every measurement performed.
LC-MS analyses of lasso peptide thermal stability, proteolytic assays of purified lasso peptides and various in vitro assays were also performed with a low resolution 1100 Series MSD (Hewlett-Packard) coupled with an Agilent 1100 HPLC system (Agilent Technologies). Separation was achieved with different C18 columns and gradients for the lasso or precursor peptides. In all cases, relative product yields were determined by integrating peak areas of the UV spectra or extracted ion chromatograms.
Purification of Paeninodin and Paeninodin-OPO32−
For the isolation of the unmodified and modified paeninodin, large scale fermentations (10 × 10 L M9 minimal medium) were performed at the optimal condition (3 days, 37 °C).
For paeninodin production, the construct pET41a-padeCAKB1B2D was expressed in E. coli BL21(DE3), and the lasso peptide was purified by two rounds of HPLC. For the first round, the following gradient of water, 0.045% formic acid (solvent A) and MeOH, 0.05% formic acid (solvent B) was used: linear increase from 20 to 30% B for 5 min, followed by linear increase from 30 to 95% B for 25 min, and then isocratic 95% B for 3 min. Paeninodin-containing elution fractions were confirmed by LC-MS and then pooled and evaporated at 40 °C and reduced pressure. The semipure lasso peptide was applied to a second round of purification using the following gradient of water, 0.1% trifluoroacetic acid (solvent C) and MeCN, 0.1% trifluoroacetic acid (solvent D): linear increase from 10 to 35% D for 30 min, followed by linear increase from 35 to 95% D for 2 min and then isocratic 95% D for 3 min. The pure paeninodin lasso peptide was obtained with a yield of ∼0.08 mg/liter.
For paeninodin-OPO32− production, the construct pET41a-padeCA-thcoK-padeB1B2D was expressed in E. coli BL21(DE3), and the lasso peptide was purified by two rounds of HPLC. For the first round, the following gradient of water, 0.045% formic acid (solvent A) and MeOH, 0.05% formic acid (solvent B) was used: linear increase from 20 to 30% B for 30 min, followed by linear increase from 30 to 95% B for 2 min and then isocratic 95% B for 3 min. Paeninodin-OPO32−-containing elution fractions were confirmed by LC-MS and then pooled and evaporated at 40 °C and reduced pressure. The semipure lasso peptide was applied to a second round of purification using the following gradient of water, 0.1% trifluoroacetic acid (solvent C) and MeCN, 0.1% trifluoroacetic acid (solvent D): linear increase from 20 to 50% D for 30 min, followed by linear increase from 50 to 95% D for 2 min and then isocratic 95% D for 3 min. The pure phosphorylated lasso peptide was obtained with a yield of ∼0.05 mg/liter.
Thermostability Assays
To test the thermostability of paeninodin and paeninodin-OPO32−, 10 μg of each lasso peptide was redissolved in 100 μl of H2O. The samples were incubated at 20, 35, 50, 65, 80, or 95 °C for 4 h and then compared with untreated samples via LC-MS.
Differentiation between Lasso and Branched-cyclic Topology
To study the topology of paeninodin, the lasso peptide was first incubated at 65 °C for 4 h and then analyzed by LC-MS. Under these conditions, the highest amount of branched-cyclic paeninodin can be obtained by thermally induced unthreading. Generally, the tail of a heat-sensitive lasso peptide may unthread from the macrolactam ring to yield a branched-cyclic isomer with the same mass but a different retention time. Hence, the observation of a new peak corresponding to the same mass but different retention time than the proposed lasso peptide is an indicator for this topology.
After incubating 10 μg of paeninodin at 65 °C for 4 h, the sample was directly applied to a carboxypeptidase Y assay (0.5 units of carboxypeptidase Y per reaction in MES buffer (50 mm MES,1 mm CaCl2, pH 7.0)). After incubation for 4 h at room temperature, samples were analyzed by LC-MS.
For ion mobility-mass spectrometric (IM-MS) analysis of paeninodin before and after incubation at 65 °C for 4 h in the presence of formic acid and the supercharging agent sulfolane, a hybrid quadrupole ion-mobility time-of-flight mass spectrometer (Waters Synapt G2-Si) equipped with an ESI source operated in positive-ion mode was employed. Before analysis, the samples were dissolved in 50% MeCN containing an additional 4% formic acid and 0.5% sulfolane as supercharging agent. The measurements were performed with previously described instrument settings (33).
Antibacterial Assays
The antibacterial activity of paeninodin and paeninodin-OPO32− was examined against Bacillus subtilis MR168, Micrococcus flavus ATCC 10240, Burkholderia rhizoxinica HKI454, Sphingobium japonicum UT26, Xanthomonas citri pv. mangiferaeindicae, Bacillus cereus, Bacillus megaterium WSH-002, and Bacillus amyloliquefaciens by spot-on-lawn assays. Soft agar was prepared by addition of 0.7% agar to liquid LB. Soft agar (10 ml, 48 °C) was then inoculated with 100 μl of overnight culture of each organism (A600 ≈0.01) and spread onto LB agar plates. Then, 10 ng of paeninodin or paeninodin-OPO32− was spotted on the soft agar plate, followed by incubation at 30 or 37 °C for 1–3 days.
Mutagenesis of the Paeninodin Gene Cluster
Mutagenesis of the paeninodin gene cluster was achieved by site-directed ligation-independent mutagenesis (SLIM) (34, 35). Mutagenesis of padeA and deletion of padeK were carried out using the standard SLIM protocol with pET41a-padeCAKB1B2D as template (see supplemental Table S3 for primers). Generally, after PCR with Phusion high fidelity DNA polymerase using GC buffer, the resulting two products (one generated with primers P1 and P4 and the other with P2 and P3) were digested with DpnI for 2 h at 37 °C and combined for further treatment according to the SLIM protocol. After transforming into E. coli TOP10, plasmids were prepared from individual transformants and confirmed by DNA sequencing. E. coli BL21(DE3) cells were retransformed with correctly sequenced mutant plasmids for subsequent expression, which was carried out with the same protocol as with pET41a-padeCAKB1B2D.
To complement the kinase deletion mutant, the padeK gene was cloned into the pACYCDuet-1 vector (see supplemental Table S3 for primers), which yielded the plasmid pACYC-padeK. pET41a-padeCAB1B2D and pACYC-padeK were then co-transformed into E. coli BL21(DE3). The resulting E. coli BL21(DE3) pET41a-padeCAB1B2D + pACYC-padeK strain was grown in LB medium containing kanamycin (50 μg/ml) and chloramphenicol (34 μg/ml) at 37 °C with shaking. Heterologous expression of this strain was carried out with the same protocol as with pET41a-padeCAKB1B2D.
To exchange padeK with thcoK in the paeninodin gene cluster, the thcoK gene was amplified by PCR from genomic DNA, and the vector backbone was amplified from pET41a-padeCAKB1B2D (see supplemental Tables S4 and S6, respectively, for primers). PCR products were purified and then ligated with Gibson Assembly Master Mix.
Mutagenesis of the kinase active site was accomplished via SLIM. The conserved His-Lys-Asp-Asp residues of PadeK and ThcoK were identified by sequence alignments and mutated using the standard SLIM protocol with the pET41a-padeCAKB1B2D and pET41a-padeCA-thcoK-padeB1B2D plasmids as templates, respectively (see supplemental Table S5 for primers).
Cloning, Expression, and Purification of PadeK, ThcoK, and PadeA
The padeK gene (GenBankTM accession number WP_006678397) was amplified by PCR from P. dendritiformis C454 genomic DNA (see supplemental Table S6 for primers). For the vector backbone, pETMBP-1a was mutated to exchange the tobacco etch virus protease site with an HRV-3C protease site and to exchange the NcoI restriction endonuclease site with an NdeI one. PCR products were digested with NdeI and XhoI, purified, and ligated with Gibson Assembly Master Mix.
The thcoK gene (GenBankTM accession number WP_015255975) was amplified by PCR from T. composti KWC4 genomic DNA (see supplemental Table S6 for primers). The pET-41a(+) plasmid was digested with NdeI and XhoI and purified. The backbone and insert DNA were ligated with Gibson Assembly Master Mix.
The padeA gene was amplified by PCR from P. dendritiformis C454 genomic DNA, and the vector backbone was amplified from pET-48b(+) (see supplemental Tables S7 and S5, respectively, for primers). The backbone amplicon was treated with DpnI and then ligated with the insert using Gibson Assembly Master Mix.
Large scale protein expression was carried out in 6 liters of LB medium. 2-Liter baffled culture flasks, each containing 600 ml of LB medium with 50 μg/ml kanamycin, were inoculated from an overnight culture to A600 ≈0.01. The cells were grown at 37 °C to A600 ≈0.4 in about 2 h, then cooled to 16 °C for another 1 h, and induced with 0.05 mm IPTG for 16 h. Cells were harvested by centrifugation (8000 rpm, 15 min, 4 °C), resuspended in 30 ml of HEPES buffer A (50 mm HEPES, 300 mm NaCl, 30 mm imidazole, 5% glycerol, pH 8.0), and lysed with a French press (SLM Aminco). Cellular debris was then removed by ultracentrifugation (17,000 rpm, 1 h, 4 °C), followed by filtration of the supernatant with a Filtropur S filter (0.2 μm). Ni-NTA affinity chromatography was performed on an ÄKTApurifier FPLC (Amersham Biosciences). The Ni-NTA column was equilibrated with HEPES buffer A, and after applying the sample (flow rate of 1 ml/min), the protein was eluted with increasing concentrations of imidazole in HEPES buffer A. Purified proteins were identified by SDS-PAGE, combined, concentrated, and buffer-exchanged into HEPES buffer B (10 mm HEPES, 150 mm NaCl, 5% glycerol, pH 7.5) in an Amicon Ultra filter. Size-exclusion chromatography was performed on the same FPLC system equipped with a HiLoad Superdex 200 16/60 prep grade column (GE Healthcare) equilibrated with HEPES buffer B. Pure protein fractions were flash-frozen in liquid nitrogen and stored at −80 °C until further use. ThcoK was handled at room temperature, as it tended to precipitate at lower temperatures.
