

Abstract
Neoantigens unique to each patient's tumor can be recognized by autologous T cells through their T-cell receptor (TCR) but the low frequency and/or terminal differentiation of mutation-specific T cells in tumors can limit their utility as adoptive T-cell therapies. Transfer of TCR genes into younger T cells from peripheral blood with a high proliferative potential could obviate this problem. We generated a rapid, cost-effective strategy to genetically engineer cancer patient T cells with TCRs using the clinical Sleeping Beauty transposon/transposase system. Patient-specific TCRs reactive against HLA-A*0201-restriced neoantigens AHNAKS2580F or ERBB2H473Y or the HLA-DQB*0601-restricted neoantigen ERBB2IPE805G were assembled with murine constant chains and cloned into Sleeping Beauty transposons. Patient peripheral blood lymphocytes were coelectroporated with SB11 transposase and Sleeping Beauty transposon, and transposed T cells were enriched by sorting on murine TCRβ (mTCRβ) expression. Rapid expansion of mTCRβ+ T cells with irradiated allogeneic peripheral blood lymphocytes feeders, OKT3, interleukin-2 (IL-2), IL-15, and IL-21 resulted in a preponderance of effector (CD27−CD45RA−) and less-differentiated (CD27+CD45RA+) T cells. Transposed T cells specifically mounted a polyfunctional response against cognate mutated neoantigens and tumor cell lines. Thus, Sleeping Beauty transposition of mutation-specific TCRs can facilitate the use of personalized T-cell therapy targeting unique neoantigens.
Introduction
Mutation-specific T cells likely play a key role in mediating long-term tumor regressions in adoptive T-cell therapy using tumor-infiltrating lymphocytes (TIL).1,2,3 In melanoma, ~20% of the patients treated with TIL and interleukin-2 (IL-2) following a nonmyeloablative conditioning regimen achieved durable, complete regression of metastatic disease.4,5 Retrospective analysis of these infused T cells revealed that TIL recognized patient-specific, somatic, non-synonymous mutations expressed by tumors.6,7,8 Prospective administration of TIL specifically reactive with ERBB2IPE805G neoantigen resulted in a durable regression of metastatic cholangiocarcinoma indicating that mutation-specific T cells could be used as a treatment for common epithelial cancers.2,9,10 Most cancers have tumor-derived mutations which could serve as neoantigens for T cells.11,12 Therefore, highly tumor-specific T-cell treatments could be potentially generated for any patient with T cells that recognize tumor mutations.
However, the direct use of TIL with desired antigen specificity is not always feasible. Our current method relies on screening multiple independently grown TIL microcultures for reactivity against the patient mutanome, which can be problematic if the tumor/mutation-specific TIL are infrequent or in late/terminal differentiation stages with limited in vivo expansion capacity.13,14,15 Alternatively, T-cell receptors (TCRs) from these patient's TIL could be transferred into autologous peripheral blood T cells with a younger phenotype and administered as treatment. This strategy would also allow for a more direct way to test the hypothesis that T cells recognizing somatic mutations can mediate objective tumor regressions.
Genetic transfer of patient-specific TCRs will likely require a rapid, flexible, safe, and cost-effective approach. The Sleeping Beauty transposon/transposase system is a candidate for this application because it uses DNA plasmids, which are inexpensive to manufacture and easy to manipulate.16,17 Sleeping Beauty transposition was originally developed from fish undergoing their evolutionary maturation and has been adapted for genetic transfer into human cells.18,19 Cotransfer of two Sleeping Beauty DNA plasmids leads to stable transgene expression. The Sleeping Beauty transposase plasmid transiently expresses transposase enzyme that digests the second plasmid, the Sleeping Beauty transposon, at inverted/direct repeats and ligates the transposon cassette containing the gene of interest, i.e., TCR, into TA dinucleotide repeats within the genome.
Sleeping Beauty plasmids have been approved for use in clinical trials evaluating the ability of T cells modified with chimeric antigen receptors to treat B-cell malignancies (NCT00968760, NCT01497184, NCT01362452, NCT01653717, NCT02194374).20,21,22 T cells genetically-modified with mutation-specific TCRs have not yet been infused into humans nor have TCRs been introduced into T cells with the Sleeping Beauty system in clinical trials. Prior studies established means to express TCRs targeting shared antigens, e.g., Wilm's tumor-1 (WT-1), melanocyte antigen recognized by T cells (MART-1) and p53, with modest efficiencies and with plasmids not developed for clinical use.23,24 We have built upon this experience by developing Sleeping Beauty transposons directly approved for clinical translation for the expression of unique, patient-derived mutation-specific TCRs and expanded transposed T cells with a schema streamlined for use in clinical protocols at the NCI Surgery Branch. Furthermore, we engineered our TCRs to express mouse constant regions which enabled the enrichment to high purities of TCR transposed T cells by sorting based on mouse TCRβ (mTCRβ). Thus, Sleeping Beauty transposition is a candidate platform for infusion of personalized TCR gene therapy.
