

Abstract
Background
Microglial activation has been reported to be involved in traumatic brain injury (TBI). Nuclear factor erythroid 2-related factor 2 (Nrf2) plays a significant role in protecting against TBI-induced secondary brain injury. However, the exact mechanism is not clearly understood. The present study aimed to explore whether Nrf2 protects against TBI partly by regulating microglia function.
Material/Methods
Microglia cells were isolated from C57BL/6 mouse brains (postnatal day 1–3). The expression of Nrf2 was suppressed by transfection with Nrf2-specific small interfering RNA (siRNA), and overexpressed by transfections with pcDNA3.1-Nrf2. The expression of Nrf2 was confirmed by real-time PCR and Western blotting. After transfection, cell viability, phagocytic ability, and the expression of pro-inflammatory cytokines (tumor necrosis factor (TNF)-α and interleukin (IL)-6) were determined by 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) colorimetric assay, phagocytosis assay, and enzyme-linked immunosorbent assay (ELISA), respectively.
Results
mRNA and protein expression levels of Nrf2 were significantly reduced by transfection with Nrf2-specific siRNA (both P<0.05) but were elevated by transfection with pcDNA3.1-Nrf2 (both P<0.01). The cell viability, phagocytic ability, and the expression of TNF-α and IL-6 were all significantly reduced by overexpression of Nrf2 but were significantly increased by silencing of Nrf2 compared with the control group.
Conclusions
Our results suggest that Nrf2 protects against TBI, at least part by regulating microglia function.
MeSH Keywords: Brain Injuries, Microglia, NF-E2-Related Factor 2
Background
Traumatic brain injury (TBI), a neurologic disorder, is characterized by damage to the cerebral tissue caused by abrupt contact (violent impact or acceleration-deceleration) or a penetrating wound to the brain tissue. It is the leading cause of death and disability in people under the age of 45 years worldwide [1]. Also, TBI imposes a tremendous financial burden on the health care system, as well as physical and emotional burden to individuals. However, the pathophysiology of TBI is very complicated and not yet fully understood. Recently, activation of the immune system in the central nervous system (CNS) has gained wide acceptance as a critical component of the pathology and progression of numerous neurological disorders, including TBI [2,3]. Microglia, the resident macrophages in the brain, are the key regulators of the immune defense system in the CNS and are responsible for subsequent inflammatory responses [4–6]. Microglia react to injury within minutes, and are chronically activated [7, 8]. Once activated, microglia can be recognized as a pro-inflammatory state and promote cell-mediated immunity [3,9–11]. It has been well demonstrated that levels of tumor necrosis factor (TNF)-α and interleukin (IL)-6 are elevated after TBI [12]. However, excessive activation of the microglia cells results in destruction of healthy neurons as well as damaged neurons. Therefore, inhibition of microglia cells activation and inflammatory factor release protect against and improve the prognosis of TBI.
Nuclear factor erythroid 2-related factor 2 (Nrf2), a basic leucine zipper (bZIP) redox-sensitive transcription factor that modulates the cellular antioxidant response, has been reported to play a number of roles in regulating acute inflammatory response [13]. It upregulates cytoprotective and antioxidant-detoxifying genes expression to rescue the cells/tissues injury [14]. In recent years, involvement of Nrf2 has been well documented in several CNS diseases [15]. It has been suggested that Nrf2 plays a significant role in protecting against secondary brain injury induced by TBI, possibly by modulating pro-inflammatory cytokines and inducing antioxidant-detoxifying enzymes [16,17]. In addition, Nrf2 −/− mice have shown increased microglial cells infiltration and high levels of inflammatory markers such as TNF-α and IL-6 in the hippocampus in response to lipopolysaccharide (LPS) [18]. However, the exact mechanism by which Nrf2 protects against TBI is not clearly understood.
Therefore, we assumed that Nrf2 plays a protective role in TBI and that this effect is regulated at least in part through the effect of Nrf2 on microglia. In the present study, the expression of Nrf2 was suppressed or upregulated, and then the effect of transfection on cell viability, phagocytic ability, and the expression of TNF-α and IL-6 were determined.
