

Abstract
Epithelial-Mesenchymal Transition (EMT) is a normal developmental program that is considered to also play an important role in cancer metastasis. Ultimate inducers of EMT are transcriptional repressors that individually can induce experimental EMT, yet in many cells, particularly cancer cells, multiple inducers are expressed simultaneously. Why, and if and how they interact to regulate EMT is unanswered. Using RNAi technology to effect protein knockdown and avoid potential over-expression artifact coupled with transient TGFβ treatment to better mimic in vivo conditions we show, in both non-tumorigenic and tumorigenic epithelial cancer cells, that Snail1 is uniquely required for EMT initiation, while Twist1 is required to maintain late EMT. Twist1, present in resting epithelial cells, is dispensable for EMT initiation. Mechanistically, in response to transient TGFβ treatment, transient Snail1 expression represses Twist1 transcription directly, which is subsequently upregulated, as Snail1 levels decrease, to sustain E-cadherin downregulation and growth arrest of EMT. Persistent Twist1 expression is associated with a p38 and ERK signal feedback loop that sustains growth-inhibitory signals characteristic of quiescent micrometastatic tumors. This Snail1-Twist1 temporal and spatial cooperation was also observed in vivo during human breast cancer progression to metastasis. Twist1 level, but not Snail1 level, and Twist1:Snail1 ratio in disseminated micrometastatic bone marrow tumor cells was found to correlate with survival and treatment resistance, and is highly predictive of metastatic or recurrent disease.
Keywords: Snail1, Twist1, EMT, transient TGFβ1, Micrometastasis
Introduction
Cancer metastasis remains the leading cause of cancer mortality. The metastatic process has been shown to share many parallels with Epithelial to Mesenchymal Transition (EMT), a normal and reversible morphogenetic program that facilitates cell movements during development (1). In both developmental and cancer EMT, signals initiating and regulating this process emanate from the tissue microenvironment and include TGFβ, Wnt, HGF, EGF, and hypoxia (1). In vivo, whether these signals are transiently or continuously present to initiate and maintain EMT is unclear (1). However, intravital microscopy of invading xenografted tumor cells found that at least for TGFβ signals are both transient and spatially restricted, yet affected cells appear to retain their EMT status long after they have migrated away from the signaling source (2). This suggests that there exists mechanism to not only initiate but to also maintain EMT. These signals could be cell autonomous, cell non-autonomous, or both. Importantly, whether the molecular regulation of EMT at these different stages is similar or different is unknown.
Despite the disparate nature of EMT stimulating signals, they all converge upon a limited set of transcriptional repressors that are considered to be the ultimate inducers of EMT. These include the Zinc finger transcription repressors Snail1 (Snail), Snail2 (Slug), and ZEB1/2, and the basic helix-loop-helix (bHLH) proteins Twist1/2 that directly bind E-boxes in promoters, and other unrelated factors such as Goosecoid and Foxc2 that repress transcription indirectly through unknown mechanisms (3, 4). Snail1 has been shown to be critical for cell movement during gastrulation in many organisms and elevated levels of Snail1 in primary breast cancers independently predict for decreased relapse-free survival (5, 6). However, several EMT inducers are often expressed at the same time and in the same cells, especially in cancers (7, 8). The functional reason for the concurrent presence of multiple EMT inducers is unknown.
One possibility is that they serve redundant or overlapping functions. This is supported by the observation that when individually overexpressed, all EMT inducers induce indistinguishable EMT (1). But the levels achieved in these experiments are much greater than those observed during biologically [e.g., gastrulation (9)] or pathologically relevant (e.g., cancer (1, 2, 10)) EMT and the ensuing EMT occurs independent of any added extrinsic factors. Most cell based EMT studies, in the absence of over-expressed EMT inducer, have relied upon continuous treatment with environmental factors for days to weeks to force epithelial cells into EMT (8, 11). Whether continuous exposure to EMT signals truly represents conditions in vivo is a concern. An alternative model to explain the concurrent presence of multiple EMT inducers in cells is that the various EMT inducers functionally cooperate at different temporal (e.g., initiation versus maintenance) and spatial (primary tumor versus metastases) stages of EMT.
To determine why cancer cells concurrently express multiple EMT inducers we determined and contrasted the endogenous expression patterns of several EMT inducing factors in multiple human epithelial and carcinoma cell lines. All cell lines studied undergo EMT in response to traditional continuous TGFβ1 treatment, but also when exposed to short (4h), transient TGFβ1 treatment. To determine the roles of the various EMT inducing factors in the EMT process, in response to TGFβ, and avoiding potential over-expression artifact, we shRNAi-depleted each factor individually and determined the effect upon EMT initiation and maintenance. We found that Snail1 was uniquely required for the initiation of EMT while Twist1 cooperated temporally to maintain late EMT. This was only apparent when cells were treated with transient, not continuous, TGFβ. Twist1 was found to sustain late EMT-associated cell growth arrest by inducing a low ERK:p38 activity ratio. Inhibiting p38 or reducing Twist1 levels could abrogate the growth arrest. This novel Snail1-Twist1 temporal and spatial cooperation was also observed in human breast cancer metastasis. While Snail1 level in the primary tumor predicted for metastasis, Twist1 level and Twist1:Snail1 expression ratio in bone marrow micrometastatic tumor cells was found to be highly predictive for distant relapses.
Methods
Cell culture reagents and Antibodies
MCF10A were maintained in DME:F12 media supplemented with 5% horse serum (Invitrogen), 10 µg/ml insulin, 10 ng/ml EGF, 0.5 µg/ml hydrocortisone, 100 ng/ml cholera toxin (Sigma) and 100 units/ml Pen/Strep (Invitrogen). HMLER cells were maintained in serum-free media (Lonza) supplemented with bovine pituitary extract, 10 µg/ml insulin, 10 ng/ml EGF, 0.5 µg/ml hydrocortisone, and Gent/Ampho (Lonza). A549, HEK293 and HEK293T cells were maintained in DMEM supplemented with 10% FBS and 100 units/ml Pen/Strep (Invitrogen). During continuous treatment condition, TGFβ1 (Sigma) was replenished every 48h. SB203580 (Calbiochem) was replenished every 24h. All antibodies were used according to the manufacturers’ recommendations. See supplemental procedures for a complete list.
DNA constructs, lentiviral production and transduction
The retroviral vector pBABE expressing either a shRNA or Snail1 were transfected with packaging plasmids into amphotropic HEK293 cells using LT1 transfection reagent (Mirus). 48h later, viral supernatants were collected, filtered, and used to infect MCF10A cells twice for 4h each time over a 24h period. Cells were allowed to recover for 24h before experiments. See supplemental procedures for shRNA sequences.
Chromatin immunoprecipitation
ChIP assay was performed according to Upstate Biotechnology’s ChiP kit.
Isolation and analyses of primary breast tumors and BM DTCs
Informed consents were obtained accordingly to IRB policy. See supplemental experimental procedures and (12) (13) for the detailed procedure.
Results
Snail1 and Twist1 expression is inversely related during EMT initiation and maintenance
Several EMT inducing transcription factors are expressed simultaneously in cells that undergo EMT (7, 8). Why, whether they cooperate, and if so how during developmental or pathologic EMT is unknown. To explore these questions we first searched for an ex vivo culture systems of TGFβ-induced EMT in which multiple EMT inducers were present. MCF10A cells are a “normal” non-tumorigenic breast epithelial cell line that has been extensively used to study EMT. MCF10A cells began to undergo EMT within 2 days of exposure to continuous TGFβ1, as determined by a change to mesenchymal morphology, epithelial E-cadherin and ZO-1 down-regulation and mesenchymal Vimentin and N-cadherin up-regulation (Fig. S1A).
We determined and contrasted the temporal expression pattern of mRNA, and in some cases protein, of the various EMT inducers present in these cells in response to continuous TGFβ1 treatment. During these experiments cell density was equivalent (~ 90% confluent) at all points of analysis, and cells were not split. Snail1 protein peaked by 24h (Fig. 1B and C) then leveled off, while its mRNA peaked at 8h. This was significantly earlier than any other EMT inducing factors examined (Fig. 1E and F, and S1D). Surprisingly, and in contrast to other experimental systems in which Twist1 is overexpressed (14, 15), endogenous Twist1 mRNA and protein was maximal in untreated, epithelial MCF10A cells and downregulated during continuous TGFβ1 treatment (Fig. 1B, D, E, and G). The initial rate of decay of Twist1 levels appeared to parallel upregulation of Snail1 suggesting a possible inverse transcriptional link between Snail1 and Twist1 expression during EMT induction in MCF10A cells (see later).
Figure 1. Transient TGFβ1-induced EMT in MCF10A cells.
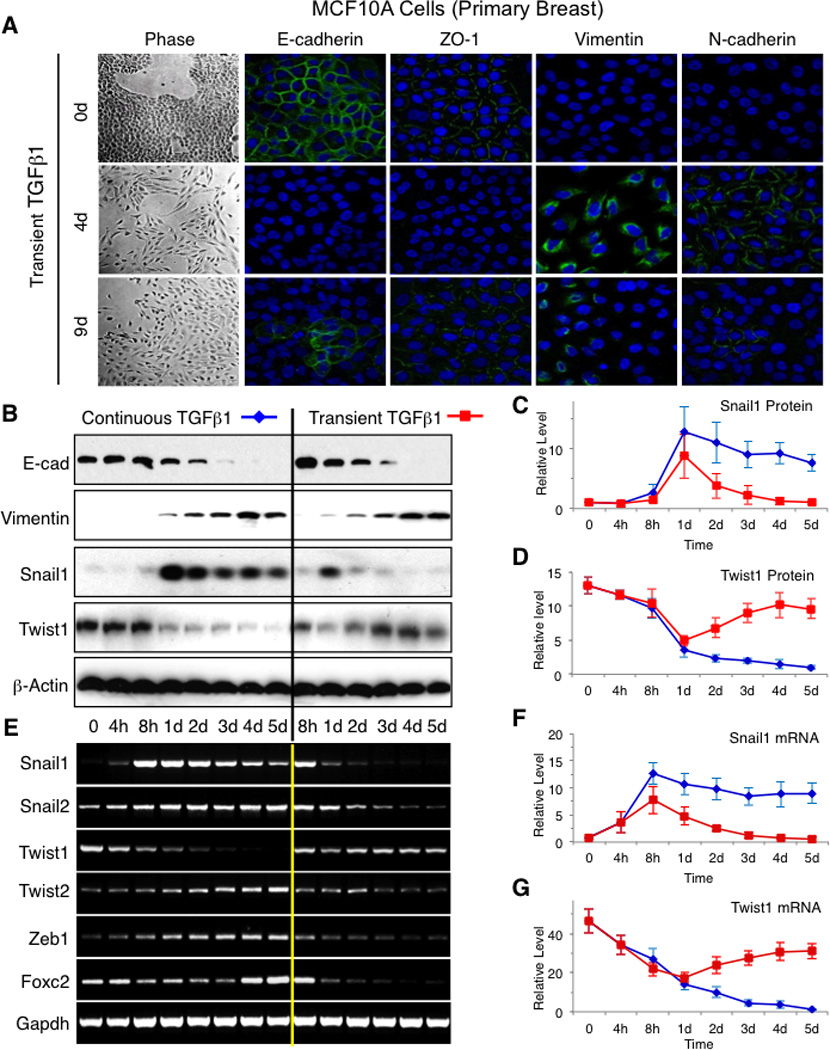
(A) Phase contrast photographs and immunofluorescent photographs for indicated proteins at indicated times of MCF10A cells treated with transient TGFβ1 for 4h. DAPI (blue) was used to detect nuclei in all panels. Representative results of 3 independent experiments are shown. (B – D) Western blotting for indicated proteins of whole cell lysates (WCL) from MCF10A cells treated with 2 ng/ml TGFβ1 either continuously or transiently for 4h and cells collected at indicated times (B) Graphs of densitometric intensity of Snail1 protein (C) and Twist1 protein (D) from MCF10A cells treated with TGFβ1 as in (B). (E – G) RT-PCR for indicated mRNA of total RNA from MCF10A cells treated with 2ng/ml TGFβ1 either continuously or transiently for 4h and cells collected at indicated times (E). Graphs of densitometric intensity of Snail1 mRNA (F) and Twist1 mRNA (G) from MCF10A cells treated with TGFβ1 as in (E). Error bars represent mean +/− s.e.m. from at least 3 independent experiments. See also related Figures S1 and S2.