For PadeK, the MBP tag was cleaved by incubation with HRV-3C protease (final concentration of 0.1 mg/ml) at 4 °C for 16 h. Final purification of PadeK was achieved by Ni-NTA affinity chromatography. The Ni-NTA column was equilibrated with HEPES buffer A, and after applying the sample, proteins in the column flow-through were analyzed by SDS-PAGE, combined, and concentrated. Pure protein fractions were flash-frozen in liquid nitrogen and stored at −80 °C until further use.
For PadeA, the Trx tag was cleaved by treatment with HRV-3C protease (final concentration of 0.1 mg/ml) for 16 h at 4 °C. After cleavage, the two last amino acids of the cleavage site (Gly and Pro) remained on the N terminus of PadeA, resulting in a GP-PadeA peptide. Final purification of GP-PadeA was achieved by preparative HPLC with the following gradient of water, 0.1% trifluoroacetic acid (solvent A) and MeCN, 0.1% trifluoroacetic acid (solvent B): linear increase from 10 to 50% B for 30 min, followed by linear increase from 50 to 95% B for 2 min and then isocratic 95% B for 3 min. The fractions containing GP-PadeA were identified by LC-MS and then pooled and dried at 40 °C and reduced pressure. The semipure peptide was subsequently applied to a second round of purification using the following gradient of water, 0.1% trifluoroacetic acid (solvent A) and MeCN, 0.1% trifluoroacetic acid (solvent B): linear increase from 20 to 50% B for 30 min, followed by linear increase from 50 to 95% B for 2 min and then isocratic 95% B for 3 min. Fractions containing GP-PadeA were pooled and lyophilized. The peptide was isolated as a white powder with a yield of ∼4 mg/liter and stored at −20 °C until further use.
To generate GP-PadeA derivatives, mutagenesis of the pET48b-padeA plasmid was carried out using the SLIM protocol (see supplemental Table S8 for primers). The mutant plasmids were then used to transform E. coli BL21(DE3) cells for subsequent expression. Heterologous expression of the variants was carried out with the same protocol as for pET48b-padeA.
Precursor Peptide Modification Assays
Pure GP-PadeA or variants thereof were redissolved in H2O to a final concentration of 2.5 mm for in vitro studies. In a typical peptide modification assay, ∼5 μm recombinant PadeK or ThcoK was incubated with 50 μm peptide in 50 μl of assay buffer (50 mm Tris·HCl, 5 mm MgCl2, 1 mm ATP, pH 8.0) at 37 °C for 4 h. Reactions were quenched by addition of TFA (final concentration of 5% (v/v)) and subjected to LC-MS analysis.
Determination of Kinetic Parameters for ThcoK
For time course measurements, a standard in vitro reaction (50 μl) in assay buffer containing 5 μm enzyme was run for varying durations (10, 20, 30, 45, 60, 120, 240, and 480 min) in triplicate. Reactions were quenched by addition of TFA (final concentration of 5% (v/v)) and subjected to LC-MS analysis. The kinetic parameters of ThcoK were determined by using different concentrations of purified GP-PadeA (25, 50, 75, 100, 200, 500, and 1000 μm) while keeping ATP, Mg2+, and kinase concentrations constant, and done in triplicate. Assays were quenched by addition of TFA (final concentration of 5% (v/v)) after 20 min, which was determined to lie within the linear conversion range. Samples were then subjected to LC-MS analysis. Kinetic parameters were calculated by fitting the data to the Michaelis-Menten equation in Microsoft Excel.
Scaled-up in Vitro Assays
To determine the position of the phosphate introduced by ThcoK, scaled-up in vitro assays were performed to obtain sufficient amounts of the enzymatically modified peptide. To simplify subsequent analysis and allow chemical synthesis of the adduct, PadeA(13–23) was employed. A total of 50 mg of PadeA(13–23) was purchased from Biomatik. A typical scaled-up assay reaction (30 ml) was performed in standard assay buffer and contained 20 mg of PadeA(13–23)-peptide (∼517 μm) and 2 mg of recombinant ThcoK (∼1.67 μm). The reaction was carried out at 37 °C for 16 h with shaking at 220 rpm.
Final purification of the resulting PadeA(13–23)-Ser-23-OPO32− was achieved by preparative HPLC with the following gradient of water, 0.1% trifluoroacetic acid (solvent A) and MeCN, 0.1% trifluoroacetic acid (solvent B): linear increase from 5 to 25% B for 30 min, followed by linear increase from 25 to 95% B for 2 min, and then isocratic 95% B for 5 min. The fractions containing PadeA(13–23)-Ser-23-OPO32− were identified by LC-MS, pooled, and lyophilized. PadeA(13–23)-Ser-23-OPO32− was isolated as ∼10 mg of white powder and stored at −20 °C until further use.
Esterification Assays
An esterification assay was performed to determine the site of phosphorylation. In a typical reaction, 10 μg of peptide was redissolved in 100 μl of 2 m methanolic HCl (prepared by careful dropwise addition of 160 μl of acetyl chloride to 1 ml of ice-cold methanol while stirring, with the reaction vessel on ice) and then shaken for 4 h at 25 °C and 500 rpm. The sample was lyophilized and redissolved in 100 μl of 20% MeCN and employed for LC-MS analysis. The unmodified PadeA(13–23), chemically synthesized PadeA(13–23)-Ser-23-OPO32− (Biomatik), and enzymatically produced PadeA(13–23)-Ser-23-OPO32− were tested separately. Esterification assays were also performed with unmodified and modified paeninodin isolated from heterologous expression cultures.
NMR Analysis of Synthetic and Enzymatically Generated PadeA(13–23)-Ser-23-OPO32−
For NMR measurements, 3.3 mg of enzymatically generated PadeA(13–23)-Ser-23-OPO32−, 3.2 mg of chemically synthesized PadeA(13–23)-Ser-23-OPO32− (Biomatik), and 3.0 mg of PadeA(13–23) (Biomatik) were dissolved in 0.2 ml of H2O/D2O (9:1), respectively. For 1H signal assignment, DQF-COSY, TOCSY, and NOESY spectra were recorded on a Bruker Avance 600 MHz spectrometer equipped with an inverse triple-resonance 1H-13C-15N probe with z-gradient. The temperature effect on the spectra was surveyed by recording 1H spectra in a range of 283–308 K. Consequently, all two-dimensional spectra were recorded at 298 K. TOCSY spectra were recorded with a mixing time of 80 ms, whereas for NOESY measurements mixing times of 150 and 300 ms were applied. Water suppression was achieved by using excitation sculpting. The one-dimensional 31P and two-dimensional 1H-31P long range correlation spectra of the phosphorylated compounds were recorded on a Bruker Avance III 500 MHz spectrometer with a broadband observation probe with z-gradient. For the latter, a standard pulse program for heteronuclear multiple quantum correlation was used (36), with a gradient ratio of 70:30:80 adjusted to the observation of 1H-31P correlation and a long range coupling constant of 7 Hz. One-dimensional 1H spectra were acquired with 65,536 data points, and two-dimensional 1H-1H spectra were collected using 4096 points in the F2 dimension and 512 increments in the F1 dimension. For two-dimensional spectra, 32 transients were used, with a relaxation delay of 3.0 s. The one-dimensional 31P spectra were recorded with 12,288 scans, and two-dimensional 1H-31P spectra were recorded with 128 transients and 128 increments in the F1 dimension. Chemical shifts of 1H were referenced to 2,2-dimethyl-2-silapentanesulfonic acid. All spectra were processed with Bruker TOPSPIN 3.2.
Results
Genome Mining Reveals a Novel Lasso Peptide Biosynthetic Gene Cluster Type in Firmicutes
As only lasso peptides originating from proteo- and actinobacteria have been characterized (3, 4, 7, 25, 31), we sought to uncover interesting gene clusters in other bacterial phyla. By employing our previously described B protein-centric genome mining approach (7), a new family of gene clusters predominantly found in Bacillus und Paenibacillus spp. was identified. These clusters differ from previously predicted (25) firmicute lasso peptide gene clusters (which have not been investigated nor have their natural products been confirmed) and contain homologs of the core conserved genes of known lasso peptide biosynthetic gene clusters (e.g. genes encoding A, B1, B2, C, and D homologs), in addition to a gene coding for a putative kinase. Interestingly, the cluster organization here differs from that of hypothetical proteobacterial lasso peptide clusters in which a similar kinase-encoding gene was detected (Fig. 2, a and b) (7).
FIGURE 2.

a, overview of the types of lasso peptide biosynthetic gene clusters identified in firmicutes. ORFs containing K, NT, and ST encode putative kinases, nucleotidyltransferases, and sulfotransferases, respectively. b, alignment of the corresponding precursor peptides. c, vector map of the plasmid for heterologous production of paeninodin. d, LC-MS analyses of pellet extracts of cultures expressing an empty vector control, the complete cluster in pET41a, the padeK knock-out cluster, and the knock-out plasmid supplemented with pACYC-padeK.