Material and Methods
Animals
Thirty C57BL/6 mice (postnatal day 1–3, 15: 15 male: female) were obtained from Shanghai SLAC Laboratory Animal Co. Ltd. (Shanghai, China). All the animals were housed in a temperature-controlled room (24±2°C) with a relative humidity of 60±5% on a 12-h light/dark cycle. The mice were euthanized via decapitation and their brains were harvested. All procedures were approved by the Ethics Committee on Animals of our University, and the animal care and use was in line with the standards in the “Guide for the Care and Use of Laboratory Animals” published by the National Institutes of Health.
Primary microglia culture
The methods of harvest and culture of microglia were performed as described before [19]. After decapitation, the cortices were collected, isolated, trypsinized, triturated, and plated onto tissue culture plastic in Dulbecco’s Modified Eagles Medium (DMEM)/F12 with L-glutamine (Invitrogen, Carlsbad, CA) supplemented with 20% heat-inactivated fetal bovine serum (FBS). After 24 h, the media was discarded and new fresh media was added. At 7 days, half the media was replaced and these cells were cultured for another 7 days. Thereafter, microglia cells were collected from the mixed glial culture and counted. Microglia cells were collected again after fresh media was added to the remaining mixed glia for 7 days.
Transfection
These cells were randomly assigned to a control group, a siNrf2 group, and an overexpression Nrf2 group. The cells in the control group received no additional treatment. For the siNrf2 group, the expression of Nrf2 was suppressed using small interfering RNA (siRNA). The plasmids specifically targeting Nrf2 were designed and synthesized by GenePharma Co (Shanghai, China). For the overexpression Nrf2 group, an Nrf2 expression vector (pcDNA3.1-Nrf2) was constructed and confirmed by sequencing. Cell transfections were performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol.
Cell viability
The cell viability was assessed using 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) colorimetric assay. Briefly, the concentration of cells in the 3 groups was adjusted to 5×104/mL, washed with phosphate-buffered saline (PBS), and seeded in a 96-well plate. After culturing for 24, 48, and 72 h, 20 μL of fresh medium with 0.5 mg/mL MTT was added to each well and incubated for 4 h at 37°C. The absorbance at 492 nm optical density (OD) was measured using a microplate reader (Bio-Tek, USA). The experiment was repeated 3 times.
Phagocytosis assay
Phagocytosis assay was performed using a Phagotest kit (OPREGEN Pharma; BD Biosciences, Oxford, UK) containing fluorescein-labeled opsonized Escherichia coli (E. coli-FITC) according to manufacturer’s protocol. Briefly, cells (5×104/mL) were seeded in a 96-well plate and incubated with 0.25 mg/mL FITC-E coli Bioparticles for 6 h at 37°C. Thereafter, the media was removed and the cells were washed with 0.25 mg/mL trypan blue in PBS to quench the surface-bound fluorescent signal. Intracellular fluorescence was read using a fluorescent plate reader (BMG Labtech, Durham, NC, USA) at 480 nm excitation and 520 nm emission.
Enzyme-linked immunosorbent assay (ELISA)
After transfection, the media was collected and centrifuged. Quantitative analyses of supernatants levels for TNF-α and IL-6 were performed using a commercial ELISA kit (Biocheck®, USA) according to the manufacturer’s instructions. All samples were prepared at least in triplicate.
Real-time PCR (RT-PCR)
Total RNA was isolated from cells using Trizol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. Complementary DNA (cDNA) was synthesized using SuperscriptTM III Reverse Transcriptase (Invitrogen, Carlsbad, CA, USA) and quantified by using a QuantTtect SYBR green RT-PCR Kit (Qiagen, Valencia, CA). The relative expressions of Nrf2 were determined by normalizing to GAPDH using the 2−ΔΔCT method. Reactions were performed in triplicate.