Results
Construction of Sleeping Beauty transposons encoding mutation-specific TCRs
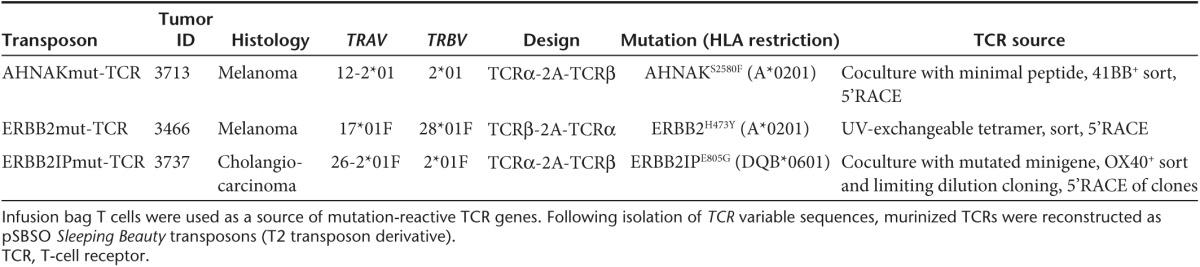
We modeled the generation of personalized TCR gene therapy with three mutation-reactive TCRs (Table 1). Genes encoding the TCRs were identified from infusion bag TIL administered to patients with metastatic cancer who experienced complete regression (CR) or partial regression (PR) of disease. The ERBB2mut-TCR was identified in patient 3466 (melanoma; CR; ongoing 59 months) from CD8+ T cells bound to HLA-A*0201/ERBB2H473Y peptide tetramer.25 The AHNAKmut-TCR was identified in patient 3713 (melanoma; CR; ongoing 39 months) following AHNAKS2580F peptide coculture and CD8+41BB+ T-cell sorting. Similarly, the ERBB2IPmut-TCR was identified in patient 3737 (cholangiocarcinoma; PR; ongoing 27 months) after ERBB2IPE805G gene coculture and CD4+OX40+ T-cell sorting.9 TCRα and TCRβ variable regions were sequenced after 5' RACE and fused to murine constant TCRα and TCRβ sequences, respectively, as has been done for other TCRs.26 Murinized TCRα and TCRβ were linked with furin cleavage site (RAKR), flexible region (SGSG), and porcine teschovirus-1 2A (P2A) ribosomal skip sequence27 so that equimolar amounts of TCRα and TCRβ polypeptides would be expressed. The AHNAKmut-TCR and ERBB2IPmut-TCR were constructed with TCRα-2A-TCRβ orientation and the ERBB2mut-TCR was designed with the TCRβ-2A-TCRα configuration. Murinized TCR genes were cloned into the clinical Sleeping Beauty transposon DNA plasmid, pSBSO.22,28 Human elongation factor 1α promoter drives translation of the TCR in this clinical plasmid and inverted/direct repeat integration sites flank the entire locus.29 Thus, mutation-specific TCRs could be identified from patients with metastatic cancer and used to generate Sleeping Beauty transposons for TCR expression in peripheral blood-derived T cells.
Table 1. Transposons used to introduce TCRs into peripheral T cells of patients with metastatic cancer.
Stable expression of mutation-specific TCRs achieved through Sleeping Beauty transposition, mTCRβ-enrichment, and REP
Peripheral blood mononuclear cells (PBMC) from patients with advanced cancer were co-electroporated with SB11 transposase16,30 and Sleeping Beauty transposons to express AHNAKmut-TCR, ERBB2mut-TCR, or ERBB2IPmut-TCR. Parallel electroporations were performed without DNA/TCR (mock) as a negative control. The day after electroporation (designated day+1), expression of mutation-specific TCRs could be detected in T cells (CD3+) by staining with anti-mTCRβ antibody (Figure 1a (top panels) and Figure 1b). Minimal detection (0.4 ± 0.1%; mean ± standard error of the mean (SEM); n = 6) of mTCRβ+ T cells in mock electroporation supported the specificity of staining. We took advantage of TCR murinization to develop a strategy for the enrichment of T cells expressing recombinant TCRs by staining with fluorescently-labeled mTCRβ-specific antibody and capturing labeled T cells with antifluorochrome magnetic beads (Figure 1a (middle panels) and Figure 1b). We chose to select the T cells prior to stimulation at a time when both integrated and transient (episomal) mTCRβ expression would be present rather than at a later time point when only integrated, stable mTCRβ would be expressed, e.g., day +14, because multiple stimulations could result in terminal differentiation of T cells for therapy which was undesired. This was an effective strategy as the cumulative frequency of CD3+mTCRβ+ T cells increased from 12.3 ± 1.4% to 80.8 ± 1.8% (mean ± SEM; n = 18) before and after mTCRβ enrichment, respectively. The recovery of mTCRβ+ T cells from magnetic bead enrichment was 13.5 ± 3.5% (mean ± SEM; n = 18) of the theoretical yield (% mTCRβ+ times total viable cells prior to the sort) and viability was 74.5 ± 2.2% (mean ± SEM; n = 18). mTCRβ-enriched T cells were then stimulated with a rapid expansion protocol (REP) consisting of a one-time addition of ≥200-fold excess of irradiated allogeneic PBMC feeders and agonistic anti-CD3 antibody (OKT3) along with supplementation every 2–3 days with IL-2, IL-15, and IL-21. At the end of the REP (day+22), stable expression of mutation-specific TCRs could be detected through mTCRβ staining, which was absent in mock/No TCR culture (Figures 1a (bottom panels) and Figure 1b). mTCRβ expression at day+1 was likely from a combination of integrated and episomal/transient transposon because the frequencies of mTCRβ+ T cells decreased following the REP. A hierarchy of mTCRβ+ T cells was detected amongst the three TCRs that followed AHNAKmut-TCR<ERBB2mut-TCR<ERBB2IPmut-TCR (Figure 1b far right). The kinetics of mTCRβ expression also followed a predictable pattern where mTCRβ-enriched T cells experienced a sharp decrease in frequency after 1 week of the REP (day+8), which is likely representative of the excess feeder PBMC population still present in the culture, but normalized to stable mTCRβ+ T-cell frequencies a week later (day+15) that were maintained during the last week (Figure 1c). The absolute numbers of T cells initiated in culture was <105 mTCRβ+ T cells and logarithmic growth of mTCRβ+ T cells over the 22-day period was observed suggesting that this strategy could be amenable to generating large numbers of mutation-reactive T cells (Figure 1d). We started with small T-cell numbers (<105 on average) because this was a proof-of-principle study where large yields were not required, and optimization of the expansion will be likely needed to treat patients with >109 T cells as is currently performed in some other T-cell therapies. In summary, nonviral Sleeping Beauty transposition, enrichment of mTCRβ+ T cells and REP were effective in generating T cells with stable mutation-specific TCR expression.
Figure 1.