Other lasso peptide systems identified here feature a putative sulfotransferase-encoding gene at the beginning of the biosynthetic operon and/or a putative nucleotidyltransferase-encoding gene between those coding for the B2 and D homologs. Table 1 provides an overview of these genes, their putative roles, and arrangements within the clusters. Notably, the similarity of the precursor peptides among these systems indicates a common and recent ancestor (Fig. 2b). In contrast to the lasso peptides that were previously predicted in firmicutes (all of which bear an N-terminal Gly) (25), those predicted here feature an N-terminal Ala, a property only recently observed for a series of lasso peptides from Caulobacter sp. K31 (11). Because of their distinct N-terminal residue and gene cluster organization, the lasso peptides detected in this study appear to constitute a new family.
TABLE 1.
Proposed functions of ORFs in the putative lasso peptide biosynthetic gene clusters identified by genome mining in firmicutes
| Protein | Length (amino acids) | GenBankTM accession no. | Proposed function | Conserved domain analysis |
|---|---|---|---|---|
| PadeA | 42 | Precursor peptide | ||
| ThcoA | 42 | AGA59244.1 | ||
| BaceA | 41 | EJR65203.1 | ||
| PapoA | 42 | YP_008910696.1 | ||
| PadeB1 | 99 | WP_006678398.1 | Maturation enzyme | PqqD superfamily, |
| ThcoB1 | 98 | AGA59242.1 | B1 part of “split B” | Coenzyme PQQ synthesis protein D |
| BaceB1 | 101 | EJR65205.1 | Protease | |
| PapoB1 | 100 | YP_008910698.1 | ||
| PadeB2 | 142 | WP_006678399.1 | Maturation enzyme | Transglutaminase-like superfamily |
| ThcoB2 | 163 | AGA59241.1 | B2 part of split B | |
| BaceB2 | 157 | EJR65206.1 | Protease | |
| PapoB2 | 161 | YP_008910699.1 | ||
| PadeC | 649 | WP_006678396.1 | Maturation enzyme | Glutamine amidotransferases class-II (GATase) asparagine synthase B type |
| ThcoC | 645 | AGA59245.1 | Adenylation | |
| BaceC | 645 | EJR65202.1 | Cyclization | |
| PapoC | 641 | YP_008910695.1 | ||
| PadeD | 607 | WP_006678400.1 | Export | ABC-type multidrug transport system |
| ThcoD | 607 | AGA59240.1 | Immunity protein | |
| BaceD | 599 | EJR65208.1 | ||
| PapoD | 601 | YP_008910701.1 | ||
| PadeK | 313 | WP_006678397.1 | Putative kinase | HPrK-related kinase |
| ThcoK | 310 | AGA59243.1 | ||
| BaceK | 328 | EJR65204.1 | ||
| PapoK | 351 | YP_008910697.1 | ||
| BaceNT | 400 | EJR65207.1 | Putative nucleotidyltransferase | Uncharacterized nucleotidyltransferase |
| PapoNT | 506 | YP_008910700.1 | ||
| PapoST | 422 | YP_008910694.1 | Putative sulfotransferase | Sulfotransferase family |
To test for homologous production of lasso peptides from firmicutes under laboratory conditions, strains of P. dendritiformis, T. composti, B. cereus, and Paenibacillus polymyxa (see Fig. 2a) were cultivated in LB medium or M9 minimal medium (both containing XAD16 resin) for 3 days. Pellet extracts were then screened for the molecular masses of the predicted lasso peptides by high resolution LC-FT-MS. As all cultures failed to yield lasso peptides, we turned to a heterologous host. For further investigation, we chose the putative lasso peptide biosynthetic gene cluster from P. dendritiformis C454, which, compared with previously characterized clusters, contains the additional kinase-encoding gene (padeK).
Heterologous Production, Isolation, and Characterization of the Novel Lasso Peptide Paeninodin
The complete lasso peptide gene cluster from P. dendritiformis C454 was cloned into pET41a (Fig. 2c) and expressed in E. coli in M9 minimal medium, as described previously (6, 7, 9–11). Concentrated extracts from supernatants and cell pellets were analyzed via high resolution LC-FT-MS. Although a mass corresponding to the predicted full-length lasso peptide was detected in the cell pellet extracts, no traces of the full-length lasso peptide or truncations thereof were found in the supernatant extracts. As expression for 3 days at 37 °C yielded the highest amounts of this compound (data not shown), all further expressions were carried out under these conditions. Thus, the target compound could be isolated from a large scale fermentation and subsequently purified by two rounds of HPLC (Fig. 3a). Tandem MS of the compound, designated here as paeninodin, confirmed its primary structure (Fig. 3, c and e), which features a nine-residue macrolactam ring between Ala-1 and Asp-9.
FIGURE 3.

Chromatograms of second-round preparative HPLC purification of paeninodin (a) and paeninodin-OPO32− (b) are shown. MS2 spectra of paeninodin (c) and paeninodin-OPO32− (d) are shown. MS3 spectra of y11 fragments of paeninodin (e) and paeninodin-OPO32− (f) are shown.
Several experiments were carried out to verify that paeninodin exhibits the predicted lasso topology. First, a previously established assay in which pure lasso peptide is heat-denatured and then digested with carboxypeptidase Y was performed (6, 7, 9–11). Initial heating tests indicated that the most branched-cyclic (unthreaded) peptide accumulated after incubation at 65 °C for 4 h (Fig. 4a). That fragments of the original peptide were the major products observed is explained by the presence of an Asp-Pro sequence at the site of fragmentation, which is known to facilitate autocatalytic hydrolysis of the peptide backbone via formation of a cyclic imide intermediate (6). We next incubated the heat-denatured product with carboxypeptidase Y. Prior to heating, the threaded topology of a lasso peptide occludes access of carboxypeptidase Y to the C-terminal tail; however, following thermally induced unthreading, the C terminus of the peptide becomes accessible to carboxypeptidase Y-mediated digestion. The results show that the isolated compound is only resistant to carboxypeptidase Y prior to thermal treatment, indicating that the peptide has adopted a lasso topology (Fig. 4c).
FIGURE 4.

LC-MS analyses of unmodified (a) and phosphorylated (b) paeninodin samples after incubation at different temperatures for 4 h. c, result of carboxypeptidase Y treatment of paeninodin following incubation at 65 °C for 4 h. d, mutational analysis of padeA and its effect on paeninodin production levels. A production level of 100% refers to the sum of modified and unmodified paeninodin observed for the wild type system. Experiments were performed in triplicate (data represent mean ± S.D.). Shown above is a schematic of the primary structure of paeninodin.
To further substantiate these findings, IM-MS analysis of paeninodin was carried out in the presence of formic acid and the supercharging agent sulfolane before and after incubation at 65 °C for 4 h. In the +4 charge state, a new peak with significantly longer drift time in the IM-MS chamber was observed for the heat-treated sample compared with the untreated one (supplemental Fig. S1). In agreement with a previous study (33), these results are explained by thermally induced unthreading, which yields a branched-cyclic peptide with a larger cross-section and therefore a longer drift time compared with the more compact lasso peptide. This analysis also yielded a pair of peaks, indicating that the peptide can adopt multiple, distinct, and interchangeable conformations in its native state.
Efforts were taken to determine the three-dimensional structure of paeninodin. A solution of paeninodin in water (∼40 mg/ml) was subjected to sparse-matrix crystal screening (Qiagen), although no crystal growth was observed even after 50 days. Structure elucidation was thus attempted via NMR spectroscopy. Purified paeninodin was dissolved in five solution variants containing four different solvents as follows: H2O/D2O (9:1); CD3OH, CD3OH/CDCl3 (1:1); CD3OH/CDCl3 (3:1); and DMSO-d6. The lasso peptide concentration was varied from 4.5 to 10.0 mm, whereas temperature dependence was studied by recording 1H spectra in the range of 278–318 K. To our surprise, the spectra showed resonance signals in the amide proton region (6.5–9.5 ppm) corresponding to more than one set of paeninodin structures (the primary sequence contains 19 NH) at reduced temperature in all tested conditions (Fig. 5). Changes in signal linewidth and numbers in this region were observed at higher temperatures, which was assumed to be due to chemical exchange in solution. At first, we suspected sample degradation (e.g. unthreading or breaking of the Asp-14–Pro-15 bond, as shown in Fig. 4a) under the experimental conditions; however, LC-MS analysis, carried out on samples following NMR measurements, discounted this. Therefore, we hypothesize that paeninodin may adopt more than one stable conformation in solution. In addition, we propose that these co-existing conformations undergo rapid chemical exchange, which results in overlapping signals and hinders NMR structure determination. A few natural peptides were shown to assume multiple, stable, and distinct conformations; in these cases, NMR structure solution was possible albeit challenging (37–39). As the thermal instability of paeninodin prevented the recording of NMR spectra at higher temperatures, structure determination was deemed outside the scope of this study.
FIGURE 5.