Western blotting analysis
After 24 h of transfection, the cells in each group were harvested for protein extraction. Total protein concentration was determined using a Pierce® BCA Protein Assay Kit (Thermo Scientific, Rockford, IL, USA). Protein samples were separated on a 10–12% sodium dodecyl sulfate (SDS)-polyacrylamide gel. Next, the protein samples were blotted onto polyvinylidene difluoride (PVDF) membranes (Millipore), blocked in 5% milk in Tris-buffered saline with Tween (TBST) for 2 h, and probed with primary anti-Nrf2 antibody (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) overnight at 4°C, followed by horseradish peroxide-conjugated secondary antibodies (Santa Cruz Biotechnology). Protein detection was further visualized using enhanced chemiluminescence.
Statistical analysis
Data are shown as mean ± standard deviation (SD). All statistical analyses were performed with the use of Statistical Package for the Social Sciences (SPSS) 19.0 (SPSS Inc., Chicago, IL, USA) statistical software. The differences between groups were assessed using a one-way analysis of variance (ANOVA) followed by Tukey post hoc test. Statistical significance was set at P<0.05.
Results
The effect of transfection on expression of Nrf2 in microglia
To explore the effect on the expression of Nrf2 after transfection with pcDNA3.1-Nrf2 or the targeting sequence for Nrf2-specific siRNA into microglia, RT-PCR and Western blotting analysis were carried out to determine the mRNA and protein levels of Nrf2. The results showed that both the mRNA and protein expression levels of Nrf2 in the siNrf2 group were significantly reduced by transfection with Nrf2-specific siRNA compared to the control group (both P<0.05), but were significantly elevated by transfection with pcDNA3.1-Nrf2 (both P<0.01) (Figure 1A–1C).
Figure 1.

The effect of transfection on expression of Nrf2 in microglia. (A) Relative Nrf2 mRNA expression after transfection with Nrf2-specific siRNA or pcDNA3.1-Nrf2; (B) Relative Nrf2 protein expression after transfection with Nrf2-specific siRNA or pcDNA3.1-Nrf2; (C) Representative images of Western blotting after transfection with Nrf2-specific siRNA or pcDNA3.1-Nrf2. Nrf2 – nuclear factor erythroid 2-related factor 2; siRNA – small interfering RNA. * P<0.05 compared with the control group; ** P<0.01 compared with the control group.
The effect of transfection on cell viability in microglia
To investigate the effect of Nrf2 on microglia, cell viability was determined using MTT after 24, 48, and 72 h of transfection with Nrf2-specific siRNA or pcDNA3.1-Nrf2. As shown in Figure 2, the cell viability was significantly reduced by overexpression of Nrf2 but was significantly increased by silencing of Nrf2 compared with the control group (both P<0.05), indicating that Nrf2 can inhibit the activation of microglia.
Figure 2.
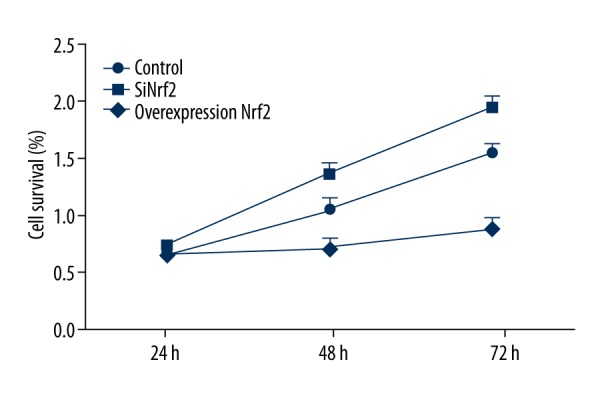
The effect of transfection with Nrf2-specific siRNA or pcDNA3.1-Nrf2 on cell viability in microglia. Nrf2 – nuclear factor erythroid 2-related factor 2; siRNA – small interfering RNA.
The effect of transfection on phagocytic ability in microglia
We further elucidated the phagocytic ability in microglia after transfection with Nrf2-specific siRNA or pcDNA3.1-Nrf2. The results demonstrated that the phagocytic ability was significantly higher by silencing of Nrf2 than that in the control group (P<0.05). In addition, the phagocytic ability was significantly lowered by overexpression of Nrf2 (P<0.05) (Figure 3). These results suggest that Nrf2 can reduce the phagocytic ability of microglia.
Figure 3.