Introduction, enrichment, and stable expression of mutation-specific T-cell receptors (TCRs) in peripheral blood T cells using Sleeping Beauty transposition. Peripheral blood leukocytes from patients with advanced cancer were coelectroporated with SB11 transposase and pSBSO Sleeping Beauty transposons (derivative of T2 transposon) containing mutation-specific TCRs fused to murine constant α and β chains. The following day, transposed T cells were enriched by capturing mouse TCRβ+ (mTCRβ) T cells with magnetic beads, and mTCRβ+ T cells were stimulated with a rapid expansion protocol (REP) supplemented with interleukin-2 (IL-2), IL-15, and IL-21. T cells electroporated without DNA/TCR (mock) were stimulated in parallel for negative control. (a) Evaluation of mTCRβ+ T cells following electroporation with (from left to right): No DNA/TCR (mock), AHNAKmut-TCR, ERBB2mut-TCR or ERBB2IPmut-TCR transposons. Day+1 pre- (top) and post- (middle) mTCRβ-enrichment and 22 days after expansion in REP (bottom) are displayed from 1 of 6 donors tested in two independent experiments. (b) Cumulative mTCRβ expression in AHNAKmut-TCR (circles), ERBB2mut-TCR (squares) and ERBB2IPmut-TCR (triangles) electroporated T cells (gated on live T cells; PInegCD3+) prior to enrichment with mTCRβ, post-mTCRβ enrichment and at the end of the REP (day+22). Each donor has a shape displayed for each TCR where means (n = 6) are displayed as lines. (c) Kinetics of mTCRβ expression during the REP of the three TCR transposon populations described in (b). Data are mean ± standard error of the mean (SEM) (n = 6) pooled from two independent experiments. (d) Kinetics of mTCRβ+ T cell expansion during the REP with the same TCR designations as in (b). mTCRβ+ T-cell counts at each time point were calculated by multiplying total cell counts by mTCRβ+CD3+ frequency. Data are mean ± SEM (n = 6) pooled from two independent experiments.
Memory phenotype of mTCRβ+ T cells and preferential expansion of CD8+ T cells
The surface phenotype of expanded mTCRβ+ T cells was evaluated to determine the potential characteristics of T cells for infusion as a personalized TCR gene therapy. Because our main goal is to generate cancer treatments, we genetically modified PBMC from patients with advanced cancer who had received multiple systemic chemotherapies such that the feasibility of this approach and the potential characteristics of the infusion product could be assessed. The initial population of mTCRβ+ T cells was dominated by CD4+ T cells but preferential expansion of CD8+ T cells during the REP resulted in a preponderance of CD8+mTCRβ+ T cells in the final product (Figure 2a). Addition of low-dose IL-2 (50 IU/ml) in combination with IL-15 and IL-21 also resulted in preferential outgrowth of CD8+ T cells in other T-cell expansion models29,31 and we did not observe the same phenomenon when culturing the T cells in IL-2 alone (data not shown). Preferential outgrowth of CD8+ T cells was noted in each of the three tested TCR populations indicating that it was not dependent upon the source of TCR, i.e., whether it was isolated from a CD4+ or CD8+ T cell (Figure 2b). Surface expression of memory markers was compared between T cells prior to electroporation (day 0) and T cells expressing mTCRβ after electroporation (day +1) to evaluate whether electroporation affected the T cells. Expression of CD27, CD57, CD62L, and CD95 were not changed but CD45RA and CD45RO were higher and lower, respectively, on mTCRβ+ T cells compared to untransfected T cells (Figure 2c). Therefore, TCR transfection was preferential in CD45RA+ T cells over CD45RO+ T cells. The same surface memory markers were compared at day +21 of the REP between mTCRβ+ T cells and mock electroporated (No DNA/TCR) T cells expanded in parallel with identical culture conditions. No DNA/TCR control T cells were predominantly CD45RO+CD95+CD57+/− (effector) phenotype. In contrast, mTCRβ+ T cells maintained similar expression of CD27, CD45RA, CD45RO, CD57, CD62L, and CD95 compared to initial (day +1) levels (Figure 2d). Thus, mTCRβ+ T cells maintained expression of surface memory molecules present in the initial PBMC composition. To characterize the T-cell phenotype further, combinations of CD45RA and CD27 expression were assessed on mTCRβ+ T cells. CD45RA−CD27−, CD45RA+CD27−, CD45RA−CD27+ and CD45RA+CD27+ cells have shown propensity to have effector, terminally differentiated (effector memory RA), memory, and naïve/minimally differentiated qualities, respectively.32,33,34,35 Using these parameters, CD4+mTCRβ+ T cells were composed primarily of effector and memory T cells (Figure 2e top) and CD8+mTCRβ+ T cells were mainly effector and minimally differentiated T cells (Figure 2e bottom). Murine models demonstrated that minimally differentiated T cells were maximally effective in adoptive T-cell therapy,32 but transfer of human TIL, which were mostly composed of effector T cells, resulted in complete regressions of metastatic melanoma.4 Furthermore, expression of CD27 on TIL correlated with clinical response to TIL therapy4 and the majority of T cells in this study expressed CD27 (Figure 2d). This platform generated mutation-specific T cells that would have characteristics of effective T cells based on murine models, i.e., minimally differentiated T cells, and based on the current experience in human clinical trials, i.e., CD27+ T cells and/or effector T cells. Thus, it would not be necessary to choose effector T cells or minimally differentiated T cells for therapy because they were both present in the final T-cell product.
Figure 2.