Recent work has suggested that tumor TGFβ1 signals, in vivo, can be transient (1), thus we asked whether MCF10A cells would undergo EMT when exposed to progressively decreasing time of TGFβ1 (Fig. S1B). We found that just 4 hours of treatment with 2ng/ml TGFβ1 (hereafter referred to as transient TGFβ1) were enough to result in EMT, morphologically, molecularly (Fig. 1A, 1B, and S1B), and functionally (i.e. increased migration in a scratch wound migration assay) (Fig. S1C), with kinetics similar to cells exposed to continuous TGFβ1. However, mesenchymal phenotype began to reverse after 9 days as evidenced by the re-expression of E-cadherin and loss of Vimentin and N-cadherin expression (Fig. 1A and S1F). This indicated that although a single transient TGFβ1 dose was sufficient to induce EMT it did not sustain the differentiation program indefinitely. Snail1 expression followed the same induction kinetics following exposure to either continuous or transient TGFβ1- very low in untreated cells followed by rapid induction of expression. However, only with transient TGFβ1 treatment did Snail1 levels rapidly disappear by day 2 (Fig. 1B, C, E and F). Despite this cells still remained mesenchymal at days 4 and 5 (Fig. 1A and B) suggesting that while Snail1 might contribute to EMT initiation other inducers were required to maintain EMT. We noted that as Snail1 levels diminished Twist1 levels began to rise and remained high throughout the 5 days of analysis, again highlighting their reciprocal relationship during EMT, (Fig. 1B, D, E, and G). These persistently high levels of Twist1 were not observed when MCF10A cells were treated with continuous TGFβ1 (Fig. 1B, D, E, and G). The level of other EMT inducing factors (Snail2, Twist2, Zeb1, and FOXC2) decreased in response to transient TGFβ1 treatment (Fig. 1E and S1E).
To determine whether the EMT response of MCF10A cells to transient TGFβ1 and the pattern of expression of EMT inducing factors observed in these cells were unique to MCF10A cells, a non-tumorigenic primary mammary epithelial cell line, we repeated the above experiments in 2 other human cell lines from 2 different organs, breast HMLER cells and lung A549 cells. The mammary epithelial cell line HMLER is tumorigenic in vivo. It is derived from the human primary mammary epithelial cells (HuMEC) that were transformed with telomerase (hTERT), large T antigen of SV40 virus and oncogenic RasV12 (16). In response to continuous TGFβ1 treatment, HMLER cells underwent EMT in culture, as expected (8). The lung cancer cell line A549 was derived from a patient with non-small cell lung adenocarcinoma. A549 cells are fully transformed, oncogenic K-RasV12-positive, and capable of forming large tumors in nude mice that are highly metastatic (17, 18). They underwent EMT within 2 days of exposure to continuous TGFβ1 (19, 20).
We treated these two cell lines with either continuous or transient TGFβ1 and profiled the expression pattern of EMT inducing factors. With differing kinetics from MCF10A cells, all underwent morphologic and molecular EMT when exposed to continuous or transient TGFβ1 – 12 days for HMLER cells and 3 days A549 cells (Fig. S2). As with MCF10A, EMT was reversible in A549 cells following transient TGFβ1 treatment (Fig. S2). For both TGFβ1 treatment regimens and cell lines, the pattern of EMT factor expression during EMT induction, maintenance and reversal was essentially similar to that observed for MCF10A cells (Fig. S2A, B, and D).
In sum, these results from both non-tumorigenic cells (MCF10A) and tumorigenic cancer cells (HMLER and A549) indicated that EMT could be induced in cells derived from different tissues and with distinct tumorigenic potentials, by only short, transient exposure to TGFβ1. Moreover, Snail1, absent in untreated cells, was the first EMT inducer to be upregulated in response to TGFβ1, and Twist1 level, present in untreated epithelial cells, exhibited an inverse correlation with Snail1 level during TGFβ1-induced (transient or continuous) EMT.
Snail1 is uniquely required for EMT initiation, in response to TGFβ
Of all EMT inducers the Snail1 expression pattern during TGFβ1-induced EMT was unique in that it was not expressed in untreated epithelial cells yet rapidly induced in response to added TGFβ1. Thus we asked whether any one EMT inducing factor alone was necessary to initiate EMT. To avoid potential artifact from overexpressing EMT inducing factors, we RNAi-depleted each EMT inducing factor individually in MCF10A cells. At least 85% depletion of each factor was obtained, as verified by Western blotting (where antibodies are available) and/or RT-PCR (Fig. 2 and S3). To ensure RNAi specificity for each EMT inducing factor, 2 or more shRNAi were utilized and gave similar results. Lentivirus shRNAi-infected cells were exposed to continuous TGFβ1 to induce EMT. Continuous TGFβ1 treatment was utilized instead of transient TGFβ in order to provide an overwhelming and prolonged signal and thereby ensure adequate EMT initiation for all MCF10A cell derivatives. Control luciferase shRNAi-transduced MCF10A cells underwent EMT with similar kinetics as observed in the parental uninfected cells (Fig, 2A, compared to Fig 1B). Only Snail1-depleted MCF10A cells were blocked in their capacity to undergo TGFβ1-induced EMT (Fig. 2A). shRNAi-based depletion of Twist1, Twist2, Snail2, and Zeb1 did not affect the ability of MCF10A cells to undergo EMT in response to continuous TGFβ1 (Fig. 2B and S3). In addition, Snail1 depletion also resulted in decreased cell numbers that was further decreased with TGFβ1 treatment (Fig. S4A), consistent with Snail1’s role as a survival factor (21). These changes were specific to Snail1 depletion as the expression patterns of other EMT inducing factors were unchanged in Snail1-depleted cells, except for a delay in Twist1 down regulation (Fig. S4B) (see later).
Figure 2. Snail1, but not Twist1, is uniquely required for the initiation of TGFβ1-induced EMT.
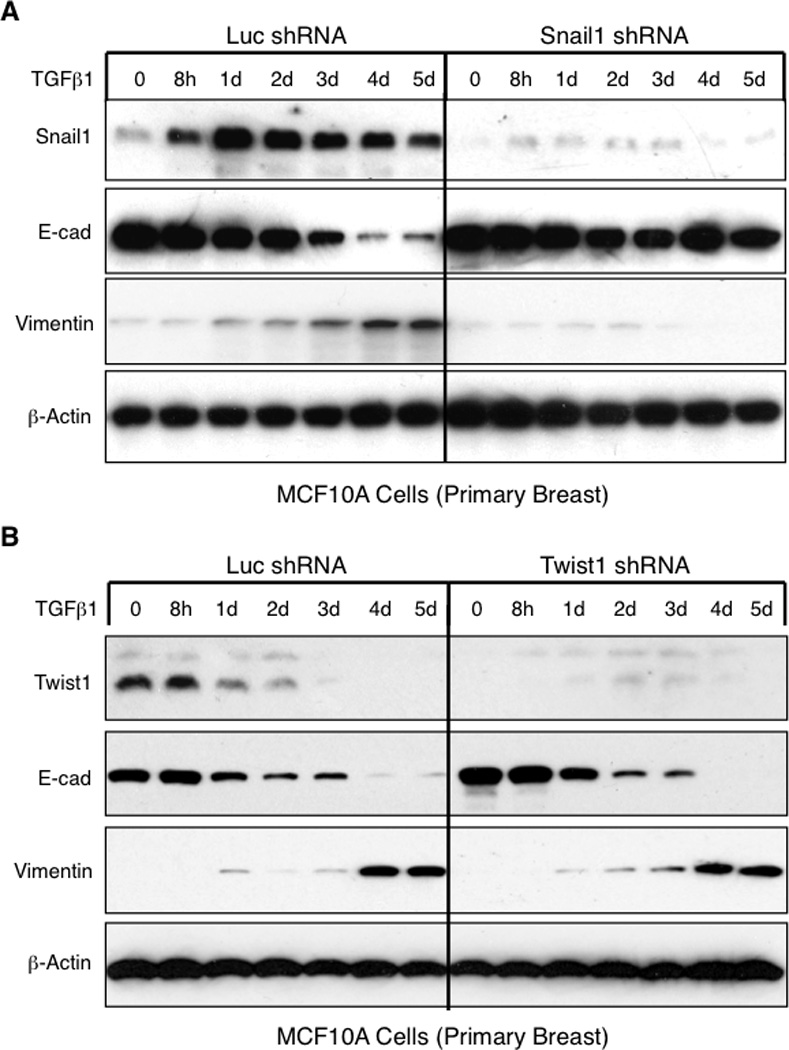
(A and B) Western blotting for indicated proteins of WCL from MCF10A cells transduced with a lentivirus expressing either the control luciferase or Snail1 shRNA (A) or Twist1 shRNA (B), then treated with 2 ng/ml TGFβ1 continuously for indicated times. Representative results of 3 independent experiments are shown. See also related Figures S3 and S4.
To confirm the requirement of Snail1 for EMT induction in tumorigenic cell lines, we shRNAi-depleted Snail1 in both HMLER and A549 cells and exposed them to continuous TGFβ1. As observed with MCF10A cells, Snail1 depletion, but not Twist1 depletion, led to complete abolition of EMT (Fig. S6A and B, left panels). Thus in three different cell lines - untransformed, transformed and tumorigenic, and of breast and lung origins, Snail1 was uniquely required to initiate TGFβ1-induced EMT in ex vivo cultured cells.