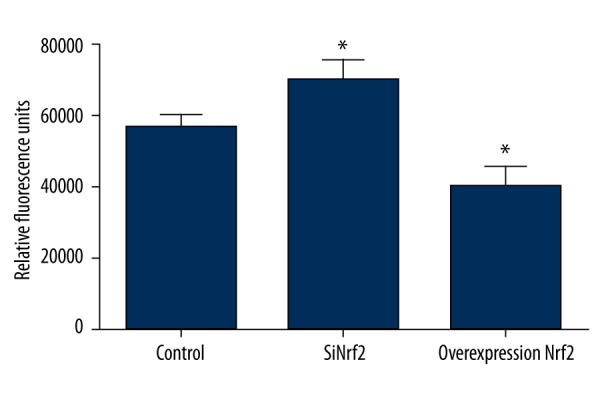
The effect of transfection with Nrf2-specific siRNA or pcDNA3.1-Nrf2 on phagocytic ability in microglia. Nrf2 – nuclear factor erythroid 2-related factor 2; siRNA – small interfering RNA. * P<0.05 compared with the control group.
The effect of transfection on the expression of TNF-α and IL-6 in microglia
The effect of transfection with Nrf2-specific siRNA or pcDNA3.1-Nrf2 on the expression of pro-inflammatory cytokines (TNF-α and IL-6) in microglia was investigated. As indicated in Figure 4, the expression of TNF-α and IL-6 were significantly increased by silencing of Nrf2 compared to the control group (both P<0.01), but were significantly reduced by overexpression of Nrf2 (both P<0.05). The results demonstrate that Nrf2 can inhibit the expression of pro-inflammatory cytokines.
Figure 4.
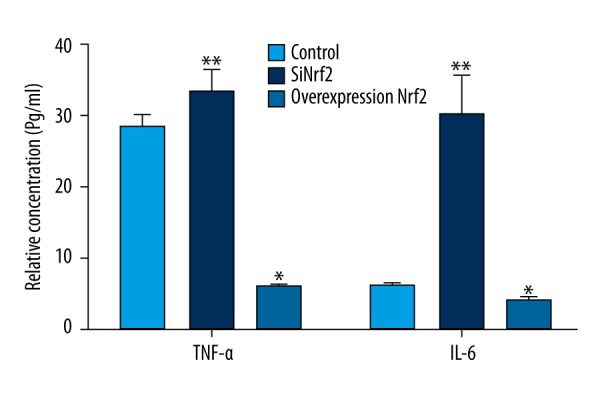
The effect of transfection with Nrf2-specific siRNA or pcDNA3.1-Nrf2 on the expression of TNF-α and IL-6 in microglia. Nrf2 – nuclear factor erythroid 2-related factor 2; siRNA – small interfering RNA; TNF – tumor necrosis factor; IL – interleukin. * P<0.05 compared with the control group; ** P<0.01 compared with the control group.
Discussion
In the present study we found that overexpression of Nrf2 inhibits the activation of microglia cells and subsequently suppresses the phagocytic ability and production of inflammatory cytokines (TNF-α and IL-6); these results were reversed when interfered by Nrf2. The results suggest the protective role of Nrf2 in microglia cells.
Microglia cells, the resident macrophages of the CNS, are involved in physiological and pathological processes of numerous CNS disorders by regulating innate immunity and adaptive immune responses [20,21]. The most remarkable and appealing characteristics of microglia cells are their activation at very early stages in response to injury and their capacity for site-directed phagocytosis. With respect to activation, microglia cells show a series of functional and morphological changes, establishing a complicated immune response network. The activated microglia cells polarize into 2 major subtypes, referred to as “M1” or “M2”. M1 is recognized as a pro-inflammatory state and stimulates cell-mediated immunity; it is involved in enhancement of phagocytosis, presentation of antigens, and secretion of reactive species, such as oxygen species (ROS) and nitric oxide (NO) [6]. M2 microglia cells inhibit inflammation, clear cellular debris, and secrete low levels of cytokines. It has been reported that microglia cells remain activated for at least 1 year [22–24], and even for as long as 17 years after TBI, as revealed by human postmortem studies [25], indicating that the persistence of a chronic inflammatory stage is regulated by microglia cells. However, excessive production of inflammatory cytokines or mediators by activated microglia cells cause the degeneration of oligodendrocytes and neuronal axons, leading to permanent neurological damage in brain tissues. Hence, some researchers have proposed that targeting microglial activation might be a prime target for therapeutic intervention in inflammation-mediated CNS disorders, including TBI and its co-morbidity factors [6].