Surface phenotype of T cells expressing mutation-specific T-cell receptors (TCRs). (a) Kinetics of CD4+mTCRβ+ (open circles) and CD8+mTCRβ+ (closed circles) frequencies during the REP. (b) Frequency of CD4+ (open bars) and CD8+ (closed bars) T cells within the mTCRβ+ gate for each TCR population on day+19. (c) Surface expression of memory markers on T cells (CD3+) on peripheral blood mononuclear cells prior to electroporation (open bars) or CD3+mTCRβ+ T cells at day+1 postelectroporation (closed bars). (d) Surface expression of memory markers on T cells (CD3+) expanded in parallel to TCR transposed T cells from mock (No DNA/TCR) electroporations (open bars) or CD3+mTCRβ+ T cells (closed bars) at day+21 postelectroporation. Student's two-tailed t-test for comparisons between mock and mTCRβ groups for each marker in (c) and (d). **P < 0.01 and ***P < 0.001 (e) Frequency of CD45RA and/or CD27 expression in CD4+mTCRβ+ (top) or CD8+mTCRβ+ (bottom) T cells. Data were mean ± standard error of the mean (n = 6 for (b) and (c) closed bars and n = 18 (six donors with three TCRs) for all other graphs). Data were pooled from two independent experiments.
Sleeping Beauty transposition of mutation-reactive TCRs redirects T cells to target tumor cell lines
The specificity of transposed T cells toward tumor cell lines was evaluated by surface expression of an activation marker (41BB) and interferon-γ (IFNγ) secretion as indicators of response. Established HLA-A*0201 tumor cell lines TC3713 (AHNAKS2580F ERBB2WT), TC3466 (AHNAKWT ERBB2H473Y) and TC4046 (AHNAKWT ERBB2WT) were coculture targets. An ERBB2IPE805G tumor cell line was unavailable, so Epstein-Barr virus (EBV)-immortalized B cells with HLA-DQB*0601 haplotype were pulsed with ERBB2IPE805G or ERBB2IPWT peptides for evaluation of the ERBB2IPmut-TCR. T cells alone (Media) or activated with OKT3 were used as negative and positive controls, respectively. AHNAKmut-TCR T cells displayed specific and CD8-restricted recognition of TC3713 (Figure 3a top panels and Figure 3b black bars). ERBB2mut-TCR T cells were specific for TC3466, which were also restricted to CD8+ T cells (Figure 3a middle panels and Figure 3b hatched bars). Reactivity to TC4046 was not detected by any of the transposed populations. ERBB2IPmut-TCR T cells showed coreceptor independent (CD4 and CD8) recognition of ERBB2IPE805G peptide with no IFNγ secretion or 41BB expression in response to ERBB2IPWT peptide (Figure 3a bottom panels and Figure 3b gray bars). Thus, Sleeping Beauty transposition of mutation-reactive TCRs conferred specific recognition of tumor cell lines.
Figure 3.

Specificity of mutation-specific T cells to tumor cells. At day 19 postelectroporation, transposed T cells expressing mutation-specific T-cell receptors (TCRs) were cocultured for 24 hours with target cells. Coculture supernatants were analyzed for interferon-γ (IFNγ) secretion by enzyme-linked immunosorbent assay and cells were evaluated by flow cytometry for 41BB expression as a marker for T-cell activation. TC3713 and TC3466 tumor cell lines endogenously express AHNAKS2580F and ERBB2H473Y, respectively. TC4046 is wild type for both genes. All three tumor cell lines were derived from patients with HLA-A*0201 haplotype. ERBB2IPWT or ERBB2IPE805G peptides were pulsed on HLA-DQB*0601 B cells for assessment of ERBB2IPmut-TCR specificity. (a) 41BB expression (y-axes) in CD8+ T cells (x-axes) of AHNAKmut-TCR (top), ERBB2mut-TCR (middle) and ERBB2IP-TCR (bottom) cocultures with targets listed above. Plots shown were gated on live (PIneg) CD3+mTCRβ+ cells. Relative frequencies are displayed in flow cytometry quadrants. (b) IFNγ secretion following coculture. Media only (no targets; T cells only) and OKT3 served as negative and positive controls, respectively. Data are mean ± standard error of the mean (n = 3 technical replicates) from one representative donor of six tested in two independent experiments.
Avidity of transposed mutation-specific TCRs can be detected below pmol/l mutated peptide concentration
After establishing that tumor cell lines could be targeted by transposed T cells expressing mutation-specific TCRs, the TCR avidity to mutated peptides was assessed. The T2 tumor cell line was used for pulsing AHNAK and ERBB2 minimal HLA-A*0201-restricted peptides because it is a tumor cell line with transporter associated with antigen processing (TAP) deficiency such that most endogenous HLA Class-I peptides were not presented on the cell surface and exogenous peptides are efficiently loaded.36 As was done for enzyme-linked immunosorbent assay (ELISA) experiments, EBV-transformed B cells (HLA-DQB*0601) were antigen presenting cells for ERBB2IP peptide testing. Cocultures of peptide-pulsed targets and transposed T cells were evaluated for IFNγ secretion by enzyme-linked immunospot (ELISPOT) assay. The AHNAKmut-TCR showed the highest avidity where IFNγ spots were off scale (>1,000 spots per 104 T cells) at 10−12.3 mol/l (0.5 pmol/l) of AHNAKS2580F peptide with recognition of AHNAKWT peptide only at high concentrations and in low frequencies (Figure 4a). Similarly, the ERBB2mut-TCR displayed avidity to ERBB2H473Y peptide to 10−12.3 mol/l (0.5 pmol/l) with minimal response to ERBB2WT peptide (Figure 4b). The ERBB2IPmut-TCR was reactive to down to 10–7.5 mol/l (31 nmol/l) ERBB2IPE805G peptide with no cross-reactivity to ERBB2IPWT peptide. Reports of other effective TCRs ranged from nmol/l to pmol/l avidities,37,38,39,40,41 which was the range for mutated neoantigen-specific TCRs used for this study. The TCR transposed T cells displayed similar avidity towards mutated peptide as TIL from which the TCRs were isolated.9,25 High avidity TCR transposed T cells were unlikely to have off-target effects in patients because (i) mutations would be present only on tumor cells, (ii) responses to wild-type peptide were only observed at high concentrations of peptide unlikely to be expressed by normal cells, and (iii) TCRs were autologous to the patient. Thus, high-avidity TCRs can be expressed on T cells to target tumor-mutated neoantigens using Sleeping Beauty transposition.