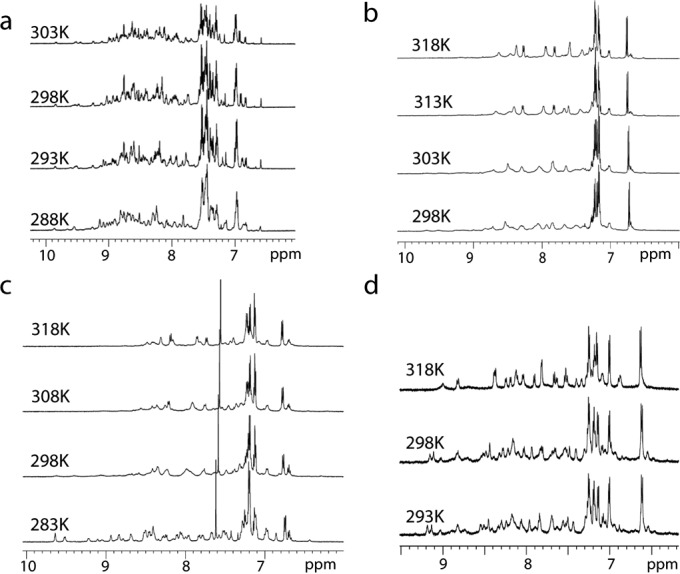
1H NMR spectra of amide protons in the 6–11 ppm region of paeninodin, collected at various temperatures in H2O/D2O (9:1) (a), CD3OH (b), CD3OH/CDCl3 (1:1) (c), and DMSO-d6 (d).
Detection of Phosphorylated Paeninodin
A small peak with similar retention time to that of paeninodin in the high resolution LC-FT-MS analysis of cell extracts did not escape our attention. Closer inspection showed that it belonged to a compound with the molecular weight of the lasso peptide plus a phosphate group (Figs. 2d and 3, b, d, and f). Based on MS2 results, we proposed that the compound results from phosphorylation of the C-terminal Ser of paeninodin by the putative kinase encoded within the cluster. Deletion of the kinase gene from the expression plasmid yielded an extract lacking the modified compound, although non-phosphorylated paeninodin was still produced at the same level as before (Fig. 2d). Co-expression of the knock-out plasmid with a pACYC vector carrying padeK restored production of the phosphorylated compound (Fig. 2d). Hence, a link between expression of the putative kinase PadeK and occurrence of the modified lasso peptide was established.
Mutational Analysis of the Precursor Peptide Provides Insight into the Biosynthesis of Paeninodin
To study the biosynthesis of paeninodin and propose the plug residues of the lasso peptide, mutagenesis studies were performed. We first analyzed the primary structure of paeninodin and performed an alanine scan of the residues that could feasibly act as plugs to maintain the lasso topology. Residues previously shown to be crucial for efficient lasso peptide processing (6, 9–11, 16, 28, 29, 40, 41) were also exchanged to compare the lasso peptide biosynthetic machinery of firmicutes with that of proteobacteria. The results of these studies are summarized in Fig. 4d.
Sequence analysis of the C-terminal region of paeninodin suggests that His-20 and Tyr-21 are the most suitable plug residues. Indeed, the H20A variant showed greatly diminished lasso peptide yield, whereas the Y21A exchange completely abolished production. In contrast, the Y21F variant was produced in amounts similar to wild type. Much smaller effects were observed for the D22A and S23A exchanges, supporting the role of His-20 and Tyr-21 as upper and lower plugs, respectively, in agreement with previous studies (6, 9, 10, 14, 27, 29). Interestingly, deletion of the C-terminal Ser-23 residue was tolerated well, although additional removal of the penultimate amino acid completely abolished production.
Results of Thr−2,4 Ala-1, and Asp-9 exchanges indicate that the paeninodin machinery employs the same biosynthetic logic as the previously investigated systems from proteobacteria (6, 9–11, 16, 29). Substitution of Thr−2 with another amino acid negatively affects lasso peptide production, with better tolerance for structurally similar residues. As shown previously for other lasso peptides, the character of the residue at position 1 is highly conserved (6, 9–11, 16, 29), with exchanges greatly diminishing lasso peptide production, presumably due to problems with enzyme recognition. Finally, exchanging the ring-forming Asp-9 with Glu completely abolishes production, also in agreement with previous studies (6, 9, 10, 16, 29). Therefore, it could be shown that the paeninodin biosynthetic machinery operates according to the same set of principles identified for proteobacterial systems.
Kinase Only Modifies the Precursor Peptide and Not the Mature Lasso Peptide
To investigate how paeninodin becomes phosphorylated, we heterologously expressed and isolated the PadeK kinase as a fusion with MBP. After expression, the MBP tag was cleaved with HRV-3C protease, and PadeK was further purified for in vitro assays (Fig. 6a). When PadeK was incubated with the lasso peptide paeninodin under assay conditions, no phosphorylation was observed. We therefore expressed the full-length precursor peptide PadeA as a thioredoxin fusion protein (42), removed the thioredoxin tag with HRV-3C, and purified the free precursor peptide (with Gly-Pro remaining at the N terminus; GP-PadeA) by HPLC (Fig. 6c). Repeating the assay with unmodified precursor peptide yielded small amounts of phosphorylated peptide, and subsequent MS2 analysis showed that modification occurs at the C-terminal Ser, in accordance with the phosphorylated lasso peptide detected earlier (Fig. 6g). This demonstrates that the kinase acts on the precursor peptide prior to processing by the lasso peptide biosynthetic enzymes. Because of difficulties isolating PadeK as well as its weak activity, we searched our genome mining results for a suitable homolog. We identified a kinase from the cluster of T. composti KWC4 (ThcoK) that could be readily isolated in high yields. The high degree of sequence similarity of precursor peptides PadeA and ThcoA (∼64% identity), lasso peptide products (∼70% identity; Fig. 2b), and associated kinases (∼41% identity, ∼58% similarity) was expected to facilitate homologous exchange. Indeed, when pure ThcoK (Fig. 6b) was subjected to in vitro assays with the lasso peptide and unmodified precursor peptide GP-PadeA, phosphorylation of the precursor peptide (∼75% conversion) but not the lasso peptide was observed (Fig. 6, d and e).
FIGURE 6.

SDS-polyacrylamide gel of purified PadeK (a) and ThcoK (b) is shown. c, chromatogram of second-round preparative HPLC purification of GP-PadeA. Phosphorylation assay using ThcoK and paeninodin lasso peptide (d) or GP-PadeA precursor peptide (e) is shown. f, LC-MS analyses of pellet extracts from cultures expressing pET41a-padeCAKB1B2D and pET41a-padeCA-thcoK-padeB1B2D (in the latter, the original kinase-encoding padeK from P. dendritiformis C454 is replaced with the kinase-encoding thcoK from T. composti KWC4). g, MS2 spectrum showing the GP-PadeA-OPO32− product of the assay with PadeK and GP-PadeA. h, exemplary time dependence of ThcoK-catalyzed phosphorylation of GP-PadeA under assay conditions. Experiments were performed in triplicate (data represent mean ± S.D.). i, Michaelis-Menten plot used to determine the Km and kcat values. Experiments were performed in triplicate (data represent mean ± S.D.).
To assess the in vivo activity of ThcoK, the whole lasso gene cluster from T. composti KWC4 was first cloned and heterologously expressed in E. coli BL21(DE3), but no lasso peptide was detected. We then replaced padeK with thcoK in our heterologous expression system to test the exchangeability of the kinases. As expected, the phosphorylated lasso peptide was found in the presence of ThcoK; surprisingly, however, it was the main product of the extract, with only minor amounts of unmodified lasso peptide (Fig. 6f). Such a system allowed for the isolation of pure phosphorylated lasso peptide for additional assays (Fig. 3b). First, incubation at elevated temperatures revealed that, although phosphorylation is stable under these conditions (Fig. 4b), it does not impact the thermal stability of the lasso peptide relative to the unmodified one. Second, the potential antimicrobial activity of both modified and unmodified paeninodin was explored with spot-on-lawn assays against B. subtilis MR168, M. flavus ATCC 10240, B. rhizoxinica HKI454, S. japonicum UT26, X. citri pv. mangiferaeindicae LMG 941, B. cereus, B. megaterium WSH-002, and B. amyloliquefaciens. However, no inhibitory effect was observed for either compound against the tested panel of bacteria (data not shown), suggesting that the lasso peptides generally either lack such an activity or only target a narrow range of organisms not covered here.
Characterization of ThcoK Kinase from T. composti KWC4
After demonstrating the much higher in vivo and in vitro phosphorylation activity of ThcoK than PadeK toward PadeA, full characterization of ThcoK was performed. First, kinetic parameters for modification of GP-PadeA were obtained by varying the substrate concentration under constant enzyme concentration (Fig. 6, h and i). In this way, the Michaelis-Menten parameters Km = 279 ± 19.7 μm and kcat = 0.41 ± 0.02 min−1 were determined.
Because ThcoK and other kinases identified in lasso peptide biosynthetic gene clusters from firmicutes are often annotated as homologs of HPr kinase (e.g. from Lactococcus casei) and phosphoenolpyruvate carboxykinase (e.g. from Thermus thermophilus Hb8), we took a closer look at the conserved regions of kinases from these families. Intriguingly, all such kinases appear to share conserved active site motifs consisting of one His, one Lys, and two Asp residues. The roles of these residues in catalysis can be derived from the co-crystal structure of L. casei HPr kinase with its substrate (the motif consists of His-140–Lys-161–Asp-178–Asp-179) (43–45). The conjugate base of Asp-179, stabilized by His-140, deprotonates the Ser-46 side chain of the HPr substrate, which then abstracts the γ-PO43− group of ATP through nucleophilic attack. Lys-161 and Asp-178 facilitate the binding of ATP by counteracting negative charges and coordinating a metal ion, respectively (43–45).