The Nrf2 system is broadly expressed in the CNS and is involved in response to acute cerebral lesions and neurodegenerative disorders. In addition to the significant regulatory role in the endogenous defense against diverse cellular stresses, Nrf2 has been suggested to play an important role in modulating neuroinflammation. Dysregulation of Nrf2 activity is thought to be one of the pathogenic mechanisms in brain injury. For example, Nrf2-knockout mice have been shown to be hypersensitive, showing increased microglial infiltration with high levels of TNF-α and IL-6 [18]. Nrf2 −/− primary cultured cortical neurons are highly sensitive to oxidative stress, and the effect can be reversed by transfection with a functional Nrf2 construct [26]. In addition, Nrf2-knockout mice administered kainite, a neurotoxin, also showed increased infiltration of microglia cells in the hippocampus [27]. Nrf2 −/− mice have been reported to show hypersensitivity to intracerebral hemorrhage (ICH) and ischemia-reperfusion-induced early brain injury [28,29], and spontaneously develop vacuolar leukoencephalopathy with widespread astrogliosis [30]. However, overexpression of Nrf2 has a critical protective role in CNS diseases. For example, Shih et al. created an activity inducer, tert-butyl-hydroquinone, that induces the expression of several Nrf2-mediated genes, and found that the activator showed a neuroprotective effect against cerebral ischemia in vivo [31]. Sulforaphane, another Nrf2 activator, has been shown to decrease brain edema following TBI [32]. Overexpression of Nrf2 in BV-2 microglia cells inhibited LPS-inducible microglial hyperactivation, contributing to inflammation-mediated neurodegenerative disorders [33]. These studies have indicated that the expression of Nrf2 is associated with protection against CNS diseases.
To identify whether Nrf2 protects against TBI partly by regulating microglia function, we established microglia cells that down-expressed and overexpressed Nrf2 by transfection with siNrf2 or pcDNA3.1-Nrf2. The mRNA and protein levels of Nrf2 were confirmed. After transfection, the results showed the viability of microglia cells was significantly inhibited by overexpression of Nrf2, demonstrating that the activation of microglia cells was suppressed. Subsequently, we explored their phagocytic activity, which is an equally remarkable function of microglia cells. The results showed that the phagocytic activity was also significantly decreased by overexpression of Nrf2, indicating the protective role of Nrf2 in microglia. Furthermore, the expression of inflammatory cytokines (TNF-α and IL-6) in microglia cells was assessed. TNF-α, a major initiator of inflammation, is released in response to inflammatory stimulus at an early stage [34]. After release of TNF-α, IL-6 is released and is regarded as an important proinflammatory cytokine, which is responsible for the pathogenesis of many inflammatory diseases [35]. Expression levels of TNF-α and IL-6 were reduced by overexpression of Nrf2, demonstrating that Nrf2 can significantly suppress the inflammatory response.
Conclusions
This study demonstrates that Nrf2 might protect against TBI partly by regulation of microglia function.
Footnotes
Conflicts of interest
There are no conflicts of interest.