Figure 4.

Avidity of T-cell receptor (TCR)-transposed T cells to tumor-derived mutated peptides. Wild-type (wt) or mutated (mut) peptides were pulsed in decreasing concentrations on either HLA-A*0201 T2 cells (AHNAK and ERBB2) or HLA-DQB*0601 B cells (ERBB2IP). Cocultures of TCR transposed T cells and peptide-pulsed targets were evaluated with interferon-γ (IFNγ) enzyme-linked immunospot (ELISPOT). Limit of enumeration was 1,000 spots. Representative ELISPOT wells are shown to the right. (a) AHNAKmut-TCR, (b) ERBB2mut-TCR, and (c) ERBB2IPmut-TCR cocultures. Representative donor from six tested in two independent experiments is shown. Data are mean ± standard error of the mean (n = 3 technical replicates).
Recognition of mutated neoantigen by TCR-transposed T cells resulted in production of multiple effector molecules
After specificity and avidity of the transposed T cells was established, their simultaneous expression of multiple effector molecules in response to mutated neoantigen peptide was examined by intracellular staining. Cocultures were initiated in the same manner as was done for the ELISPOT assays, except (i) a single peptide concentration was used, (ii) cocultures were performed in round-bottom plates instead of ELISPOT plate to facilitate staining, and (iii) inhibitors of exocytosis, i.e., GolgiStop and GolgiPlug, were added 4 hours into coculture to block secretion of effector molecules. Cocultures were stained for IL-2, CD107a (measure of degranulation),42 IFNγ and tumor necrosis factor-α (TNFα) to evaluate CD4 and CD8 T-cell-specific responses. IL-2 was specifically expressed in response to mutated antigens mainly by CD4+mTCRβ+ T cells (Figure 5a,b first panels and graph). In contrast, CD107a expression was primarily restricted to responses by CD8+mTCRβ+ T cells to mutated peptide, which may be explained by the inherent cytolytic function of CD8+ T cells (Figure 5a,b second panels and graph). Consistent with the observations from tumor cell cocultures (ELISA) and ELISPOT assays, IFNγ was expressed specifically in response to mutated peptide in a coreceptor-independent fashion in CD4 and CD8 T cells (Figure 5,b third panels and graph). TNFα was also expressed in both CD4+mTCRβ+ and CD8+mTCRβ+ T cells in response to mutated peptide with minimal reactivity to wild-type peptide (Figure 5a,b fourth panels and graph). Boolean gating, which calculates the number of subpopulations a particular cell is found in the multiparameter flow cytometry data, demonstrated that mTCRβ+ T cells simultaneously expressed multiple effector molecules in response to mutated peptide (Figure 5c). Therefore, transposed T cells could generate a multifunctional attack on tumor cells based on recognition of mutated neoantigens.
Figure 5.

Production of multiple effector molecules by T-cell receptor (TCR)-transposed T cells in response to tumor-derived mutated peptides. Wild-type (wt) or mutated (mut) peptides were pulsed on either HLA-A*0201 T2 cells (AHNAK and ERBB2) or HLA-DQB*0601 B cells (ERBB2IP). TCR transposed T cells were cocultured with peptide-pulsed targets for 15 total hours and GolgiStop and GolgiPlug were added to cocultures after the first 4 hours of coculture to block exocytosis of effector molecules. (a) Representative expression of (from left to right): interleukin-2 (IL-2), CD107a, interferon-γ (IFNγ), and tumor necrosis factor-α (TNFα) in CD4+mTCRβ+ (top) and CD8+mTCRβ+ (bottom) T cells. The example shown is from the ERBB2IPmut-TCR. Frequencies are displayed above gates. Open histograms show wild type peptide cocultures and red shaded histograms display mutated peptide cocultures. (b) Cumulative expression of IL-2, CD107a, IFNγ, and TNFα by mTCRβ+ T cells in either CD4+ or CD8+ gates. Data are mean ± standard error of the mean (n = 18; triplicate technical replicates of six donors) pooled from two independent experiments. Student's paired; two-tailed t-tests were used for statistical analysis between wt/mut cultures for CD4 or CD8 gates. *P < 0.05, **P < 0.01, ***P < 0.001 (c) Boolean gating was used to determine the number of effector functions expressed by mTCRβ+ T cells in response to peptide coculture. Data are shown following gating on either CD4 or CD8. Data were pooled from two independent experiments for six donors with triplicate technical replicates for each condition.
TCR-transposed T cells specifically lyse autologous tumor cell lines
The ability of TCR-transposed T cells to lyse tumor targets was assessed using non-radioactive lysis assay (lactate dehydrogenase release).43 Tumor cell lines which endogenously expressed AHNAKS2580F (TC3713) or ERBB2H473Y (TC3466) were used as targets for T cells expressing AHNAKmut-TCR and ERBB2mut-TCR, respectively. Mock transfected T cells (No DNA/TCR) grown in parallel to TCR transposed T cells were used as negative controls. T cells expressing AHNAKmut-TCR lysed TC3713 significantly higher than their TCRneg controls in two of three donors tested (Figure 6a). T cells expressing ERBB2mut-TCR demonstrated significant lysis of TC3466 compared to autologous T cells without TCR expression in three of three donors tested (Figure 6b). The frequency lysis was relatively low which was (i) consistent with frequencies of CD8+ T-cell degranulation (CD107a expression) in response to mutated peptide (Figure 5b second panels) and (ii) consistent with high frequencies of “young” (CD45RA+CD27+) CD8+mTCRβ+ T cells which would have limited lytic capacity (Figure 2e bottom). In sum, TCR transposed T cells were highly functional and specific for mutated neoantigen and autologous tumor cells as evidenced by (i) surface expression of 41BB, (ii) IFNγ secretion (ELISPOT and ELISA), (iii) simultaneous production of multiple effector molecules, and (iv) cell lysis. Therefore, Sleeping Beauty transposition is a candidate platform for personalized TCR gene therapy.