A similar mode of catalysis is feasible for PadeK, ThcoK, and their homologs, an alignment of which shows complete conservation of the His-Lys-Asp-Asp motif. To test this hypothesis, we generated single or quadruple Ala variants of these residues, as well as double Asp-to-Ala variants of both PadeK and ThcoK in our lasso peptide production plasmids. Expression cultures were grown under normal conditions, and their extracts were analyzed by high resolution LC-FT-MS. For each variant, no phosphorylated lasso peptide was observed, although the overall yields of lasso peptide remained unaffected. This strongly suggests that the kinase variants were rendered inactive and that each of the four residues is crucial for catalysis (Fig. 7, a–c).
FIGURE 7.
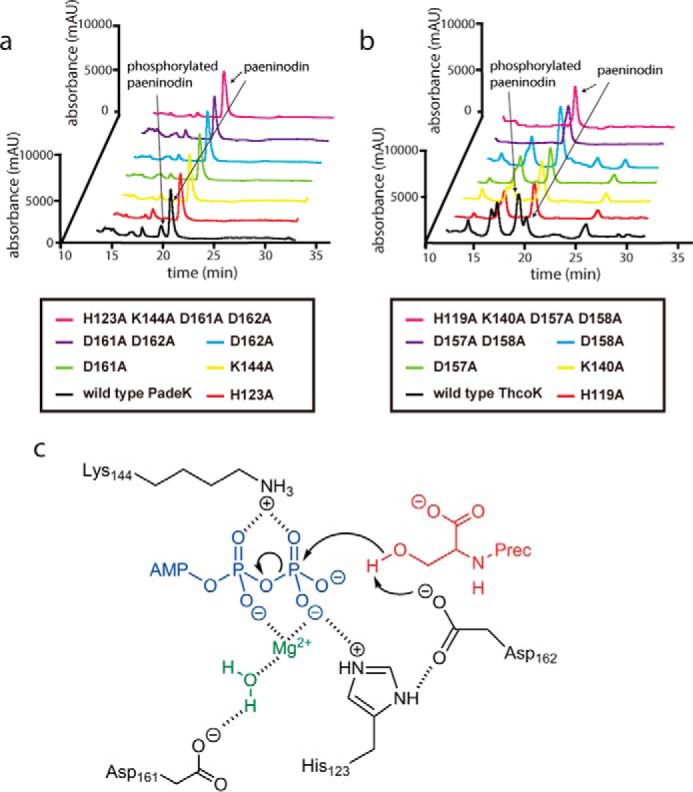
Mutational analysis of essential catalytic residues in kinases PadeK (a) and ThcoK (b) is shown. c, proposed mechanism of PadeK-catalyzed phosphorylation of the C-terminal Ser of a precursor peptide. PadeK residues are shown in black, ATP in blue, water and Mg2+ in green, and the precursor peptide in red.
Nevertheless, definitive experimental evidence for phosphorylation of the side chain of the C-terminal Ser, as opposed to its carboxylic acid group, was still missing. Hence, a thorough substrate screen was performed to determine the specificity of ThcoK.
Substrate Specificity of ThcoK
Regarding the substrate specificity of ThcoK, several questions needed to be addressed, namely the minimal length of a peptide necessary for modification, the motifs recognized by the kinase, and the precise site of phosphate addition. To address these questions, a wide variety of peptides were treated with ThcoK under our established assay conditions. Depending on length, the peptides were expressed and purified (similar to GP-PadeA) or chemically synthesized. A summary of all tested peptides and their observed conversion ratios is given in Table 2.
TABLE 2.
Results of substrate specificity assays with ThcoK
The color code is as follows: precursor peptide (green); macrolactam ring of lasso peptide (orange); ring-forming Asp of lasso peptide (red); loop and tail of lasso peptide (blue); mutation or chemical alteration (black); GP from HRV-3C protease site (purple).

* Experiments were performed in triplicate (data represent mean ± S.D.).
Surprisingly, although the overall efficiency of enzyme-catalyzed phosphorylation decreases with peptide length, even the shortest peptide consisting of the last five residues was modified to a small extent. This suggests that neither the leader peptide nor the core peptide contains any essential recognition motifs aside from the modified region.
We generated additional variants of full-length GP-PadeA wherein the three residues before Ser-23 or the seven residues before the HYDS sequence were completely exchanged with alanine. Even though long stretches of the amino acid sequence were altered in these variants, the corresponding decreases in phosphorylation efficiency were rather small. In contrast, no modification was observed for S23A substitution in GP-PadeA, emphasizing the absolutely essential role of the C-terminal Ser for enzyme activity.
As described above, the mature lasso peptide was not modified by ThcoK. However, we were curious whether the branched-cyclic peptide, whose C terminus is not shielded by the macrolactam ring, could be phosphorylated. To test this, a sample of isolated paeninodin was heated at 65 °C for 4 h and then incubated with ThcoK under assay conditions. Indeed, a significant amount of phosphorylated, branched-cyclic peptide was observed. Because the lasso fold itself appears to hinder access of the kinase to the C-terminal Ser, the only feasible route to production of phosphorylated paeninodin involves modification of the precursor peptide prior to processing.
The PadeA(13–23) peptide consisting of the last 11 residues of the precursor peptide is converted with moderate efficiency and can be quickly and easily accessed by chemical synthesis. It was therefore selected as a scaffold for more intricate exchanges of the C-terminal Ser. As for the full-length precursor peptide, S23A exchange completely abolishes enzymatic modification. This is also true of S23T and S23Y substitutions, both of which preserve the side chain hydroxyl moiety. To further assess the position of phosphate transfer, we analyzed 11-residue peptides that were methylated at the C-terminal carboxylic acid or the Ser-23 side chain. Surprisingly, phosphorylation was observed in only trace amounts or not at all, respectively.
Finally, we tested a series of Ser shift variants in which the C-terminal Ser was exchanged with Ala, and a new Ser residue was introduced at position 14, 17, 20, or 22. None of these variants were phosphorylated, confirming the importance of the C-terminal Ser for kinase activity (Table 2).
Taken together, these assays suggest the requirement of both the C-terminal carboxylic acid and Ser-23 side chain for enzymatic recognition and modification. No other Ser side chains or carboxylic acid moieties of Asp or Glu residues were phosphorylated.
Phosphorylation Occurs at the C-terminal Ser-23 Side Chain
To definitively prove the site of phosphorylation, NMR analysis and chemical modification assays were employed. First, an optimized modification assay was performed on a large scale on PadeA(13–23) to obtain pure phosphorylated peptide. Additionally, a version of the 11-residue peptide already carrying a phosphorylated Ser side chain (PadeA(13–23)-Ser-23-OPO32−) was chemically synthesized as a reference.
For the chemical modification assay, a protocol to selectively methylate all carboxylic acid moieties by use of methanolic HCl was employed (46). The methylation reaction was carried out on the unmodified and enzymatically modified 11-residue peptide, as well as the chemically synthesized peptide carrying a phosphorylated Ser side chain. In all three cases, the mass of six additional methyl moieties was observed by LC-MS, corresponding to methylation of all five Asp/Glu side chains and the C-terminal carboxylic acid group (Fig. 8a). Additional experiments on modified and unmodified paeninodin also showed complete methylation of Asp/Glu side chains and the C-terminal carboxylic acid moiety (Fig. 8b). Tandem mass spectrometry of all reaction products confirmed methylation and (if already present at the start of the assay) phosphorylation of the C-terminal Ser (data not shown). Taken together, these results provide strong evidence that side chain phosphorylation occurs both in vitro and in vivo, although the C-terminal carboxylic acid group remains unmodified.
FIGURE 8.
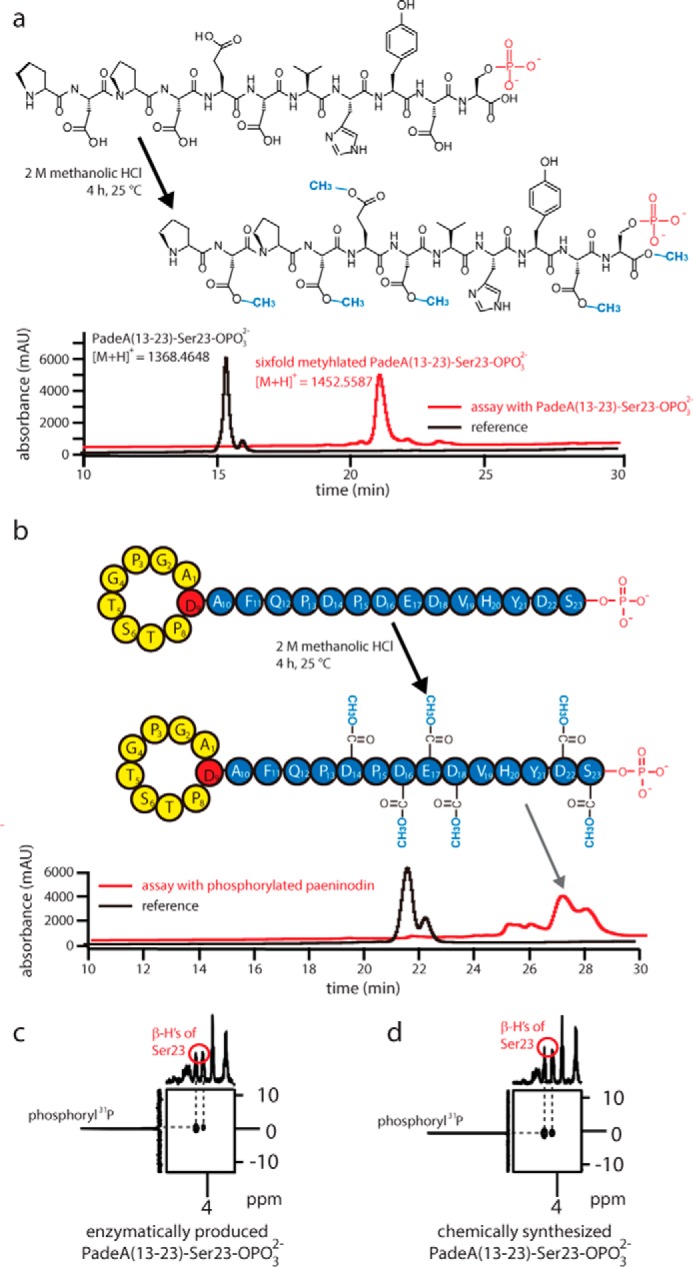
Results of the esterification assays of phosphorylated 11-residue peptide (a) and paeninodin lasso peptide (b). Shown is the primary structure before and after the reaction (see black and red traces, respectively). Excerpts from 1H-31P heteronuclear multiple-bond correlation spectra of enzymatically produced (c) and synthetically derived (d) PadeA(13–23)-Ser-23-OPO32−, showing phosphoryl 31P-Ser-23 β-H cross-peaks.