Source of support: Departmental sources
References
- 1.Werner C, Engelhard K. Pathophysiology of traumatic brain injury. Br J Anaesth. 2007;99:4–9. doi: 10.1093/bja/aem131. [DOI] [PubMed] [Google Scholar]
- 2.Balu R. Inflammation and immune system activation after traumatic brain injury. Curr Neurol Neurosci Rep. 2014;14:484. doi: 10.1007/s11910-014-0484-2. [DOI] [PubMed] [Google Scholar]
- 3.Schwarzmaier SM, Plesnila N. Contributions of the immune system to the pathophysiology of traumatic brain injury – evidence by intravital microscopy. Front Cell Neurosci. 2014;8:358. doi: 10.3389/fncel.2014.00358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Venkatesan C, Chrzaszcz M, Choi N, Wainwright MS. Chronic upregulation of activated microglia immunoreactive for galectin-3/Mac-2 and nerve growth factor following diffuse axonal injury. J Neuroinflammation. 2010;7:32. doi: 10.1186/1742-2094-7-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Loane DJ, Byrnes KR. Role of microglia in neurotrauma. Neurotherapeutics. 2010;7:366–77. doi: 10.1016/j.nurt.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hernandez-Ontiveros DG, Tajiri N, Acosta S, et al. Microglia activation as a biomarker for traumatic brain injury. Front Neurol. 2013;4:30. doi: 10.3389/fneur.2013.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gehrmann J. Microglia: A sensor to threats in the nervous system? Res Virol. 1996;147:79–88. doi: 10.1016/0923-2516(96)80220-2. [DOI] [PubMed] [Google Scholar]
- 8.Koshinaga M, Katayama Y, Fukushima M, et al. Rapid and widespread microglial activation induced by traumatic brain injury in rat brain slices. J Neurotrauma. 2000;17:185–92. doi: 10.1089/neu.2000.17.185. [DOI] [PubMed] [Google Scholar]
- 9.Gehrmann J, Matsumoto Y, Kreutzberg GW. Microglia: Intrinsic immuneffector cell of the brain. Brain Res Brain Res Rev. 1995;20:269–87. doi: 10.1016/0165-0173(94)00015-h. [DOI] [PubMed] [Google Scholar]
- 10.Mosser DM. The many faces of macrophage activation. J Leukoc Biol. 2003;73:209–12. doi: 10.1189/jlb.0602325. [DOI] [PubMed] [Google Scholar]
- 11.Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–64. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 12.Frugier T, Morganti-Kossmann MC, O’Reilly D, McLean CA. In situ detection of inflammatory mediators in post mortem human brain tissue after traumatic injury. J Neurotrauma. 2010;27:497–507. doi: 10.1089/neu.2009.1120. [DOI] [PubMed] [Google Scholar]
- 13.Kim J, Cha YN, Surh YJ. A protective role of nuclear factor-erythroid 2-related factor-2 (Nrf2) in inflammatory disorders. Mutat Res. 2010;690:12–23. doi: 10.1016/j.mrfmmm.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 14.Lee JM, Johnson JA. An important role of Nrf2-ARE pathway in the cellular defense mechanism. J Biochem Mol Biol. 2004;37:139–43. doi: 10.5483/bmbrep.2004.37.2.139. [DOI] [PubMed] [Google Scholar]
- 15.van Muiswinkel FL, Kuiperij HB. The Nrf2-ARE Signalling pathway: Promising drug target to combat oxidative stress in neurodegenerative disorders. Curr Drug Targets CNS Neurol Disord. 2005;4:267–81. doi: 10.2174/1568007054038238. [DOI] [PubMed] [Google Scholar]
- 16.Jin W, Wang HD, Hu ZG, et al. Transcription factor Nrf2 plays a pivotal role in protection against traumatic brain injury-induced acute intestinal mucosal injury in mice. J Surg Res. 2009;157:251–60. doi: 10.1016/j.jss.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 17.Jin W, Wang H, Yan W, et al. Disruption of Nrf2 enhances upregulation of nuclear factor-kappaB activity, proinflammatory cytokines, and intercellular adhesion molecule-1 in the brain after traumatic brain injury. Mediators Inflamm. 2008;2008:725174. doi: 10.1155/2008/725174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Innamorato NG, Rojo AI, Garcia-Yague AJ, et al. The transcription factor Nrf2 is a therapeutic target against brain inflammation. J Immunol. 2008;181:680–89. doi: 10.4049/jimmunol.181.1.680. [DOI] [PubMed] [Google Scholar]