Figure 6.

Specific lysis of tumor cell lines by T-cell receptor (TCR)-transposed T cells. Four-hour nonradioactive lysis assays (lactate dehydrogenase assay) were performed with transposed T cells and established tumor cell lines expressing the corresponding mutated neoantigen. (a) AHNAKmut-TCR-transposed T cells or No DNA/TCR (mock) T-cell cultures were cocultured with TC3713 (HLA-A*0201; AHNAKS2580F) at an effector to target (E:T) ratio of 10:1. (b) ERBB2mut-TCR transposed T cells or No DNA/TCR (mock) T-cell cultures were cocultured with TC3466 (HLA-A*0201; ERBB2H473Y) at an E:T ratio of 10:1. Three donors were evaluated for each TCR as indicated on the x-axes. Student's paired, two-tailed t-tests were used for statistical analysis between autologous TCR transposed T cells and No DNA/TCR T cells. *P < 0.05 and **P < 0.01
Discussion
TIL recognition of patient-specific, tumor-derived mutations through their TCR likely contributed to long-term, complete tumor regressions following adoptive T-cell therapy.1,10 Identification and transfer of mutation-reactive TCRs into “younger” T cells from peripheral blood could lead to T-cell therapies regardless of the abundance or phenotype of the TCR cell source, e.g., TIL. Because mutations and TCRs will be unique for each individual, a personalized approach is warranted. Lentiviral and γ-retroviral supernatants which are used to generate T-cell treatments for many patients with the same gene, e.g., CD19-specific chimeric antigen receptors, currently require expensive and time consuming safety testing and are thus limited in their ability to fit this therapeutic need. Sleeping Beauty transposition is approved by the Food and Drug Administration and is a candidate platform for personalized application because of the low cost and speed at which TCRs can be synthesized and cloned into DNA transposons.22 Herein, we establish that the clinical Sleeping Beauty transposon/transposase system can be used for stable, nonviral expression of recombinant TCRs in T cells from patients with advanced cancer. TCR-modified T cells were specific for mutated peptide and mutated tumor cell lines. Thus, this approach is a rapid, inexpensive, noninfectious approach to personalize TCR gene therapy.
The scalability of the Sleeping Beauty transposition and REP system may need to be optimized before patients are treated. In this research study, a fold expansion of 205 ± 41 (mean ± SEM; n = 18) was observed from <105 cells at the start of REP (Figure 1d). One of the ways to increase final cell counts would be to increase the number of cells that go into the REP. Starting mTCRβ+ T cells numbers could be increased by electroporating multiple cuvettes, e.g., 10 cuvettes of 2 × 107 PBMC to start with 2 × 108 cells,17 or by electroporating large cell numbers in one step, e.g., with the MaxCyte system. The initial expression of TCR (day+1) could be improved upon by (i) using ultraclean DNA which improves transgene transfer efficiency by electroporation22 or (ii) modifying transposons with alternate promoters or other untranslated elements to maximize production and stability of transgene expression.44,45 Another option would be to enrich for mTCRβ at the end of one REP (TCR expression will be stable) and perform a second REP to boost cell numbers and further improve frequency of mTCRβ+ T cells. However, this may promote the differentiation of transposed T cells and we chose to forego this strategy in this study to limit these potential complications. Some TCRs have more robust expression than do others (Figure 1), which is likely mediated by their variable regions because TCRs were fused to the same murine constant regions, and the levels of TCR expression may affect therapeutic efficacy.
The transfer of more than one TCR gene modified population may be desirable. As passenger mutations unique to each tumor are likely targets of TCRs, the probability of successfully eliminating the tumor could increase by infusing T cells with multiple specificities. Furthermore, mutation-specific TCR gene therapy could also be combined with TCRs targeting shared antigens, i.e., cancer germline antigens, to achieve a similar effect. In addition to TCRs-targeting neoantigens, we have already successfully transposed T cells with TCRs targeting MAGEA3 and MART-1 shared antigens (data not shown). During the course of this study, a total of 41 different TCRs were expressed at >20% mTCRβ+ using Sleeping Beauty transposons in a total of 16 cancer patient donor PBMC (all of the data not shown; three representative TCRs detailed in this report) indicating that this could be a widely applicable strategy for personalized TCR gene therapy. Capture of mTCRβ+ T cells, which would be electroporated and expanded separately in parallel to abrogate mis-pairing from cotransfection of multiple TCRs into the same T cell, could enrich different transposed populations with a common reagent and minimize dilution effects of mixing populations with variable frequencies of recombinant TCR. Production of mTCRβ-specific antibody at good manufacturing protocol standards is needed for the clinical implementation of this strategy. Alternatively, selection markers could be introduced into the transposon, e.g., truncated nerve growth factor receptor, for isolation of transposed T cells with magnetic beads already produced for clinical use.46 The ease of DNA plasmid manipulation lends itself to a tailored TCR gene therapy approach.