Twist1 is required to maintain EMT
When MCF10A, HMLER and A549 cells were exposed to transient TGFβ1 Snail1 expression was rapidly induced and then quickly disappeared while EMT was maintained (Fig. 1 and S2), indicating that although Snail1 was required for EMT initiation it was dispensable for maintaining EMT. One explanation for the expendability of Snail1 in the maintenance phase of EMT is that another EMT inducing factor takes over to maintain EMT. Based upon the reciprocal relationship between Snail1 and Twist1 levels and the fact that Twist1 level increased late in EMT following transient TGFβ signals (Fig. 1 and S2) we suspected that Twist1 could be that factor. To test this possibility, Twist1, Twist2, or Snail2 were individually shRNAi-depleted in MCF10A cells. Cells were then treated with TGFβ1 either continuously or transiently. Similar to previous results (Fig. 2 and S3), depletion of Twist1, Twist2 or Snail2 did not alter E-cadherin downregulation when cells were treated with continuous TGFβ1 (Fig. 3A and B, and S5), likely because Snail1 levels remained high. However, when cells were treated with transient TGFβ1, E-cadherin repression on day 4 did not occur in Twist1-depleted cells (Fig. 3A). In Twist2- or Snail2-depleted cells E-cadherin suppression persisted, like in control cells (Fig. 3B and S5).
Figure 3. Twist1, but not Twist2, is required for EMT maintenance in MCF10A cells.
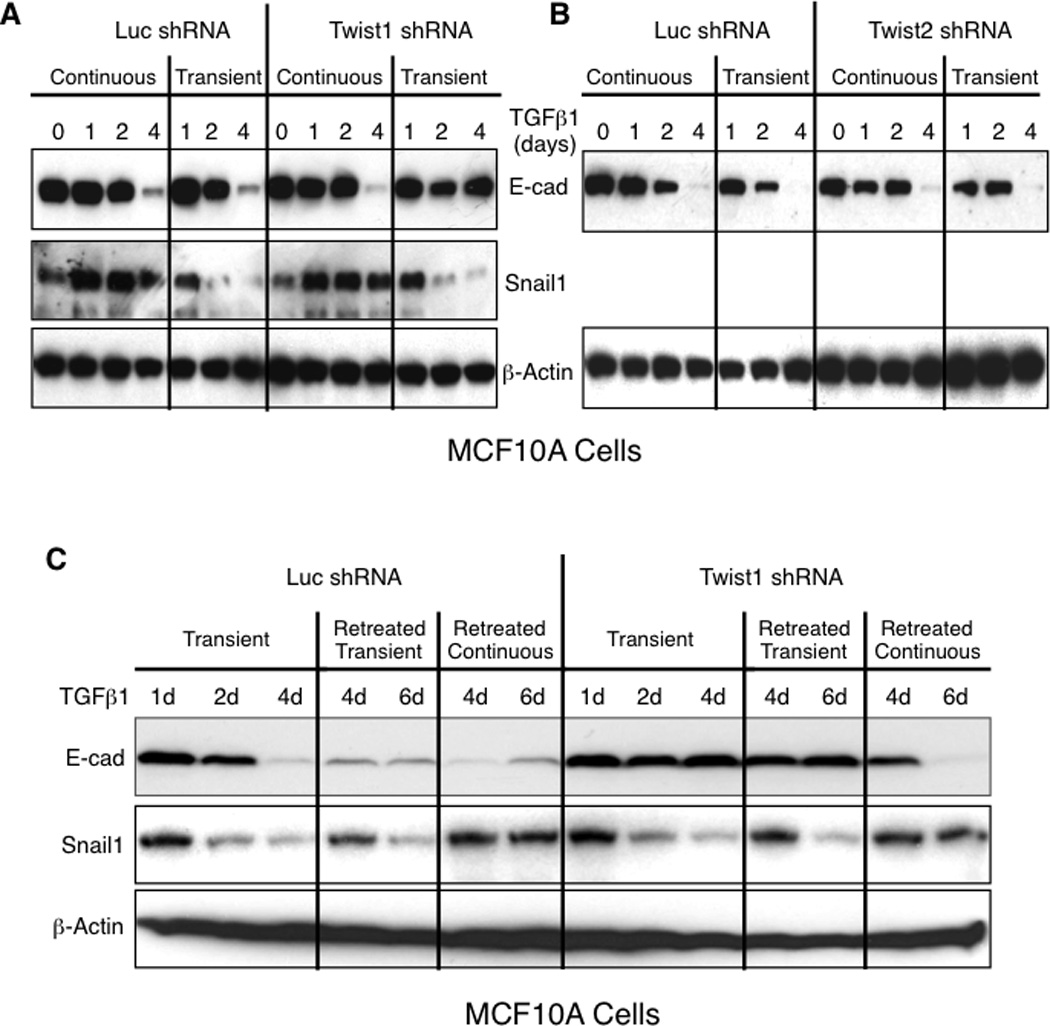
(A and B) Western blotting for indicated proteins of WCL from MCF10A cells transduced with a lentivirus expressing either the control luciferase shRNA or Twist1 shRNA (A) or Twist2 shRNA (B), and treated with 2 ng/ml TGFβ1 either continuously or transiently for 4h, and cells collected at indicated times. Representatives of 3 independent experiments are shown. See also related Figure S4. (C) Western blotting for indicated proteins of WCL from MCF10A cells transduced with a lentivirus expressing either the control luciferase or Twist1 shRNA, treated with 2ng/ml TGFβ1 transiently for 4h, and then either not retreated or retreated on day 3 with 2ng/ml TGFβ1 either transiently for 4h or continuously, and cells collected at indicated times. Representatives of 3 independent experiments are shown. See also related Figures S5–S7.
We next asked whether Twist1 was similarly required to maintain or sustain EMT in HMLER and A549 cells treated with transient TGFβ1. Twist1 depletion specifically resulted in the absence of E-cadherin repression and Vimentin upregulation at day 12 and 4 in HMLER and A549 cells, respectively, whereas in control luciferase shRNAi-treated cells EMT was maintained (Fig. S6 and S7). As with MCF10A cells, this was only observed when cells were treated with transient TGFβ1, as Snail1 expression persisted in HMLER and A549 cells when treated with continuous TGFβ1 (Fig. S6 and S7).
In sum, these findings indicated that Twist1, specifically, was required to sustain or maintain E-cadherin repression and Vimentin upregulation in the later phase of EMT. Importantly, this function of Twist1 was masked under conditions of continuous TGFβ1 treatment, likely due to persistent Snail1 expression.
Since TGFβ stimulation during tumor invasion, in vivo, can be transient and localized (2) tumor cells may be exposed to repeated waves of intermittent TGFβ either in the primary tumor, en route to, or in the metastatic organ. Therefore to determine whether the requirement of Twist1 to maintain E-cadherin repression during EMT is reversible, we treated Twist1-depleted MCF10A cells with transient TGFβ1, washed it away, and then repeated TGFβ1 treatment at day 3 either transiently (4h) or continuously (3 days). Recurrent exposure to transient TGFβ1 did not relieve the requirement for Twist1 to maintain E-cadherin repression, as evidenced by constantly high levels of E-cadherin at days 4 and 6 (Fig. 3C, lanes 11 and 12), likely due to the absence of Snail1 expression during EMT maintenance phase (Fig. 3C, lanes 11 and 12 of the Snail1 panel). However, when cells were re-exposed to continuous TGFβ1, Twist1 was no longer necessary to maintain E-cadherin repression, presumably due to the sustained expression of Snail1 in cells treated with continuous TGFβ1 (Fig. 3C, lanes 13 and 14). In control cells (luciferase shRNA), re-exposure to TGFβ1, regardless of whether transient or continuous, E-cadherin expression remained suppressed (Fig. 3C, lanes 4–7).
Taken together, these data revealed a temporal regulation of EMT by Snail1 and Twist1. Snail1 was required for the initiation of EMT while Twist1, dispensable for EMT initiation, was required to maintain EMT. This temporal cooperation between Snail1 and Twist1 was only evident under conditions of transient TGFβ1 treatment.
Snail1 binds to the Twist1 promoter to repress its transcription
We observed a reciprocal pattern of Snail1 and Twist1 expression, suggesting the possibility that Snail1 may transcriptionally regulate Twist1 expression. To test this, we first asked if Snail1 overexpression resulted in Twist1 suppression, which would be predicted if Twist1 was a transcriptional target of the repressor Snail1. Indeed, Twist1 level was significantly reduced in MCF10A cells overexpressing Snail1 (Fig. 4). As observed when Twist1 was overexpressed in MCF10A cells (Fig. S11A), constitutive overexpression of Snail1 resulted in robust EMT, independent of added TGFβ1 (Fig. 4).
Figure 4. Snail1 transcriptionally represses Twist1 expression.