- 19.Floden AM, Combs CK. Microglia repetitively isolated from in vitro mixed glial cultures retain their initial phenotype. J Neurosci Methods. 2007;164:218–24. doi: 10.1016/j.jneumeth.2007.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garden GA, Moller T. Microglia biology in health and disease. J Neuroimmune Pharmacol. 2006;1:127–37. doi: 10.1007/s11481-006-9015-5. [DOI] [PubMed] [Google Scholar]
- 21.Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- 22.Nagamoto-Combs K, Combs CK. Microglial phenotype is regulated by activity of the transcription factor, NFAT (nuclear factor of activated T cells) J Neurosci. 2010;30:9641–46. doi: 10.1523/JNEUROSCI.0828-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jacobowitz DM, Cole JT, McDaniel DP, et al. Microglia activation along the corticospinal tract following traumatic brain injury in the rat: A neuroanatomical study. Brain Res. 2012;1465:80–89. doi: 10.1016/j.brainres.2012.05.008. [DOI] [PubMed] [Google Scholar]
- 24.Jin X, Ishii H, Bai Z, Itokazu T, Yamashita T. Temporal changes in cell marker expression and cellular infiltration in a controlled cortical impact model in adult male C57BL/6 mice. PLoS One. 2012;7:e41892. doi: 10.1371/journal.pone.0041892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ramlackhansingh AF, Brooks DJ, Greenwood RJ, et al. Inflammation after trauma: microglial activation and traumatic brain injury. Ann Neurol. 2011;70:374–83. doi: 10.1002/ana.22455. [DOI] [PubMed] [Google Scholar]
- 26.Lee JM, Shih AY, Murphy TH, Johnson JA. NF-E2-related factor-2 mediates neuroprotection against mitochondrial complex I inhibitors and increased concentrations of intracellular calcium in primary cortical neurons. J Biol Chem. 2003;278:37948–56. doi: 10.1074/jbc.M305204200. [DOI] [PubMed] [Google Scholar]
- 27.Kraft AD, Lee JM, Johnson DA, et al. Neuronal sensitivity to kainic acid is dependent on the Nrf2-mediated actions of the antioxidant response element. J Neurochem. 2006;98:1852–65. doi: 10.1111/j.1471-4159.2006.04019.x. [DOI] [PubMed] [Google Scholar]
- 28.Shah ZA, Li RC, Thimmulappa RK, et al. Role of reactive oxygen species in modulation of Nrf2 following ischemic reperfusion injury. Neuroscience. 2007;147:53–59. doi: 10.1016/j.neuroscience.2007.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang J, Fields J, Zhao C, et al. Role of Nrf2 in protection against intracerebral hemorrhage injury in mice. Free Radic Biol Med. 2007;43:408–14. doi: 10.1016/j.freeradbiomed.2007.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hubbs AF, Benkovic SA, Miller DB, et al. Vacuolar leukoencephalopathy with widespread astrogliosis in mice lacking transcription factor Nrf2. Am J Pathol. 2007;170:2068–76. doi: 10.2353/ajpath.2007.060898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shih AY, Li P, Murphy TH. A small-molecule-inducible Nrf2-mediated antioxidant response provides effective prophylaxis against cerebral ischemia in vivo. J Neurosci. 2005;25:10321–35. doi: 10.1523/JNEUROSCI.4014-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao J, Moore AN, Clifton GL, Dash PK. Sulforaphane enhances aquaporin-4 expression and decreases cerebral edema following traumatic brain injury. J Neurosci Res. 2005;82:499–506. doi: 10.1002/jnr.20649. [DOI] [PubMed] [Google Scholar]
- 33.Koh K, Kim J, Jang YJ, et al. Transcription factor Nrf2 suppresses LPS-induced hyperactivation of BV-2 microglial cells. J Neuroimmunol. 2011;233:160–67. doi: 10.1016/j.jneuroim.2011.01.004. [DOI] [PubMed] [Google Scholar]
- 34.Hesse DG, Tracey KJ, Fong Y, et al. Cytokine appearance in human endotoxemia and primate bacteremia. Surg Gynecol Obstet. 1988;166:147–53. [PubMed] [Google Scholar]
- 35.Kishimoto T. IL-6: from its discovery to clinical applications. Int Immunol. 2010;22:347–52. doi: 10.1093/intimm/dxq030. [DOI] [PubMed] [Google Scholar]