As an additional control, NMR studies were performed with the synthetically and enzymatically phosphorylated 11-residue peptides (supplemental Figs. S2–S4 and supplemental Table S9). For both peptides, essentially identical NMR spectra were obtained, and after signal assignment a long range correlation between the β-hydrogen of Ser-23 and phosphorus could be observed in 1H-31P heteronuclear multiple-bond correlation spectra (Fig. 8c). Hence, it could be unambiguously determined that ThcoK exclusively transfers a phosphate group from ATP to the side chain of the C-terminal Ser, as proposed earlier.
Despite the strict requirement for a C-terminal Ser with free hydroxyl and carboxyl groups, the enzyme shows a remarkable degree of promiscuity toward its substrates to the extent that their primary structures may be otherwise completely flexible. To our knowledge, no other kinase has been shown to phosphorylate a C-terminal Ser, whereas those that phosphorylate an internal Ser residue are quite common in nature. This unique combination of relaxed substrate specificity and highly selective modification of a C-terminal Ser makes these kinases interesting candidates for applications in synthetic biology, and could nicely complement the chemical toolbox available for in vitro peptide synthesis and modification. Future studies could aim at evolving one of these kinases for robustness, speed, and efficiency for such purposes.
Bioinformatic Analysis of Putative Lasso Peptide Biosynthetic Gene Clusters Featuring Kinase-encoding Genes
To investigate how these lasso peptide precursor-modifying kinases are distributed throughout the bacterial domain, we used our protein-centric genome mining approach, employing ThcoK and PadeK as the search input. Many related proteins in firmicutes were identified (Table 3) in addition to homologs in proteobacterial lasso peptide gene clusters. The latter, which feature an A B1 B2 C K D organization, belong to the same family of clusters predicted in our B protein-centric genome mining study of proteobacteria (7). Analysis of the corresponding precursor peptides revealed that the precursor peptides from firmicutes share significant similarity with each other. The same is true when comparing the proteobacterial precursor peptides to one another (data not shown). Interestingly, comparison of precursor peptides from all of these clusters revealed little similarity between the different cluster types, with the exception of the completely conserved C-terminal Ser. This observation suggests that kinases of both firmicutes and proteobacteria strictly modify a C-terminal Ser in the same manner, although future studies must confirm this experimentally. Employing the MEME algorithm, a total of 25 kinases identified from firmicutes and an additional 10 from proteobacterial clusters were investigated for the presence of conserved motifs. Interestingly, two motifs were found in all 35 kinases, with one motif expectedly containing the essential catalytic motif His-Lys-Asp-Asp (data not shown). In addition, a third motif was identified in the kinases from firmicutes but not proteobacteria.
TABLE 3.
Kinase-harboring lasso peptide gene clusters from firmicutes and proteobacteria
N, nucleotidyltransferase; K, kinase; S, sulfotransferase.
| No. | Strain | GenBankTM accession no. | Gene cluster organization |
|---|---|---|---|
| 1 | P. dendritiformis C454 | AHKH01000064.1 23026 … 28726 | CAKB1B2D |
| 2 | B. cereus VD136 | AHFC01000001.1 329527 … 336459 | CAKB1B2ND |
| 3 | Bacillus mycoides DSM 2048 | ACMU01000105.1 37016 … 43947 | CAKB1B2ND |
| 4 | Bacillus thuringiensis | NZ_AYPV01000024.1 395713 … 402624 | CAKB1B2ND |
| 5 | Bacillus weihenstephanensis | NC_010184.1 3435993 … 3442907 | CAKB1B2ND |
| 6 | Paenibacillus riograndensis | NZ_AGBD01000087.1 30500 … 36208 | CAKB1B2ND |
| 7 | T. composti KWC4 | NC_019897.1 3352916 … 3358626 | CAKB1B2D |
| 8 | P. polymyxa | NZ_JWJJ01000002.1 548177 … 557005 | SCAKB1B2ND |
| 9 | Paenibacillus terrae HPL-003 | NC_016641.1 3033233 … 3041545 | SCAKB1B2ND |
| 10 | Paenibacillus peoriae | NZ_AGFX01000048.1 27851 … 35346 | CAKB1B2D |
| 11 | Bacillus macauensis | NZ_AKKV01000020.1 16218 … 23006 | CAKB1B2ND |
| 12 | Paenibacillus harenae DSM 16969 | NZ_KE383843.1 594303 … 599983 | CAKB1B2D |
| 13 | Bacillus subterraneus | NZ_JXIQ01000098.1 20476 … 27361 | CAKB1B2ND |
| 14 | Bacillus kribbensis | NZ_KE387239.1 41261 … 48195 | CAKB1B2ND |
| 15 | B. megaterium WSH-002 | CP003017.1 3899438 … 3905391 | CAKB1B2D |
| 16 | Bacillus flexus | NZ_JANV01000106.1 11574 … 17331 | CAKB1B2D |
| 17 | Bacillus oceanisediminis | NZ_ALEG01000067.1 32511 … 40721 | CAKB1B2ND |
| 18 | Bacillus vietnamensis | NZ_CCDN010000009.1 39263 … 46139 | CAKB1B2ND |
| 19 | Bacillus aryabhattai | NZ_JPIE01000001.1 482105 … 489018 | CAKB1B2ND |
| 20 | Paenibacillus alginolyticus | NZ_AUGY01000047.1 37379 … 44267 | CAKB1B2ND |
| 21 | Bacillus endophyticus | NZ_ALIM01000014.1 895870 … 901612 | CAKB1B2D |
| 22 | Paenibacillus algorifonticola | NZ_LAQO01000025.1 22190 … 29253 | CAKB1B2ND |
| 23 | Paenibacillus wulumuqiensis | NZ_KQ040793.1 393438 … 401239 | CAKB1B2ND |
| 24 | Paenibacillus borealis | NZ_CP009285.1 998071 … 1003776 | CAKB1B2D |
| 25 | Bacillus akibai | BAUV01000025.1 34686 … 41595 | CAKB1B2ND |
| 26 | Sphingobium yanoikuyae | AGZU01000016.1 543969 … 549488 | AB1B2CKD |
| 27 | Mesorhizobium loti | NZ_KB913026.1 6199725 … 6205323 | AB1B2CKD |
| 28 | Mesorhizobium opportunistum | CP002279.1 267289 … 273124 | AB1B2CKD |
| 29 | Mesorhizobium ciceri | NZ_AWZS01000002.1 71030 … 76676 | AB1B2CKD |
| 30 | Mesorhizobium amorphae | AGSN01000187.1 19062 … 24852 | AB1B2CKD |
| 31 | Mesorhizobium australicum | NC_019973.1 265394 … 271267 | AB1B2CKD |
| 32 | Rhodobacter sphaeroides | CP001150.1 875732 … 881323 | AB1B2CKD |
| 33 | Sphingomonas sp. Y57 | NZ_LDES01000074.1 26756 … 32158 | AB1B2CKD |
| 34 | Mesorhizobium sp. WSM1293 | NZ_KI911320.1 331530 … 337146 | AB1B2CKD |
| 35 | Altererythrobacter atlanticus | NZ_CP011452.1 1803258 … 1808418 | AB1B2CKD |
To obtain more information about the relationship of all previously characterized lasso peptide biosynthetic gene clusters with the new ones containing kinases, a phylogenetic analysis was performed with C protein sequences as the basis (precursor peptides are too short and variable and B proteins may be split, making them unsuitable for such an analysis). Sequences of representative C proteins accessible from published functional clusters (6, 7, 10, 11, 18, 22–25), in addition to those of the six C proteins from kinase-containing clusters, were used as input. The phylogenetic analysis shows clear separation of gene clusters into five clades, coinciding with the bacterial phylum of origin as well as the organization of genes within the clusters (Fig. 9a).
FIGURE 9.

a, phylogenetic tree of C proteins from different lasso peptide biosynthetic gene clusters. Red asterisks mark kinase-containing clusters. b, phylogenetic tree of the 35 kinases identified from lasso peptide biosynthetic gene clusters.
Hence, there is one group of actinobacterial clusters, one of firmicute clusters and three of proteobacterial clusters. The latter is divided into clusters with a simple A B C organization adjacent to a gene encoding a lasso peptide isopeptidase, clusters containing an additional gene encoding an ABC transporter (D protein), and clusters featuring genes coding for both a D protein and a putative kinase.