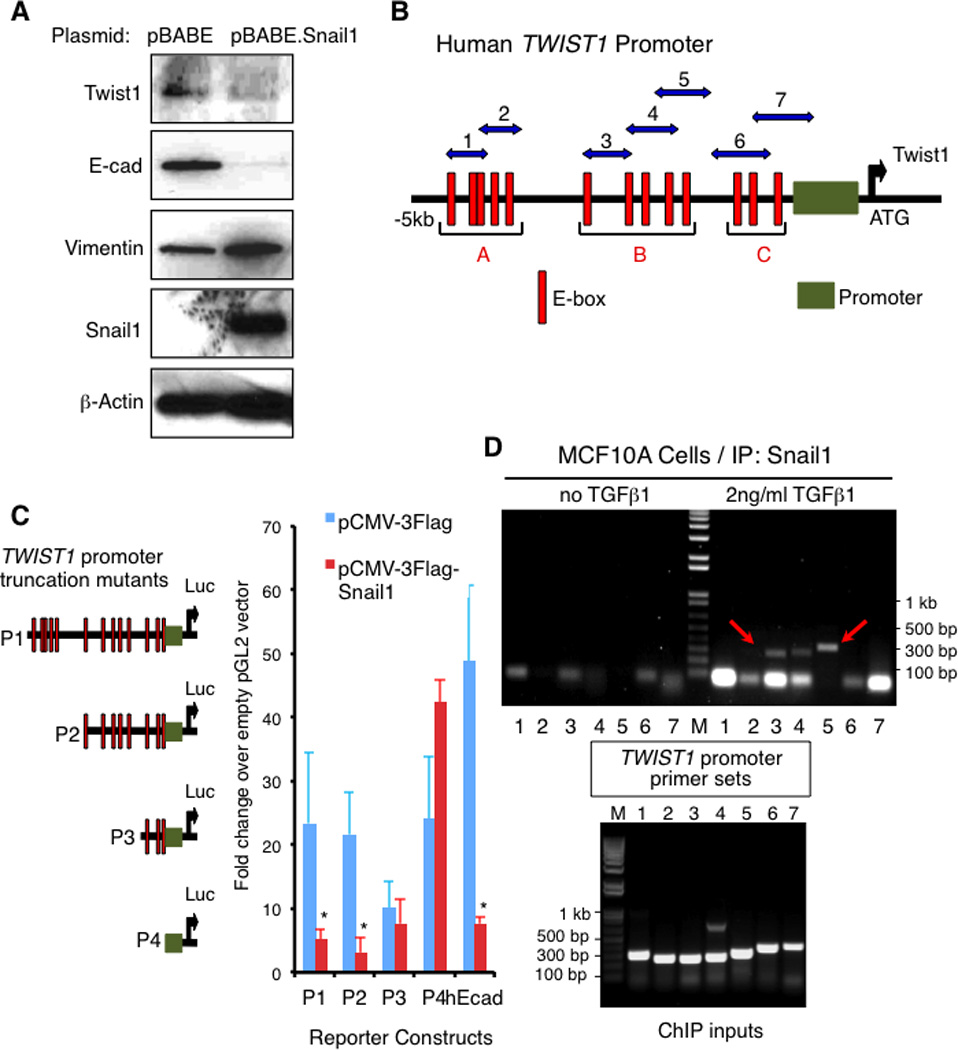
(A) Western blotting for indicated proteins of WCL from MCF10A cells transduced with a lentivirus expressing either the vector pBABE or Snail1. (B) Schematic of the human Twist1 promoter showing 13 consensus E-boxes grouped into 3 main clusters A to C. Green box: minimal Twist1 promoter; Red box: E-box; ATG: Transcription start site; Numbered blue double arrows: primer sets 1–7. (C) Luciferase reporter assay in HEK293T cells using mutants (P1-P4) of the human Twist1 promoter fused with the luciferase reporter plasmid pGL2 (left), and co-transfected with either a pCMV-3Flag-Snail1 plasmid or the empty vector (right). hEcad: Human E-cadherin promoter used as a positive control for Snail1-dependent repression. Fold changes in luciferase activity over the empty pGL2 vector are shown. Each experiment was performed in triplicate. Error bars represent means +/− s.e.m. Representatives of 3 independent experiments are shown. *, p < 0.05, Student’s T-test. (D) Upper: Chromatin immunoprecipitation using a Snail1 monoclonal antibody in nuclear extracts from MCF10A cells either untreated or treated with 2ng/ml TGFβ1 for 2 days. Immunoprecipitated chromatins were subjected with PCR using primer sets 1 to 7 as shown in (B). Lower: PCR using primer sets 1 to 7 of total chromatin inputs. Representatives of 3 experiments are shown. See also related Figure S8.
DNA sequence analysis of the human Twist1 promoter region revealed the presence of 13 consensus E-boxes within 5 kilobases of the transcriptional start site that could be grouped into 3 main clusters upstream of the minimal promoter (22) (Fig. 4B). To determine whether Snail1 could repress transcriptional activity of the Twist1 promoter and if so to identify potential binding sites of Snail1 in the Twist1 promoter, successive truncation mutants of the Twist1 promoter (P1–P4) were fused to a luciferase reporter (Fig. 4C, left panel), co-transfected with either a pCMV-Snail1 plasmid or control empty vector into HEK293 cells, and Twist1 promoter function measured as luciferase activity. The human E-cadherin promoter luciferase reporter was used as a positive control for a Snail1-repressed promoter. Similar to its effect on the E-cadherin promoter (7-fold repression), Snail1 decreased the transcriptional function of the Twist1 promoter mutants P1-P2 by 4-fold (Fig. 4C). However, this ability was lost when the E-box cluster B was deleted (mutant P3, Fig. 4C). To confirm that endogenous Snail1 binds the endogenous Twist1 promoter at the same region, in a TGFβ1-dependent manner, we performed chromatin immunoprecipitations (ChIP) from nuclear extracts of MCF10A cells either untreated or treated with TGFβ1 for 2 days. Consistent with reporter mapping, the E-box cluster B region of the Twist1 promoter was immunoprecipitated with Snail1 monoclonal antibodies but only in cells treated with TGFβ1 (Fig. 4D), using three different primer sets covering the B cluster of E-boxes (Fig. 4B). A primer set specific for the known Snail1-binding site on the human E-cadherin promoter (23) was used as a positive control for a Snail1-binding promoter, and showed similar TGFβ1-dependent promoter binding (Fig. S8).
These results indicated that Snail1 could repress Twist1 transcription by binding to the Twist1 promoter. Therefore when Snail1 levels are induced (initial EMT), Twist1 is repressed. As Snail1 expression returns to a low baseline level, after TGFβ1 withdrawal, Twist1 transcription becomes de-repressed and its level begins to rise. We cannot exclude the possibility of additional Snail1-independent TGFβ1 effects on early Twist1 expression, however.
Temporal and spatial cooperativity of Snail1 and Twist1 in breast cancer metastasis: Twist1:Snail1 expression ratio in bone marrow micrometastases is highly predictive for distant recurrences in human breast cancer
Whether cancer EMT contributes to the survival and temporary quiescence of micrometastases in secondary organs is not known. In multiple studies Snail1 levels in primary breast tumors is predictive for metastatic relapse (6, 24). Our new results, led us to ask whether Twist1, through its temporal cooperation with Snail1 in EMT maintenance, might also cooperate spatially with Snail1 during breast cancer progression to contribute to the survival and relative dormancy of micrometastic breast cancer in secondary organs.
The temporal cooperation of Snail1 and Twist1 in EMT of breast epithelial cells (MCF10A) and breast cancer cells (HMLER), in culture, might predict for the following in vivo consequences: 1) since EMT initiation, a Snail1-dependent process, occurs in primary tumors Snail1 expression in primary breast tumors would correlate with distant recurrences. However, in micrometastases, such as bone marrow (BM) disseminated tumor cells (DTCs) that are present in a significant number of clinically asymptomatic patients, Snail1 would no longer be necessary as these tumor cells would be in a late or maintenance stage of EMT as well as possibly dormant. 2) Twist1 expression in primary tumors would not correlate with distant recurrence as Twist1 is dispensable for EMT initiation, whereas in BM DTCs it should since Twist1 is required to maintain EMT. 3) If (1) and (2) are indeed true then the Twist1:Snail1 expression ratio in BM DTCs could be a strong predictor of tumor relapse in foreign organs (i.e., metastasis) as the ratio takes into account the inverse relationship and temporal cooperation between these two EMT inducing factors.
To test these predictions we interrogated samples derived from a unique, previously reported cohort of 100 stage II or III breast cancer patients (i.e. locally invasive tumors without overt evidence of distant metastases) 30 of whom had evidence for clinically silent BM DTCs. Paired specimens of primary tumors and BM DTCs were both collected at the time of diagnosis (12). None of these patients had received any radiation or chemotherapy prior to tumor sample acquisition. Of these 30 patients, 15 remained relapse-free with an average follow-up of 5 years, and the other 15 had distant recurrences within 1.5 years of the initial surgery with the most common metastatic sites being bone, lung, liver and brain. To minimize sampling bias, the 2 groups of patients (nonrecurrent and recurrent) were matched for age and the status of tumor biomarkers including Estrogen Receptor (ER), Progesterone receptor (PR) and her2/Neu amplification, and subsequently received identical adjuvant chemotherapy (12). Rare BM DTCs were enriched from bone marrow using EpCAM immunoselection [for the detailed procedure, refer to(12) (13)]. Although EpCAM is considered an epithelial cell surface marker, it has been detected in non-epithelial cells [e.g. pluripotent stem cells (25) and germ cells (26)] and associated with poor prognosis and metastatic disease development in many cancers including breast cancer (27), unlike the more classic epithelial marker Cytokeratin (e.g. CK19) that has no significant prognostic implication in breast cancer in our studies (12, 13). Importantly, EpCAM expression has been found on putative CD44high/CD24low breast cancer tumor-initiating cells (28), which have been proposed to be derived from breast cancer EMT (8). In fact, in MCF10A cells exposed to transient TGFβ1 treatment, EpCAM expression diminished only modestly as cells entered later stages of EMT (Fig. S1F). However, EpCAM antigen may not be expressed on all clinically relevant DTC subpopulations. To rule out this possibility, we compared the molecular signatures of EpCAM-enriched DTCs to those of whole bone marrow and found that they were similar. Thus, EpCAM-based immune enrichment was effective at isolating a reasonable sampling of DTCs with varying degrees of epithelial or mesenchymal phenotypes (i.e. partial EMT or MET) and presumably including some of the putative cancer stem cells (12) (13) (29).
Total mRNAs were prepared from both primary tumors and BM DTCs and subjected to microarray analyses for Snail1, Twist1, and Snail2 (internal control) using Affymetrics platforms. Due to the relative rarity of BM DTCs making microarray analyses prone to potential artifact, we validated the microarray results for BM DTCs using quantitative reverse transcription-PCR (qRT-PCR) for Snail1, Twist1, and Snail2, which will be presented here instead. Student’s t-test was used to compare standard deviations of the 2 independent sample groups.
Snail1 mRNA level in primary breast tumors of the 30 patients with BM DTCs showed a trend toward correlation with metastasis, it did not reach statistical significance in this small patient cohort (p=0.056) (Fig. 5A). Thus we analyzed primary tumors from a larger cohort of patients (not matched with bone marrow sampling for DTCs) and in this group confirmed that Snail1 expression in primary tumor correlated with distant relapses (Fig. S9A), consistent with previous reports (6, 24). However, in BM DTCs, Snail1 levels were not correlated with metastasis (Fig. 5B).
Figure 5. The Twist1:Snail1 ratio in bone marrow micrometastases is highly predictive for distant recurrences in human breast cancer.