The type of T cell used for genetic engineering could also impact therapeutic efficacy. One of the advantages of Sleeping Beauty transposition over other gene transfer platforms which require cell division for transgene integration is its ability to transfer genes into cells without division, including minimally differentiated T cells. Mouse models demonstrated that T cells earlier in the differentiation cascade, e.g., stem cell memory or central memory, had superior antitumor activity compared to effector and effector memory T-cell counterparts.32,47,48 A syngeneic model for mutated p68 has been generated40 which opens opportunity to test the effects of T-cell differentiation on targeting mutated neoantigens in an immunologically replete host. The generation of xenogeneic models for human T cells expressing individualized TCRs targeting patient-specific mutations could allow for investigation of tumor eradication capabilities by sorted memory T-cell subsets. Surface phenotype of mTCRβ+ T cells produced from PBMC of patients with advanced cancer by Sleeping Beauty transposition and REP with IL-2/IL-15/IL-21 was consistent with both effector (CD27-CD45RA-) and early differentiation (CD27+CD45RA+) stages (Figure 2). This phenotypic composition mirrored that of T cells before expansion, suggesting that the ability to generate TCR transposed T cells with a particular surface phenotype will depend, at least in part, on the original composition of the patient PBMC. Nonetheless, retrospective studies of melanoma patients treated with TIL revealed that infusion of larger numbers of CD8+CD27+ T cells correlated for response to immunotherapy.4 The infusion of CD8+CD27+mTCRβ+ T cells could test if a similar memory phenotype could mediate tumor regressions in the gene-modified setting. The initial selection could also be modified to include isolation of CD62L+ T cells with magnetic beads to increase frequencies of T cells at earlier stages of differentiation.49 Alternatively, the platform for T-cell propagation could be changed to tailor T cells towards a particular phenotype. Intracellular signaling critical for memory maintenance could be modulated by engaging costimulatory (41BB or CD28) or coinhibitory (PD-1 or CTLA-4) pathways with agonistic/antagonistic antibodies added in the REP50,51,52,53,54,55 or through addition of irradiated artificial antigen presenting cells expressing the costimulatory ligand(s).29,56,57 Molecules that perturb T-cell differentiation signaling cascades, e.g., AKT or GSK3β inhibitors,13,32 could be added to the REP to limit differentiation. Sleeping Beauty transposes genes in nondividing cells,44,58 which gives it a capacity for infusion of T cells immediately after gene transfer. In this scenario, PBMC with full repertoire of T-cell memory populations, including unmanipulated stem cell memory T cells, could be genetically modified with TCRs and directly infused into patients where “young” T cells could capitalize on homeostatic cytokines immediately after infusion.
Translation of Sleeping Beauty transposition of TCRs into clinical trials can test whether T cells can directly mediate tumor regressions by recognition of somatic mutations. The identification and direct use of mutation-reactive TCRs is a feasible approach because T cells with specificity for mutated neoantigens have been reproducibly identified from a number of tumor histologies and HLA haplotypes.2,7,9,59,60 Some of the techniques used to find these TCRs include but are not limited to: (i) TIL coculture with mutated genes/peptides, (ii) isolation with tetramers, e.g., ultraviolet light-exchangeable tetramers,25,61 or (iii) pairing of TCRs based on frequency in T-cell subpopulations, e.g. PD-1+ or 41BB+, directly from tumor digests or patient blood.15,62 Approaches to isolate mutation-reactive TCRs directly from tumors or blood, i.e., bioinformatics-based pairing of TCRα and TCRβ (pairSEQ) or sorting subsets with FACS and identifying pairs by single-cell PCR,63,64 could decrease the time required to make TCR transposed T cells. Mutation-specific T cells are likely to result in tumor elimination9 which could justify spending more time and effort to identify and transfer the neoantigen-specific TCRs into autologous T cells. If these studies can be performed prior to standard of care then personalized TCR gene therapy can be waiting for patients if conventional care fails to cure them of disease. Using this platform, the hypothesis that T cells can mediate tumor regressions through the recognition of mutated tumor antigens by TCRs can be directly evaluated.
Materials and Methods
Patient samples. Blood and tumor specimens were obtained after written, informed consent was granted. Mutations were identified by exomic sequencing as previously described.2,6,9,25 PBMC from apheresis were isolated by Ficoll-Hypaque and cryopreserved. An immortalized antigen presenting cell line was generated from patient 3737 (autologous to ERBB2IPmut-TCR; HLA-DQB*0601) from B cells initially expanded on irradiated NIH-3T3-CD40L feeder cells (+IL-4) then infected with EBV according to standard protocols.
Reagents. Antibodies specific for human CD3, CD4, CD8, CD27, CD28, CD45RA, CD45RO, CD62L, CD57, CD95, and 41BB were purchased from BD Biosciences (San Jose, CA). Anti-human CD107a, IL-2, IFNγ, and TNFα antibodies were acquired from Biolegend (San Diego, CA). Anti-mouse TCRβ antibody was purchased from eBioscience (San Diego, CA). Cells were stained at 4 °C for 30 minutes and washed with PBS, 0.5% fetal bovine serum and 5 mmol/l ethylenediaminetetraacetic acid, pH 8.0. AHNAKS2580F (FMPDFDLHL), AHNAKWT (SMPDFDLHL), ERBB2H473Y (ALIHHNTYL), ERBB2WT (ALIHHNTHL), ERBB2IPE805G (TSFLSINSKEETEHLENGNKYPNLE), and ERBB2IPWT (TSFLSINSKEETGHLENGNKYPNLE) peptides were purchased from Peptide 2.0 (Chantilly, VA).9,25 Peptides were solubilized in dimethyl sulfoxide (Sigma, St. Louis, MO) at 10 mg/ml. T-cell REP media was formulated with 50% AIM-V media (Gibco; Life Technologies, Grand Island, NY), 40% RPMI-1640 with L-glutamine media (Lonza, Basel, Switzerland), 10% human AB serum (Valley Biomedical, Winchester, VA), and antimicrobial agents (penicillin, streptomycin, gentamycin, and Fungizone; Gibco). Transfection media was phenol-free RPMI-1640 media (Lonza), Glutamax-100 (Gibco), 20% human AB serum and antimicrobial agents as above. Tumor cell line cultures were maintained in RPMI-1640 with L-glutamine, 10% fetal bovine serum, and antimicrobial agents detailed above.