Deeper analysis of the 35 kinase-containing clusters shows clear separation into two clades, one from firmicutes and one from proteobacteria (Fig. 9b). Intriguingly, only the firmicute clusters contain putative transferases, which presumably serve as tailoring enzymes for their respective lasso peptides. These enzymes, once characterized, could further expand the available tools for lasso peptide engineering.
It appears that the five lasso peptide clades evolved by divergence from a common ancestor. This is reasonable when considering the known and presumed biological functions of these peptides. The isopeptidase-containing clusters from proteobacteria, which lack an ABC transporter, are able to catabolize their lasso peptides, indicating that the peptides play a role in signaling or function in scavenging through a catch-and-release mechanism (8, 26). However, lasso peptide gene clusters from actinobacteria and proteobacteria, which feature an ABC transporter, often produce lasso peptides with antibacterial properties, indicating that the molecules are synthesized and then exported for competition (3, 4). That the organization of these gene clusters is different indicates that they arose independently. The newly identified kinase-featuring clusters from firmicutes and the potential kinase-featuring clusters from proteobacteria represent two new families of lasso peptide gene clusters. Although both clusters contain a kinase, there is little similarity between their precursor peptides other than the C-terminal serine. Thus, it seems that firmicutes and proteobacteria independently evolved a similar strategy to produce phosphorylated lasso peptides.
Discussion
In this study, we have shown that lasso peptides are not limited to proteobacteria and actinobacteria, but are also produced by clusters found in firmicutes. Interestingly, these clusters possess a unique organization not observed for other lasso peptide systems. One such lasso peptide, paeninodin, was isolated and characterized, and its biosynthetic machinery was probed by mutational analysis. Additionally, it was shown that a kinase, encoded by a central gene in this cluster, phosphorylates the side chain of the C-terminal Ser-23 of the precursor peptide PadeA, thus leading to production of a phosphorylated lasso peptide (Fig. 10). Herein, we identified and thoroughly characterized the responsible enzyme. Descriptions of lasso peptide tailoring in the literature are notably rare (the only examples are C-terminal methylation of lassomycin (20) and Trp hydroxylation of some RES-701 family lasso peptides).
FIGURE 10.
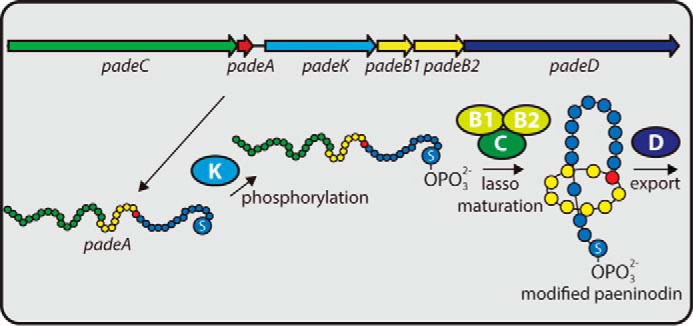
Proposed schematic of paeninodin biosynthesis. Precursor peptide PadeA is phosphorylated at its C-terminal Ser by PadeK and then processed into the mature phosphorylated lasso peptide by PadeB1, PadeB2, and PadeC. Cellular export of the lasso peptide is accomplished by ABC transporter PadeD.
The roles of paeninodin and its phosphorylated form remain elusive, and further studies will be necessary to validate their natural functions. Phosphorylated paeninodin could serve as a signal molecule or participate in self-immunity in the producing organism. Nevertheless, the in vitro studies performed on PadeK and ThcoK revealed these kinases to be intriguing enzymes. They are highly selective for modification of the C-terminal Ser yet relaxed with regard to the size and sequence of the rest of the substrate. Furthermore, although Ser phosphorylation is quite common (e.g. in modulating protein activities or in other RiPP systems, such as certain classes of lanthipeptides), there is to our knowledge no other kinase capable of modifying a C-terminal Ser.
Thus, our study not only broadens the existing knowledge of lasso peptides and their biosynthetic systems by proving that predicted lasso peptide clusters from firmicutes can indeed be functional but also uncovers a family of enzymes with interesting properties and potential for in vitro applications.
Author Contributions
S. Z., J. D. H., C. D. F., M. Z., and M. A. M. designed the study and wrote the paper. S. Z. carried out the experiments. J. D. H., C. D. F., and M. Z. provided experimental support. X. X. performed and analyzed NMR experiments and assisted with NMR figure preparation. U. L. provided technical assistance for MS experiments and supervised IM-MS experiments.
Supplementary Material
Acknowledgments
We thank Eshel Ben-Jacob and Alin Finkelshtein (Tel Aviv University, Israel) for providing a sample of P. dendritiformis C454.
This work was supported in part by the Deutsche Forschungsgemeinschaft and the LOEWE Center for Synthetic Microbiology. The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Table S1–S9 and Figs. S1–S4.
Thr−2 indicates that this residue is found at the −2 position of the precursor peptide (e.g. two residues before the first Ala of the core peptide).
- RiPP
- ribosomally synthesized and post-translationally modified peptide
- ABC
- ATP-binding cassette
- FT
- Fourier transform
- IM
- ion mobility
- MBP
- maltose-binding protein
- IPTG
- isopropyl 1-thio-β-d-galactopyranoside
- Ni-NTA
- nickel-nitrilotriacetic acid
- mAU
- milliabsorbance units.
References
- 1. Arnison P. G., Bibb M. J., Bierbaum G., Bowers A. A., Bugni T. S., Bulaj G., Camarero J. A., Campopiano D. J., Challis G. L., Clardy J., Cotter P. D., Craik D. J., Dawson M., Dittmann E., Donadio S., et al. (2013) Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature. Nat. Prod. Rep. 30, 108–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Burkhart B. J., Hudson G. A., Dunbar K. L., and Mitchell D. A. (2015) A prevalent peptide-binding domain guides ribosomal natural product biosynthesis. Nat. Chem. Biol. 11, 564–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hegemann J. D., Zimmermann M., Xie X., and Marahiel M. A. (2015) Lasso peptides: an intriguing class of bacterial natural products. Acc. Chem. Res. 48, 1909–1919 [DOI] [PubMed] [Google Scholar]
- 4. Maksimov M. O., Pan S. J., and James Link A. (2012) Lasso peptides: structure, function, biosynthesis, and engineering. Nat. Prod. Rep. 29, 996–1006 [DOI] [PubMed] [Google Scholar]
- 5. Xie X., and Marahiel M. A. (2012) NMR as an effective tool for the structure determination of lasso peptides. ChemBioChem. 13, 621–625 [DOI] [PubMed] [Google Scholar]
- 6. Hegemann J. D., Zimmermann M., Xie X., and Marahiel M. A. (2013) Caulosegnins I–III: a highly diverse group of lasso peptides derived from a single biosynthetic gene cluster. J. Am. Chem. Soc. 135, 210–222 [DOI] [PubMed] [Google Scholar]
- 7. Hegemann J. D., Zimmermann M., Zhu S., Klug D., and Marahiel M. A. (2013) Lasso peptides from proteobacteria: genome mining employing heterologous expression and mass spectrometry. Biopolymers 100, 527–542 [DOI] [PubMed] [Google Scholar]
- 8. Maksimov M. O., and Link A. J. (2013) Discovery and characterization of an isopeptidase that linearizes lasso peptides. J. Am. Chem. Soc. 135, 12038–12047 [DOI] [PubMed] [Google Scholar]
- 9. Zimmermann M., Hegemann J. D., Xie X., and Marahiel M. A. (2013) The astexin-1 lasso peptides: biosynthesis, stability, and structural studies. Chem. Biol. 20, 558–569 [DOI] [PubMed] [Google Scholar]
- 10. Hegemann J. D., Zimmermann M., Zhu S., Steuber H., Harms K., Xie X., and Marahiel M. A. (2014) Xanthomonins I–III: a new class of lasso peptides with a seven-residue macrolactam ring. Angew. Chem. Int. Ed. Engl. 53, 2230–2234 [DOI] [PubMed] [Google Scholar]
- 11. Zimmermann M., Hegemann J. D., Xie X., and Marahiel M. A. (2014) Characterization of caulonodin lasso peptides revealed unprecedented N-terminal residues and a precursor motif essential for peptide maturation. Chem. Sci. 5, 4032–4043 [Google Scholar]
- 12. Hegemann J. D., Fage C. D., Zhu S., Harms K., Di Leva F. S., Novellino E., Marinelli L., and Marahiel M. A. (2016) The ring residue proline 8 is crucial for the thermal stability of the lasso peptide caulosegnin II. Mol. Biosyst. 12, 1106–1109 [DOI] [PubMed] [Google Scholar]
- 13. Clarke D. J., and Campopiano D. J. (2007) Maturation of McjA precursor peptide into active microcin MccJ25. Org. Biomol. Chem. 5, 2564–2566 [DOI] [PubMed] [Google Scholar]
- 14. Yan K.-P., Li Y., Zirah S., Goulard C., Knappe T. A., Marahiel M. A., and Rebuffat S. (2012) Dissecting the maturation steps of the lasso peptide microcin J25 in vitro. ChemBioChem. 13, 1046–1052 [DOI] [PubMed] [Google Scholar]