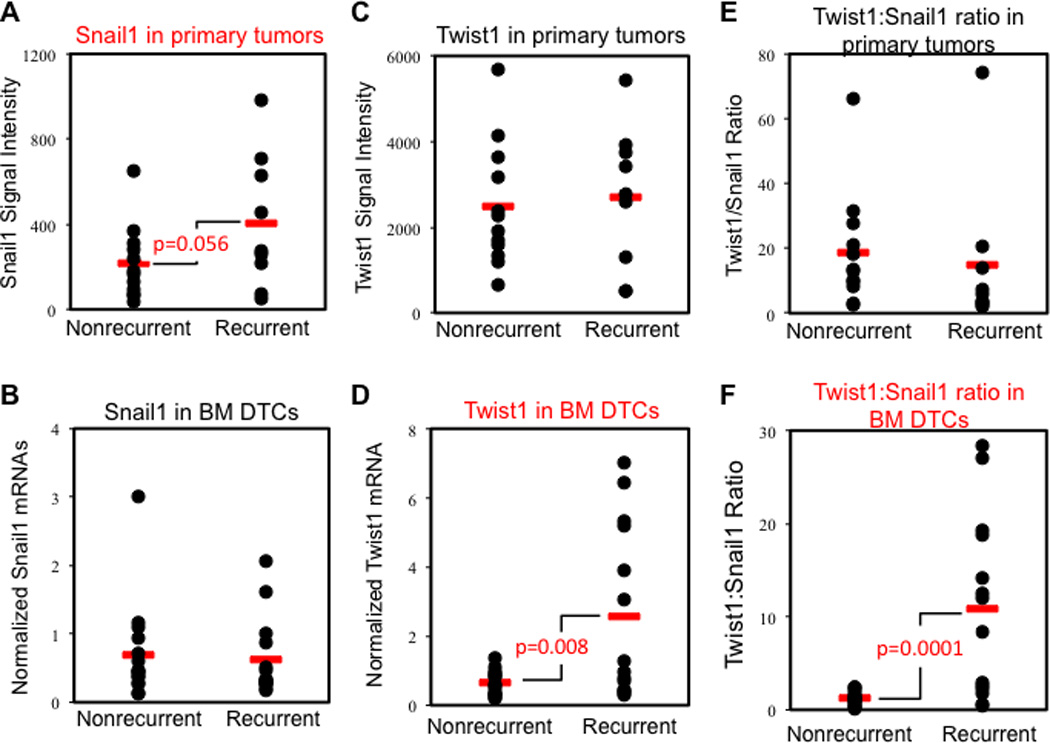
Total RNAs were isolated from paired samples of primary tumors and BM DTCs (micrometastases) and subjected to microarray analyses. Quantitative RT-PCR (qRT-PCR) for Snail1, Twist1, and Snail2 (internal control) mRNA was used to validate DTC data, which was normalized against Gapdh mRNA. See also related Figure S9.
(A) Snail1 array signals in primary tumors showed a trend toward correlation with distant recurrences (p= 0.056).
(B) Normalized Snail1 mRNA in BM DTCs did not correlate with distant recurrences.
(C) Twist1 array signals in primary tumors did not correlate with distant recurrences.
(D) Normalized Twist1 in BM DTCs correlates with distant recurrences (p= 0.008).
(E) Twist1:Snail1 ratio in primary tumors does not correlate with distant recurrences.
(F) Twist1:Snail1 ratio in BM DTCs highly correlates with distant recurrences (p=0.0001).
Twist1 mRNA levels were similar in the primary tumors from both the recurrent and non-recurrent patients (Fig. 5C). In contrast, high Twist1 expression was detected in BM DTCs and was strongly associated with metastatic recurrences (p=0.008) (Fig. 5D) and treatment resistance (12, 13). Although the calculated Twist1:Snail1 ratio in the 30 primary tumors of those patients with evidence for BM DTCs bore no connection with disease outcomes (Fig. 5E), its value in BM DTCs showed a striking correlation with distant metastasis (p=0.0001) (Fig. 5F). The ratio was more significant than either Twist1 alone in BM DTCs or Snail1 alone in primary tumors. As an internal control, we calculated the Snail2:Snail1 ratio and found that it did not correlate with metastatic outcome regardless of location (Fig. S9F).
The small sample size did not provide enough power to confidently reject the possibility that Twist1, the Twist1:Snail1 ratio or the Snail2:Snail1 ratio in primary tumors may have prognostic value. To overcome this statistical limitation, we collected an additional 92 pre-treatment primary tumor samples (13). Of these patients, 56 had remained relapse-free for 5 years and 36 recurred within 1.5 years of diagnosis. With the larger sample size, we confirmed that higher Snail1 mRNA levels in primary tumors were more frequently found in patients who later relapsed (p=0.0015) (Fig. S9A). On the other hands, Twist1 mRNA, the Twist1:Snail1 ratio and the internal control Snail2:Snail1 ratio in primary tumor all failed to show any significant link with distant recurrences (Fig. S9B–D). Thus the temporal Snail1-Twist1 cooperation observed in breast epithelial cell and breast cancer cell culture EMT was found to correlate with different stages of human breast cancer metastasis and was strongly predictive for distant recurrences.
Twist1-induced growth arrest is associated with, and requires, a low ERK:p38 MAPK activity ratio
One of the hallmarks of EMT is G0/G1 cell cycle arrest that allows cells to differentiate along mesenchymal lineages and become motile and invasive. More recently EMT has been suggested to also give rise to cancer stem cells that are often growth arrested (1, 8, 30). The cell cycle arrest of EMT is associated with, in part, the activation of the stress-related p38 MAPK, the inhibition of the mitogenic ERK1/2 MAP kinase, and induction of p53 and its downstream effector p21(30–32). Thus, we asked whether Twist1 was required to maintain or sustain growth arrest during late EMT by determining the number of Twist1-depleted MCF10A cells remaining in culture in the presence of continuous or transient TGFβ1 treatment. Twist1-depleted cells, in which EMT is not maintained, exhibited steady growth and reached 100% confluence within 4 days after transient TGFβ1 treatment (Fig. 6A). In contrast, control shRNAi-treated cells and Twist2-depleted cells underwent EMT and growth arrest in response to transient TGFβ1 (Fig. 6A). These results indicated that, in addition to Twist1’s unique function to maintain late E-cadherin downregulation, it was also required to sustain growth arrest in response to transient TGFβ1.
Figure 6. Twist1 induces growth arrest and is associated with a low ERK:p38 activity ratio.
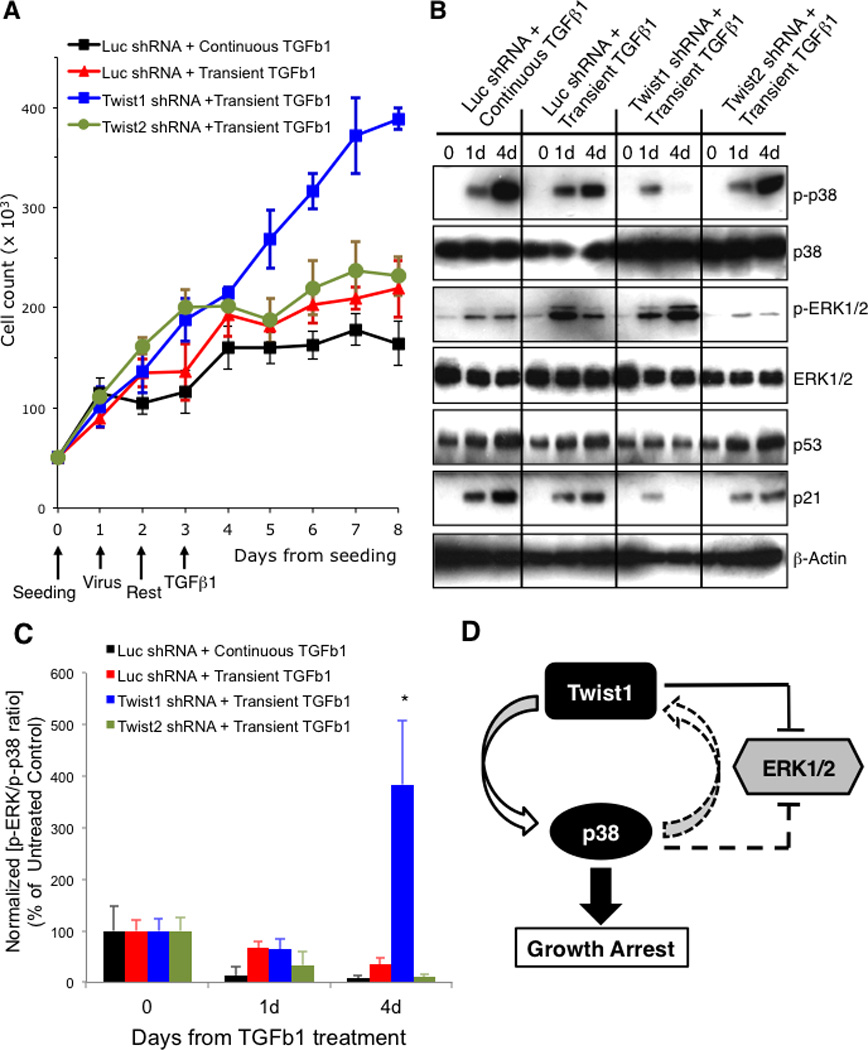
(A) Cell counts in triplicate in a 24-well plate at indicated times from seeding of MCF10A cells transduced with a lentivirus expressing either the control luciferase, Twist1, or Twist2 shRNA, then treated with 2 ng/ml TGFβ1 continuously or transiently for 4h, and cell numbers determined at indicated times. Error bars represent mean +/− s.e.m. Representatives of 3 independent experiments are shown. (B and C) Western blotting for indicated proteins of WCL (B) from MCF10A cells similarly manipulated as in (A). The ratio of p-ERK1/2 normalized to total ERK1/2 over p-p38 normalized to total p38 was determined by densitometry (B) and shown as percent of the ratio in untreated control cells (C). Error bars represent mean +/− s.e.m. from 3 independent experiments. Student’s T-test, * p<0.05 for each comparison between Twist1 shRNA and each other experimental condition. (D) Schematic of a potential Twist1-p38-ERK signal feedback loop. Directions where direct evidence is provided are represented as solid lines. The p38 to Twist1 direction is proposed and shown as broken arrow. The p38 to ERK1/2 inhibitory crosstalk has been reported elsewhere. See also related Figure S10–S12.
We determined the activity profile for p38, ERK1/2, p53 and p21 following transient TGFβ1 treatment. Although Twist1-depleted cells were able to activate p38 at day 1, as measured by phospho-p38 (Fig. 6B, lane 8), they failed to maintain p38 activity at day 4 (lane 9), as compared to control luciferase shRNAi or Twist2-depleted cells (lanes 6 and 12, respectively). The dampening of p38 activity associated with Twist1 depletion was also associated with a blunted activation and upregulation of p53 and p21 (Fig. 6B, lanes 7–9). Moreover, ERK1/2 activity on day 4, as represented by phospho-ERK1/2, was greater in Twist1-depleted cells (Fig. 6B and C). Of note, only in response to transient TGFβ1 did we observe high ERK and low p38 activity in Twist1-depleted cells. Exposing Twist1-depleted cells to continuous TGFβ1 produced low ERK and high p38 activity, EMT, and growth arrest (Fig. 6A and S10). Thus late Twist1 expression was required to maintain a low ERK:p38 activity ratio in cells undergoing EMT in response to transient TGFβ1.
In light of the observed association between the presence of Twist1 in late EMT, a low ERK:p38 activity ratio, and sustained growth arrest despite only transient exposure to TGFβ1, we asked whether Twist1 might facilitate the establishment of a positive feed-forward loop that enhances p38 activity while weakening ERK1/2 activity. This could then amplify and propagate the growth arrest program critical for the long-term maintenance of EMT (Fig. 6D). If so, then small changes in either Twist1 level or p38 activity would be predicted to impact the continued growth arrest of EMT. Other TGFβ1 signals, in addition to or independent of its effect upon EMT inducing factors (e.g., Twist1), could also contribute to this regulatory loop.
To test this possibility, we stably overexpressed Twist1 in MCF10A cells (2 independent clones). Twist1 was overexpressed so as to achieve maximal Twist1 effects without the confounding problem of added TGFβ1, since Twist1 overexpression results in TGFβ-independent EMT (Fig. S11A). Consistent with the presence of a positive feedback loop, p38 activity increased and the ERK:p38 activity ratio was at least 2–3 fold lower in Twist1 overexpressing cells than control MCF10A cells (Fig. S11A and B). Twist1 overexpressing cells also exhibited greater growth arrest than control cells (Fig. S11C). To ensure that the heightened p38 activity, present in Twist1 overexpressing cells, was not due to cell selection bias, we shRNAi-depleted both exogenously expressed and endogenous Twist1, and asked if this would rescue growth arrest and the low ERK:p38 activity ratio. Greater than 90% depletion of both exogenous and endogenous Twist1 was achieved (Fig. S12A) and this resulted in diminished p38 activity, increased ERK1/2 activity (increased ERK:p38 activity ratio) (Fig. S12A) and reverted growth arrest (Fig. S12B).
If Twist1 triggered growth arrest was associated with changes in p38 activity, then Twist1-dependent growth arrest should be reversed with effective inhibition of p38 kinase activity. To this end, we treated Twist1-overexpressing and control MCF10A cells with increasing concentrations of SB203580, a specific and potent p38 inhibitor (33), and determined cell numbers on day 4. Twist1-overexpressing cells treated with SB203580 produced increased cell numbers and this effect of SB203580 was dose-dependent (Fig. S11D), resulting in 2–2.5 fold higher cell numbers with 1µM SB203580 compared to the solvent DMSO. Control MCF10A cells showed no appreciable change in cell numbers under the same condition (Fig. S11D). Taken together, these results confirmed the existence and functional importance of a Twist1-p38/ERK feedback loop that maintained growth arrest critical for the late, maintenance phase of EMT.
Discussion
EMT changes, in many epithelial derived tumors, are now well appreciated to be a critical initial event in cancer metastasis (local invasion). However, what shapes or sustains the later stages of EMT and metastasis (dissemination) is unclear. The concept of distinct phases of EMT (e.g., initiation and maintenance) is suggested from in vivo observations that EMT inducing cytokines are often produced locally and transiently during both developmental and cancer EMT (2, 34), yet defining EMT changes such as E-cadherin repression persist even after the initial cytokine signal has dissipated. Whether and how the regulation of these 2 distinct phases of EMT differs remains poorly understood. Using transient TGFβ1 treatment to more closely mimic possible in vivo conditions of EMT, and then RNAi-manipulating the cellular level of various EMT inducers so as to avoid potential overexpression artifact revealed a regulatory hierarchy in which Snail1 precedes all other EMT inducing factors and is uniquely required for the initiation of EMT in cultured cells, including cancer cells. Twist1 supplants Snail1’s function to maintain late EMT in the absence of persistent TGFβ1. Importantly this temporal cooperation of Snail1 and Twist1 was observed during tumor metastasis in human breast cancer progression. The Twist1:Snail1 ratio in BM DTCs was found to be highly predictive for distant metastatic recurrences (Fig. 7).
Figure 7. Model of the temporal and spatial cooperation of Snail1 and Twist1 during cancer EMT and the metastatic cascade.
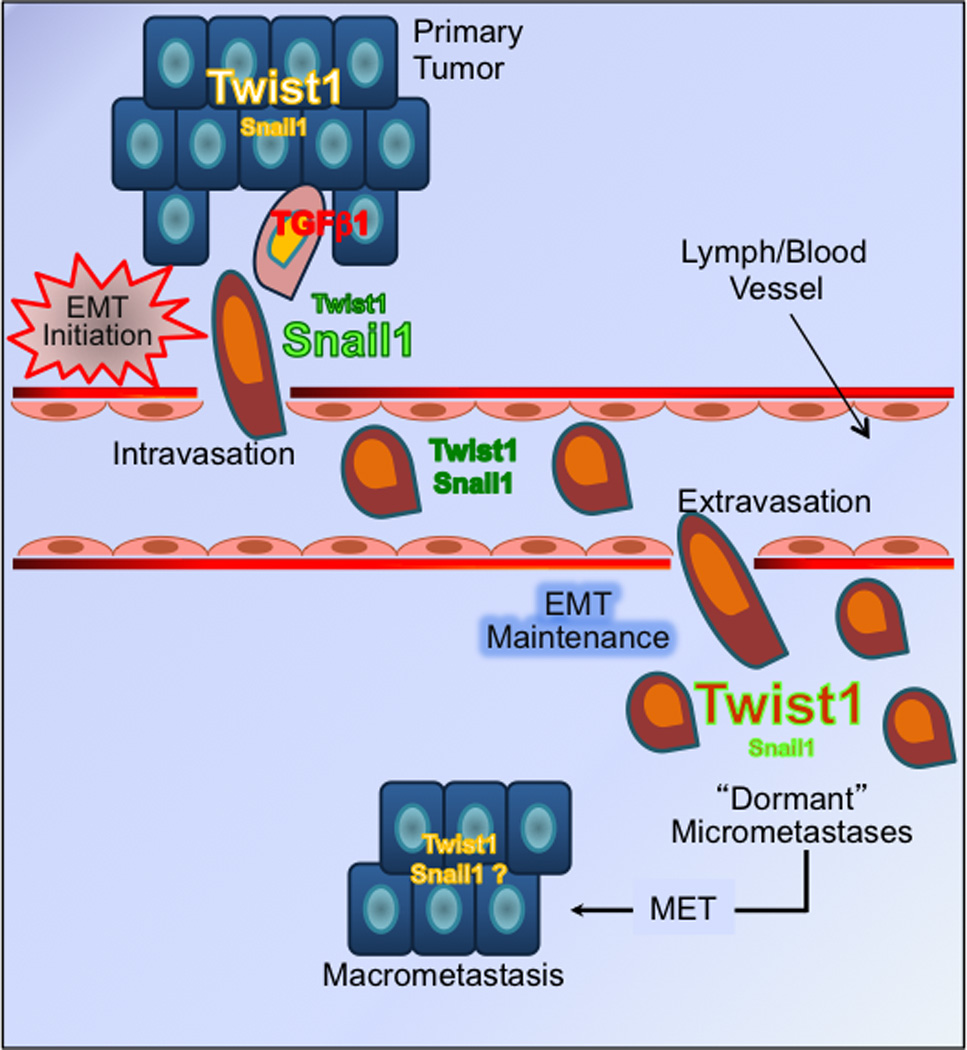
In response to a transient EMT stimulating signal (e.g. TGFβ1), Snail1 level rises briefly to initiate EMT in cancer cells near or at the invasive, leading edge of a primary tumor. As metastatic cells migrate away from the primary tumor, they lose Snail1 expression. In contrary, Twist1, present throughout the primary tumor, first experiences transient repression by Snail1 at the invasive edge during EMT initiation, then begins to rise again in late stages of EMT to maintain cancer EMT, and possibly dormant micrometastases. One potential scenario for the emergence of macrometastases is the downregulation of Twist1 expression in dormant micrometastases, thus relieving growth arrest associated with Twist1 and EMT maintenance.
These findings provide a new framework on which to expand and dissect complex interactions between the many EMT inducing factors that are often expressed concurrently. Consistent with its role as an EMT initiator, we confirmed previous findings that Snail1 expression in the primary tumor predicts for late recurrences and poor outcome (1, 5, 6). Yet, Snail1 expression outside the primary tumor is both lower and sporadic, and lacks any prognostic power. In contrast Twist1 expression in micrometastatic tumor cells, but not primary tumors, is highly predictive for disease relapse. When coupled with in vitro observations, this suggests that Twist1 is likely an important EMT maintenance factor that cancer cells employ to complete the metastatic process and possibly survive in secondary organs as micrometastases until suitable environmental conditions are present that reactivate tumor cell outgrowth (Fig. 7). Our data are not inconsistent with recent studies (14, 15) in which Twist1 depletion in a highly metastatic murine breast cancer cell line prior to its implantation in the mammary gland led to reduced numbers of macrometastatic foci in the lung. In the same mouse model, Twist1 depletion was found to correlate with decreased numbers of blood borne CTCs as measured by the ability of isolated CTCs to form colonies in ex vivo cultures. This observation was taken to indicate that Twist1 may be required for intravasation of metastatic tumor cells (e.g. early EMT), however our data would suggest that Twist1 depletion would decrease colony formation ability or survival of isolated CTCs in ex vivo cultures. While DTCs in secondary organs have been shown to be a strong predictor of disease-free survival, the prognostic utility of blood borne CTCs in early stage cancer is unclear as the majority of CTCs are unable to complete extravasation and are eventually removed from the circulation (35).
Recently, Snail2 was shown to be a transcriptional target of and required for Twist1 functions (36). These results predict that Snail2 depletion would interfere with Twist1-dependent EMT maintenance, which was not observed in our various cell systems. Moreover, Snail2 expression in micrometastatic DTCs offered no prognostic value for cancer relapse or progression. One possible explanation for this discrepancy is that Casas et al. described the Twist1-Snail2 connection through primarily Twist1 overexpression, an approach that produces robust signal-independent EMT, as opposed to our approach of siRNA-based protein depletion in cells that then remain responsive to TGFβ1 treatment for EMT induction, a condition likely to more accurately represent the in vivo setting. If Snail2 is transcriptionally activated by Twist1 and required for its function, one would predict that the levels of these 2 proteins would be concordant at all stages of cellular EMT and tumor metastasis, and that high levels of Snail2 would overcome low levels of Twist1 and correlate with distant metastases. Throughout all stages of transient TGFβ1-induced EMT the level of Snail2 and Twist1 were actually discordant. In human breast cancer samples, we observed high Snail2 expression in both primary tumors and BM DTCs in all patients regardless of their recurrence status (See Table S2). This was in contrast to Twist1 that exhibited high expression only in bone marrow DTCs and in patients who develop distant metastases. This suggests that high expression of Snail2 in bone marrow DTCs in non-recurrent patients was not able to rescue low Twist1 expression and is unrelated to metastatic rates in these patients (Fig. 5 and S9).