- 15. Ducasse R., Yan K.-P., Goulard C., Blond A., Li Y., Lescop E., Guittet E., Rebuffat S., and Zirah S. (2012) Sequence determinants governing the topology and biological activity of a lasso peptide, microcin J25. ChemBioChem. 13, 371–380 [DOI] [PubMed] [Google Scholar]
- 16. Pavlova O., Mukhopadhyay J., Sineva E., Ebright R. H., and Severinov K. (2008) Systematic structure-activity analysis of microcin J25. J. Biol. Chem. 283, 25589–25595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cheung W. L., Pan S. J., and Link A. J. (2010) Much of the microcin J25 leader peptide is dispensable. J. Am. Chem. Soc. 132, 2514–2515 [DOI] [PubMed] [Google Scholar]
- 18. Solbiati J. O., Ciaccio M., Farías R. N., González-Pastor J. E., Moreno F., and Salomón R. A. (1999) Sequence analysis of the four plasmid genes required to produce the circular peptide antibiotic microcin J25. J. Bacteriol. 181, 2659–2662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Choudhury H. G., Tong Z., Mathavan I., Li Y., Iwata S., Zirah S., Rebuffat S., van Veen H. W., and Beis K. (2014) Structure of an antibacterial peptide ATP-binding cassette transporter in a novel outward occluded state. Proc. Natl. Acad. Sci. U.S.A. 111, 9145–9150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gavrish E., Sit C. S., Cao S., Kandror O., Spoering A., Peoples A., Ling L., Fetterman A., Hughes D., Bissell A., Torrey H., Akopian T., Mueller A., Epstein S., Goldberg A., et al. (2014) Lassomycin, a ribosomally synthesized cyclic peptide, kills mycobacterium tuberculosis by targeting the ATP-dependent protease ClpC1P1P2. Chem. Biol. 21, 509–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Metelev M., Tietz J. I., Melby J. O., Blair P. M., Zhu L., Livnat I., Severinov K., and Mitchell D. A. (2015) Structure, bioactivity, and resistance mechanism of streptomonomicin, an unusual lasso peptide from an understudied halophilic actinomycete. Chem. Biol. 22, 241–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Inokoshi J., Matsuhama M., Miyake M., Ikeda H., and Tomoda H. (2012) Molecular cloning of the gene cluster for lariatin biosynthesis of Rhodococcus jostii K01-B0171. Appl. Microbiol. Biotechnol. 95, 451–460 [DOI] [PubMed] [Google Scholar]
- 23. Elsayed S. S., Trusch F., Deng H., Raab A., Prokes I., Busarakam K., Asenjo J. A., Andrews B. A., van West P., Bull A. T., Goodfellow M., Yi Y., Ebel R., Jaspars M., and Rateb M. E. (2015) Chaxapeptin, a lasso peptide from extremotolerant Streptomyces leeuwenhoekii strain C58 from the hyperarid atacama desert. J. Org. Chem. 80, 10252–10260 [DOI] [PubMed] [Google Scholar]
- 24. Li Y., Ducasse R., Zirah S., Blond A., Goulard C., Lescop E., Giraud C., Hartke A., Guittet E., Pernodet J.-L., and Rebuffat S. (2015) Characterization of Sviceucin from Streptomyces provides insight into enzyme exchangeability and disulfide bond formation in lasso peptides. ACS Chem. Biol. 10, 2641–2649 [DOI] [PubMed] [Google Scholar]
- 25. Maksimov M. O., Pelczer I., and Link A. J. (2012) Precursor-centric genome-mining approach for lasso peptide discovery. Proc. Natl. Acad. Sci. U.S.A. 109, 15223–15228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Maksimov M. O., Koos J. D., Zong C., Lisko B., and Link A. J. (2015) Elucidating the specificity determinants of the AtxE2 lasso peptide isopeptidase. J. Biol. Chem. 290, 30806–30812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rosengren K. J., Clark R. J., Daly N. L., Göransson U., Jones A., and Craik D. J. (2003) Microcin J25 has a threaded sidechain-to-backbone ring structure and not a head-to-tail cyclized backbone. J. Am. Chem. Soc. 125, 12464–12474 [DOI] [PubMed] [Google Scholar]
- 28. Pan S. J., Rajniak J., Maksimov M. O., and Link A. J. (2012) The role of a conserved threonine residue in the leader peptide of lasso peptide precursors. Chem. Comm. 48, 1880–1882 [DOI] [PubMed] [Google Scholar]
- 29. Knappe T. A., Linne U., Robbel L., and Marahiel M. A. (2009) Insights into the biosynthesis and stability of the lasso peptide capistruin. Chem. Biol. 16, 1290–1298 [DOI] [PubMed] [Google Scholar]
- 30. Hegemann J. D., De Simone M., Zimmermann M., Knappe T. A., Xie X., Di Leva F. S., Marinelli L., Novellino E., Zahler S., Kessler H., and Marahiel M. A. (2014) Rational improvement of the affinity and selectivity of integrin binding of grafted lasso peptides. J. Med. Chem. 57, 5829–5834 [DOI] [PubMed] [Google Scholar]
- 31. Maksimov M. O., and Link A. J. (2014) Prospecting genomes for lasso peptides. J. Ind. Microbiol. Biotechnol. 41, 333–344 [DOI] [PubMed] [Google Scholar]
- 32. Sirota-Madi A., Olender T., Helman Y., Brainis I., Finkelshtein A., Roth D., Hagai E., Leshkowitz D., Brodsky L., Galatenko V., Nikolaev V., Gutnick D. L., Lancet D., and Ben-Jacob E. (2012) Genome sequence of the pattern-forming social bacterium Paenibacillus dendritiformis C454 chiral morphotype. J. Bacteriol. 194, 2127–2128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jeanne Dit Fouque K., Afonso C., Zirah S., Hegemann J. D., Zimmermann M., Marahiel M. A., Rebuffat S., and Lavanant H. (2015) Ion mobility–mass spectrometry of lasso peptides: signature of a rotaxane topology. Anal. Chem. 87, 1166–1172 [DOI] [PubMed] [Google Scholar]
- 34. Chiu J., Tillett D., Dawes I. W., and March P. E. (2008) Site-directed, ligase-independent mutagenesis (SLIM) for highly efficient mutagenesis of plasmids greater than 8kb. J. Microbiol. Methods 73, 195–198 [DOI] [PubMed] [Google Scholar]
- 35. Chiu J., March P. E., Lee R., and Tillett D. (2004) Site-directed, ligase-independent mutagenesis (SLIM): a single-tube methodology approaching 100% efficiency in 4 h. Nucleic Acids Res. 32, e174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Willker W., Leibfritz D., Kerssebaum R., and Bermel W. (1993) Gradient selection in inverse heteronuclear correlation spectroscopy. Magn. Reson. Chem. 31, 287–292 [Google Scholar]
- 37. Bystrov V. F., and Arseniev A. S. (1988) Diversity of the gramicidin A spatial structure: two-dimensional 1H NMR study in solution. Tetrahedron 44, 925–940 [Google Scholar]
- 38. Klages J., Neubauer C., Coles M., Kessler H., and Luy B. (2005) Structure refinement of cyclosporin A in chloroform by using RDCs measured in a stretched PDMS-gel. ChemBioChem 6, 1672–1678 [DOI] [PubMed] [Google Scholar]
- 39. Kessler H. (1990) Reinvestigation of the conformation of cyclosporin A in chloroform. Helv. Chim. Acta 73, 1818–1832 [Google Scholar]
- 40. Al Toma R. S., Kuthning A., Exner M. P., Denisiuk A., Ziegler J., Budisa N., and Süssmuth R. D. (2015) Site-directed and global incorporation of orthogonal and isostructural noncanonical amino acids into the ribosomal lasso peptide capistruin. ChemBioChem 16, 503–509 [DOI] [PubMed] [Google Scholar]
- 41. Piscotta F. J., Tharp J. M., Liu W. R., and Link A. J. (2015) Expanding the chemical diversity of lasso peptide MccJ25 with genetically encoded noncanonical amino acids. Chem. Commun. 51, 409–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wieckowski B. M., Hegemann J. D., Mielcarek A., Boss L., Burghaus O., and Marahiel M. A. (2015) The PqqD homologous domain of the radical SAM enzyme ThnB is required for thioether bond formation during thurincin H maturation. FEBS Lett. 589, 1802–1806 [DOI] [PubMed] [Google Scholar]
- 43. Fieulaine S., Morera S., Poncet S., Mijakovic I., Galinier A., Janin J., Deutscher J., and Nessler S. (2002) X-ray structure of a bifunctional protein kinase in complex with its protein substrate HPr. Proc. Natl. Acad. Sci. U.S.A. 99, 13437–13441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nessler S. (2005) The bacterial HPr kinase/phosphorylase: A new type of Ser/Thr kinase as antimicrobial target. Biochim. Biophys. Acta 1754, 126–131 [DOI] [PubMed] [Google Scholar]
- 45. Poncet S., Mijakovic I., Nessler S., Gueguen-Chaignon V., Chaptal V., Galinier A., Boël G., Mazé A., and Deutscher J. (2004) HPr kinase/phosphorylase, a Walker motif A-containing bifunctional sensor enzyme controlling catabolite repression in Gram-positive bacteria. Biochim. Biophys. Acta 1697, 123–135 [DOI] [PubMed] [Google Scholar]
- 46. Ficarro S. B., McCleland M. L., Stukenberg P. T., Burke D. J., Ross M. M., Shabanowitz J., Hunt D. F., and White F. M. (2002) Phosphoproteome analysis by mass spectrometry and its application to Saccharomyces cerevisiae. Nat. Biotechnol. 20, 301–305 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.