The presence of abundant Twist1 in untreated, epithelial MCF10A, HMLER and A549 cells was surprising since overexpression of Twist1 in breast epithelial cells induces robust EMT (14, 15). In agreement with those studies, when we overexpressed Twist1 in MCF10A cells robust EMT ensued, but this occurred independent of added TGFβ1 (Fig. S11A). This suggested the possibility that Twist1 present in parental epithelial cells is inactive. Since nuclear localization of EMT inducing factors is critical for their function as EMT inducers, and is tightly regulated (37), it is possible that Twist1 in parental non-EMT MCF10A cells did not induce EMT because it was sequestered in the cytoplasm. However, this was not the case as immunofluorescence and cellular fractionation analysis demonstrated that Twist1 was predominantly nuclear in untreated MCF10A cells (Fig. S13). These contradictory observations highlight the potential artifact of protein overexpression experiments in the study of EMT. One simple explanation as to why Twist1 does not induce EMT in resting non-EMT cells is that endogenous Twist1 is expressed at too low a level, compared to that achieved with overexpression. However this is unlikely as Twist1 level at the late phase of EMT mesenchymal cells, when it rebounds from initial downregulation, is not higher than untreated starting epithelial cells and yet it is able to regulate many aspects of EMT. Rather this points to a fundamental regulatory differences between EMT induction and maintenance with Snail1 controlling the former and Twist1 the latter. It is entirely conceivable that in non-EMT cells Twist1 regulates a distinct set of processes that are unrelated to EMT. Alternatively, it may constitutively act to inhibit EMT induction in resting cells and acquire a new function in response to TGFβ1 to maintain EMT. This notion is supported by the observation that Twist1 transcription is actively repressed by Snail1 during EMT initiation. Yet, Twist1 downregulation alone is not sufficient to activate EMT (Fig. 2B) likely because the necessary EMT initiation signal is provided by Snail1 and TGFβ1, not Twist1 or Twist1 downregulation. Whether early Twist1 downregulation relieves an inhibitory signal to allow cells to respond to EMT stimulating factors remains unanswered, however. Alternatively Twist1 may be kept in an inactive conformation in non-EMT cells either through an inhibitory binding partner(s) or a post-translational event (e.g., phosphorylation or dephosphorylation). The inhibition is then relieved during the late phase of EMT by a signal possibly provided by TGFβ1 and/or earlier Snail1, which may repress expression of the inhibitory partner. A precedent for such an inhibitory switch involves the protein E47, another bHLH transcriptional repressor that usually remains inactive until being phosphorylated by p38 MAPK (38).
Lastly, how Twist1 leads to activation of p38 and which isoforms of p38 are involved is unclear. Twist1 could affect p38 kinase activity directly, or indirectly through transcriptional repression of a p38-specific phosphatase or secretion of a cytokine(s) that would stimulate a low ERK:p38 activity ratio.
Fortunately metastasis is a highly inefficient process, yet distant recurrences, which can occur years after diagnosis of the primary tumor, represent a major outstanding challenge for both clinicians and scientists alike. Experimental work has suggested that rate-limiting steps in metastasis are the ability of dormant tumor cells to resist initial systemic therapy, remain viable in a foreign tissue, and reactivate proliferation when suitable conditions are present (39, 40). Evidence for clinically silent residual disease, such as blood borne circulating tumor cells (CTCs) or disseminated tumor cells (DTCs) in bone marrow (BM), for example, has been found in clinically cancer-free patients (41–43). DTCs, in particular, represent a significant prognostic indicator of reduced disease-free and overall survival and are thought to be intermediaries in the metastatic process (41–43). Prolonged survival of some of these DTCs in secondary organs has been suggested to be associated with impaired angiogenesis, immune escape, hormonal controls, diet and autophagy (i.e., metabolism) which are likely regulated by the tumor micro-environment (41, 44–46). From the cell-intrinsic point of view, the ERK:p38 MAPK activity ratio, which has been shown to be regulated by the urokinase receptor and EGFR, appears to be important. A low ratio (i.e. low ERK and high p38 activity) favors the dormant state (31, 47, 48). Although p38 can be activated as part of the EMT program (49, 50), it is unclear whether cancer EMT, which is thought to precede cancer dormancy both temporally and spatially, contributes to the dormancy signaling pathway. The current results suggest that Twist1 may provide a potential mechanistic link between these 2 processes through its ability to maintain both the late phase of EMT and p38-dependent growth arrest.
It remains to be determined whether the potential in vivo function of Twist1 in dormant tumor cells is to promote growth arrest or survival. Our cell-based findings favor growth arrest since Twist1 depletion resulted in increased cell numbers whereas Twist1 overexpression was associated with a profound growth arrest phenotype. Additional studies using genetic mouse models of breast cancer dormancy and metastasis, which are currently not perfect, will be needed to differentiate between these 2 possibilities.
The remarkable correlation between BM DTCs’ Twist1:Snail1 ratio and late breast cancer recurrences has the potential to be developed into a powerful prognostic biomarker in breast cancer. To do so a significantly larger cohort of patients would be necessary to determine whether it complements the existing prognostic biomarkers or can serve independently. We have previously shown that the presence of Twist1, but not Snail1, in BM DTCs was also associated with a persistent DTC microarray signature (i.e. persistent presence of BM DTCs) after chemotherapy (12), (13), suggesting that the Twist1:Snail1 ratio in BM DTCs may not only have prognostic value but also predict for treatment resistance and prolonged survival of BM DTCs, and thus may have therapeutic relevance for breast cancer and perhaps other malignancies as well.
Supplementary Material
Acknowledgments
We thank Dr. Loren Michel for MCF10A, HMLER and A549 cells and Dr. Alain Puisieux for Twist1 plasmid. Members of our laboratory were thanked for their technical assistance and helpful comments. This work was supported by grants from the National Institutes of Health (G.D.L, and R.L.T.), and a T32 and the Mallinckrodt Foundation (D.D.T.).
References
- 1.Thiery JP, Acloque H, Huang RYJ, Nieto MA. Epithelial-Mesenchymal Transitions in Development and Disease. 2009;139(5):871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 2.Giampieri S, Manning C, Hooper S, Jones L, Hill CS, Sahai E. Localized and reversible TGFbeta signalling switches breast cancer cells from cohesive to single cell motility. Nat Cell Biol. 2009 Nov;11(11):1287–1296. doi: 10.1038/ncb1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007 Jun;7(6):415–428. doi: 10.1038/nrc2131. [DOI] [PubMed] [Google Scholar]
- 4.Yang M-H, Hsu DS-S, Wang H-W, Wang H-J, Lan H-Y, Yang W-H, Huang C-H, Kao S-Y, Tzeng C-H, Tai S-K, Chang S-Y, Lee OK-S, Wu K-J. Bmi1 is essential in Twist1-induced epithelial-mesenchymal transition. Nat Cell Biol. 2010;12(10):982–992. doi: 10.1038/ncb2099. [10.1038/ncb2099] [DOI] [PubMed] [Google Scholar]
- 5.Kudo-Saito C, Shirako H, Takeuchi T, Kawakami Y. Cancer Metastasis Is Accelerated through Immunosuppression during Snail-Induced EMT of Cancer Cells. 2009;15(3):195–206. doi: 10.1016/j.ccr.2009.01.023. [DOI] [PubMed] [Google Scholar]
- 6.Moody SE, Perez D, Pan TC, Sarkisian CJ, Portocarrero CP, Sterner CJ, Notorfrancesco KL, Cardiff RD, Chodosh LA. The transcriptional repressor Snail promotes mammary tumor recurrence. Cancer Cell. 2005 Sep;8(3):197–209. doi: 10.1016/j.ccr.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 7.Lindley LE, Briegel KJ. Molecular characterization of TGF[beta]-induced epithelial-mesenchymal transition in normal finite lifespan human mammary epithelial cells. Biochemical and Biophysical Research Communications. 2010;399(4):659–664. doi: 10.1016/j.bbrc.2010.07.138. [DOI] [PubMed] [Google Scholar]
- 8.Mani SA, Guo W, Liao M-J, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J, Weinberg RA. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell. 2008;133(4):704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006 Feb;7(2):131–142. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- 10.Goswami S, Sahai E, Wyckoff JB, Cammer M, Cox D, Pixley FJ, Stanley ER, Segall JE, Condeelis JS. Macrophages promote the invasion of breast carcinoma cells via a colony-stimulating factor-1/epidermal growth factor paracrine loop. Cancer Res. 2005 Jun 15;65(12):5278–5283. doi: 10.1158/0008-5472.CAN-04-1853. [DOI] [PubMed] [Google Scholar]
- 11.Maeda M, Johnson KR, Wheelock MJ. Cadherin switching: essential for behavioral but not morphological changes during an epithelium-to-mesenchyme transition. J Cell Sci. 2005 Mar 1;118(5):873–887. doi: 10.1242/jcs.01634. 2005. [DOI] [PubMed] [Google Scholar]
- 12.Watson MA, Ylagan LR, Trinkaus KM, Gillanders WE, Naughton MJ, Weilbaecher KN, Fleming TP, Aft RL. Isolation and molecular profiling of bone marrow micrometastases identifies TWIST1 as a marker of early tumor relapse in breast cancer patients. Clin Cancer Res. 2007 Sep 1;13(17):5001–5009. doi: 10.1158/1078-0432.CCR-07-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin Y, Lin S, Watson M, Trinkaus K, Kuo S, Naughton M, Weilbaecher K, Fleming T, Aft R. A gene expression signature that predicts the therapeutic response of the basal-like breast cancer to neoadjuvant chemotherapy. Breast Cancer Research and Treatment. 2009 doi: 10.1007/s10549-009-0664-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ansieau S, Bastid J, Doreau A, Morel AP, Bouchet BP, Thomas C, Fauvet F, Puisieux I, Doglioni C, Piccinin S, Maestro R, Voeltzel T, Selmi A, Valsesia-Wittmann S, Caron de Fromentel C, Puisieux A. Induction of EMT by twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence. Cancer Cell. 2008 Jul 8;14(1):79–89. doi: 10.1016/j.ccr.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 15.Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004 Jun 25;117(7):927–939. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 16.Elenbaas B, Spirio L, Koerner F, Fleming MD, Zimonjic DB, Donaher JL, Popescu NC, Hahn WC, Weinberg RA. Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes Dev. 2001 Jan 1;15(1):50–65. doi: 10.1101/gad.828901. 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Basu I, Locker J, Cassera MB, Belbin TJ, Merino EF, Dong X, Hemeon I, Evans GB, Guha C, Schramm VL. Growth and Metastases of Human Lung Cancer Are Inhibited in Mouse Xenografts by a Transition State Analogue of 5′-Methylthioadenosine Phosphorylase. Journal of Biological Chemistry. 2011 Feb 11;286(6):4902–4911. doi: 10.1074/jbc.M110.198374. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giard DJ, Aaronson SA, Todaro GJ, Arnstein P, Kersey JH, Dosik H, Parks WP. In vitro cultivation of human tumors: establishment of cell lines derived from a series of solid tumors. Journal of the National Cancer Institute. 1973 Nov;51(5):1417–1423. doi: 10.1093/jnci/51.5.1417. [DOI] [PubMed] [Google Scholar]
- 19.Kolosionek E, Savai R, Ghofrani HA, Weissmann N, Guenther A, Grimminger F, Seeger W, Banat GA, Schermuly RT, Pullamsetti SS. Expression and activity of phosphodiesterase isoforms during epithelial mesenchymal transition: the role of phosphodiesterase 4. Molecular biology of the cell. [Research Support, Non-U.S. Gov't] 2009 Nov;20(22):4751–4765. doi: 10.1091/mbc.E09-01-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu Q, Krakowski AR, Dunham EE, Wang L, Bandyopadhyay A, Berdeaux R, Martin GS, Sun L, Luo K. Dual role of SnoN in mammalian tumorigenesis. Molecular and cellular biology. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov't] 2007 Jan;27(1):324–339. doi: 10.1128/MCB.01394-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vega S, Morales AV, Ocana OH, Valdes F, Fabregat I, Nieto MA. Snail blocks the cell cycle and confers resistance to cell death. Genes Dev. 2004 May 15;18(10):1131–1143. doi: 10.1101/gad.294104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ohkuma M, Funato N, Higashihori N, Murakami M, Ohyama K, Nakamura M. Unique CCT repeats mediate transcription of the TWIST1 gene in mesenchymal cell lines. Biochemical and Biophysical Research Communications. 2007;352(4):925–931. doi: 10.1016/j.bbrc.2006.11.114. [DOI] [PubMed] [Google Scholar]
- 23.Hou Z, Peng H, Ayyanathan K, Yan KP, Langer EM, Longmore GD, Rauscher FJ., 3rd The LIM protein AJUBA recruits protein arginine methyltransferase 5 to mediate SNAIL-dependent transcriptional repression. Mol Cell Biol. 2008 May;28(10):3198–3207. doi: 10.1128/MCB.01435-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blanco MJ, Moreno-Bueno G, Sarrio D, Locascio A, Cano A, Palacios J, Nieto MA. Correlation of Snail expression with histological grade and lymph node status in breast carcinomas. Oncogene. 2002 May 9;21(20):3241–3246. doi: 10.1038/sj.onc.1205416. [DOI] [PubMed] [Google Scholar]
- 25.Sundberg M, Jansson L, Ketolainen J, Pihlajamäki H, Suuronen R, Skottman H, Inzunza J, Hovatta O, Narkilahti S. CD marker expression profiles of human embryonic stem cells and their neural derivatives, determined using flow-cytometric analysis, reveal a novel CD marker for exclusion of pluripotent stem cells. Stem Cell Research. 2009;2(2):113–124. doi: 10.1016/j.scr.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 26.Anderson R, Schaible K, Heasman J, Wylie C. Expression of the homophilic adhesion molecule, Ep-CAM, in the mammalian germ line. J Reprod Fertil. 1999 Jul 1;116(2):379–384. doi: 10.1530/jrf.0.1160379. 1999. [DOI] [PubMed] [Google Scholar]
- 27.Yamashita T, Forgues M, Wang W, Kim JW, Ye Q, Jia H, Budhu A, Zanetti KA, Chen Y, Qin LX, Tang ZY, Wang XW. EpCAM and alpha-fetoprotein expression defines novel prognostic subtypes of hepatocellular carcinoma. Cancer research. [Research Support, N.I.H., Intramural Research Support, Non-U.S. Gov't] 2008 Mar 1;68(5):1451–1461. doi: 10.1158/0008-5472.CAN-07-6013. [DOI] [PubMed] [Google Scholar]
- 28.Gadalla SE, Alexandraki A, Lindstrom MS, Nister M, Ericsson C. Uncoupling of the ERalpha regulated morphological phenotype from the cancer stem cell phenotype in human breast cancer cell lines. Biochem Biophys Res Commun. 2011 Feb 25;405(4):581–587. doi: 10.1016/j.bbrc.2011.01.072. [DOI] [PubMed] [Google Scholar]
- 29.Debnath J, Brugge JS. Modelling glandular epithelial cancers in three-dimensional cultures. Nat Rev Cancer. 2005;5(9):675–688. doi: 10.1038/nrc1695. [10.1038/nrc1695] [DOI] [PubMed] [Google Scholar]
- 30.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. The Journal of Clinical Investigation. 2009;119(6):1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adam AP, George A, Schewe D, Bragado P, Iglesias BV, Ranganathan AC, Kourtidis A, Conklin DS, Aguirre-Ghiso JA. Computational identification of a p38SAPK-regulated transcription factor network required for tumor cell quiescence. Cancer Res. 2009 Jul 15;69(14):5664–5672. doi: 10.1158/0008-5472.CAN-08-3820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bhowmick NA, Zent R, Ghiassi M, McDonnell M, Moses HL. Integrin beta 1 signaling is necessary for transforming growth factor-beta activation of p38MAPK and epithelial plasticity. J Biol Chem. 2001 Dec 14;276(50):46707–46713. doi: 10.1074/jbc.M106176200. [DOI] [PubMed] [Google Scholar]
- 33.Cuenda A, Rouse J, Doza YN, Meier R, Cohen P, Gallagher TF, Young PR, Lee JC. SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Letters. 1995;364(2):229–233. doi: 10.1016/0014-5793(95)00357-f. [DOI] [PubMed] [Google Scholar]
- 34.Zavadil J, Bottinger EP. TGF-[beta] and epithelial-to-mesenchymal transitions. Oncogene. 2005;24(37):5764–5774. doi: 10.1038/sj.onc.1208927. [DOI] [PubMed] [Google Scholar]
- 35.Vincent-Salomon A, Bidard FC, Pierga JY. Bone marrow micrometastasis in breast cancer: review of detection methods, prognostic impact and biological issues. J Clin Pathol. 2008 May;61(5):570–576. doi: 10.1136/jcp.2007.046649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Casas E, Kim J, Bendesky A, Ohno-Machado L, Wolfe CJ, Yang J. Snail2 is an essential mediator of Twist1-induced epithelial mesenchymal transition and metastasis. Cancer Res. 2011 Jan 1;71(1):245–254. doi: 10.1158/0008-5472.CAN-10-2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou BP, Deng J, Xia W, Xu J, Li YM, Gunduz M, Hung M-C. Dual regulation of Snail by GSK-3[beta]-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat Cell Biol. 2004;6(10):931–940. doi: 10.1038/ncb1173. [DOI] [PubMed] [Google Scholar]
- 38.Lluis F, Ballestar E, Suelves M, Esteller M, Munoz-Canoves P. E47 phosphorylation by p38 MAPK promotes MyoD/E47 association and muscle-specific gene transcription. EMBO J. 2005;24(5):974–984. doi: 10.1038/sj.emboj.7600528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chambers AF, Groom AC, MacDonald IC. Metastasis: Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002;2(8):563–572. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- 40.McGowan PM, Kirstein JM, Chambers AF. Micrometastatic disease and metastatic outgrowth: clinical issues and experimental approaches. Future Oncology. 2009;5(7):1083–1098. doi: 10.2217/fon.09.73. [DOI] [PubMed] [Google Scholar]
- 41.Aguirre-Ghiso JA. Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer. 2007;7(11):834–846. doi: 10.1038/nrc2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alix-Panabieres C, Riethdorf S, Pantel K. Circulating tumor cells and bone marrow micrometastasis. Clin Cancer Res. 2008 Aug 15;14(16):5013–5021. doi: 10.1158/1078-0432.CCR-07-5125. [DOI] [PubMed] [Google Scholar]
- 43.Braun S, Vogl FD, Naume B, Janni W, Osborne MP, Coombes RC, Schlimok G, Diel IJ, Gerber B, Gebauer G, Pierga JY, Marth C, Oruzio D, Wiedswang G, Solomayer EF, Kundt G, Strobl B, Fehm T, Wong GY, Bliss J, Vincent-Salomon A, Pantel K. A pooled analysis of bone marrow micrometastasis in breast cancer. N Engl J Med. 2005 Aug 25;353(8):793–802. doi: 10.1056/NEJMoa050434. [DOI] [PubMed] [Google Scholar]
- 44.Chambers AF. Influence of diet on metastasis and tumor dormancy. Clin Exp Metastasis. 2009;26(1):61–66. doi: 10.1007/s10585-008-9164-4. [DOI] [PubMed] [Google Scholar]
- 45.Eyles J, Puaux A-L, Wang X, Toh B, Prakash C, Hong M, Tan TG, Zheng L, Ong LC, Jin Y, Kato M, Pr√©vost-Blondel A, Chow P, Yang H, Abastado J-P. Tumor cells disseminate early, but immunosurveillance limits metastatic outgrowth, in a mouse model of melanoma. The Journal of Clinical Investigation. 2010;120(6):2030–2039. doi: 10.1172/JCI42002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lu Z, Luo RZ, Lu Y, Zhang X, Yu Q, Khare S, Kondo S, Kondo Y, Yu Y, Mills GB, Liao WSL, Bast RC. The tumor suppressor gene ARHI regulates autophagy and tumor dormancy in human ovarian cancer cells. The Journal of Clinical Investigation. 2008;118(12):3917–3929. doi: 10.1172/JCI35512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Aguirre-Ghiso JA, Estrada Y, Liu D, Ossowski L. ERKMAPK Activity as a Determinant of Tumor Growth and Dormancy; Regulation by p38SAPK. Cancer Res. 2003 Apr 1;63(7):1684–1695. 2003. [PubMed] [Google Scholar]
- 48.Aguirre-Ghiso JA, Liu D, Mignatti A, Kovalski K, Ossowski L. Urokinase receptor and fibronectin regulate the ERK(MAPK) to p38(MAPK) activity ratios that determine carcinoma cell proliferation or dormancy in vivo. Mol Biol Cell. 2001 Apr;12(4):863–879. doi: 10.1091/mbc.12.4.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bakin AV, Rinehart C, Tomlinson AK, Arteaga CL. p38 mitogen-activated protein kinase is required for TGF{beta}-mediated fibroblastic transdifferentiation and cell migration. J Cell Sci. 2002 Aug 1;115(15):3193–3206. doi: 10.1242/jcs.115.15.3193. 2002. [DOI] [PubMed] [Google Scholar]
- 50.Zohn IE, Li Y, Skolnik EY, Anderson KV, Han J, Niswander L. p38 and a p38-Interacting Protein Are Critical for Downregulation of E-Cadherin during Mouse Gastrulation. 2006;125(5):957–969. doi: 10.1016/j.cell.2006.03.048. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.