Sleeping Beauty transposon and transposase plasmids. AHNAKmut-TCR and ERBB2IPmut-TCRs were constructed with human TCR variable (V) and murine constant (mC) regions following: 5'—Vα/mCα/furin-SGSG-P2A/Vβ/mCβ—3'. Similarly, ERBB2mut-TCR construction followed: 5'—Vβ/mCβ/furin-SGSG-P2A/Vα/mCα—3' but the murine constant chains contained hydrophobic and cysteine modifications in order to assist pairing.65 Murinized TCRs were codon-optimized for expression in human cells, synthesized de novo (Gene Oracle, Mountain View, CA) and cloned into pMSGV1 γ-retroviral vector.9 TCRs were excised from pMSGV1 with XbaI and SalI restriction enzyme digestion and ligated into pSBSO transposon vector28,29 digested with NheI and XhoI restriction enzymes. TCR transposons and pKan-CMV-SB11 transposase plasmid16 were purified with HiSpeed Plasmid Maxi Kit including Buffer ER to remove endotoxin (Qiagen, Valencia, CA). Final plasmid preparations were adjusted to final concentration >2.0 mg/ml in endotoxin-free water. Plasmid identities were confirmed by differential restriction enzyme digestion.
Sleeping Beauty transposition and expansion of T cells. Cryopreserved metastatic cancer patient PBMCs were thawed and rested in RPMI-1640 media for 2 hours at 37 °C prior to electroporation. Nonadherent cells were harvested and spun at 200g for 10 minutes. For each electroporation reaction, 2 × 107 PBMC were adjusted to 100 μl with Human T cell Nucelofector Solution (Lonza), 15 μg SB transposon (TCR) plasmid and 5 μg SB11 transposase plasmid. T cells were electroporated on U-014 program of the Nucleofector II device (Lonza) then moved to a single well of a six-well plate containing 6 ml transfection media. Mock electroporations without DNA (no TCR) were performed in parallel as negative control. The day after electroporation (day+1), electroporated cells (~107) were stained with 10 μl anti-mTCRβ-allophycocyanin antibody for 30 minutes at 4 °C. Anti-allophycocyanin beads were added according to manufacturer's instructions (Miltenyi Biotec, San Diego, CA) and mTCRβ+ T cells were captured on LS columns (Miltenyi Biotec). mTCRβ+ T cells were stimulated the same day with REP consisting of 2 × 107 γ-irradiated allogeneic PBMC (pooled from three donors), 30 ng/ml OKT3 antibody, 50 IU/ml IL-2, 25 ng/ml IL-15, and 30 ng/ml IL-21. As negative control, 105 mock (no TCR/DNA) T cells were stimulated in parallel. Cultures were fed complete media supplemented with IL-2, IL-15, and IL-21 every 2–3 days. Six donors were tested in two independent experiments. Samples were phenotyped for mTCRβ, CD3, CD4, and CD8 on days 1, 8, 15, 19, and 22 with FACS Canto II (BD Biosciences). Flow cytometry data was analyzed on FlowJo (v10.0.6; FlowJo, Ashland, OR).
Tumor cell line coculture, IFNγ ELISA, and 41BB expression. ERBB2IP peptides (10 μg/ml) were pulsed overnight at 37 °C on HLA-DQB*0601 EBV-immortalized B cells to serve as ERBB2IPmut-TCR targets. Cocultures of tumor cell lines TC3713 (AHNAKS2580F ERBB2WT), TC3466 (AHNAKWT ERBB2H473Y), TC4046 (AHNAKWT ERBB2WT) or peptide-pulsed B cells with transposed T cells (5 × 104 of each target and effector per well) were initiated in round bottom plates and incubated overnight at 37 °C. Supernatants were evaluated for IFNγ secretion by ELISA. Cells were stained for CD3, CD4, CD8, mTCRβ, and 41BB then assessed by flow cytometry.
IFNγ ELISPOT assay of TCR avidity to mutated peptide. Wild-type and mutated peptides were serially diluted 10-fold eight times starting at 20 μg/ml and were then added to an equal volume of APCs. AHNAK and ERBB2 peptides were pulsed for 2 hours at 37 °C on T2 cells (HLA-A*0201). ERBB2IP peptides were pulsed overnight at 37 °C on HLA-DQB*0601 EBV-immortalized B cells. Cocultures of 5 × 104 target cells and 104 transposed T cells were incubated overnight in IFNγ ELISPOT wells. The following day, ELISPOT plates were developed according to manufacturer's instructions (Mabtech, Cincinnati, OH) and spots were enumerated by Immunospot (Cellular Technology Limited, Shaker Heights, OH).
Intracellular staining and analysis of polyfunctional responses. Wild-type and mutated peptides were pulsed as detailed for ELISPOT assays at 1 μg/ml (AHNAK and ERBB2) or 10 μg/ml (ERBB2IP). Transposed T cells (5 × 104 per well) were preincubated with anti-CD107a antibody for 1 hour at 37 °C then mixed with an equal number of target cells (5 × 104) for 15 hours coculture at 37 °C in round bottom plates. After 4 hours of coculture, GolgiPlug and GolgiStop (BD Biosciences) were added to inhibit exocytosis of effector molecules. Cocultures were stained for surface expression of CD4, then fixed and permeabilized with BD Cytofix/Cytoperm (BD Biosciences) and stained for expression of IL-2, IFNγ, TNFα, CD3, CD8 and mTCRβ.66 Samples were acquired on BD LSR Fortessa (BD Biosciences) and analyzed with FlowJo using Boolean gating function.
Nonradioactive lysis assay. Specific lysis was assessed by lactate dehydrogenase (LDH) Cytotoxicity Assay Kit according to manufacturer's instructions (ThermoFisher Scientific, Grand Island, NY).43 Briefly, 105 tumor cells from either TC3713 or TC3466 cell lines were cocultured with 105 TCR transposed T cells for 4 hours at 37 °C in phenol-free RPMI, 1% fetal bovine serum. Maximum lysis was achieved by addition of lysis buffer per the kit's instructions. Conditioned media (50 μl) was mixed 1:1 with assay substrate and incubated for 30 minutes at ambient temperature. Absorbance was immediately read at 490 and 680 nm. Media alone, T cells alone, and tumor cells alone were used for background subtraction. Specific lysis was calculated by the following equation: % lysis = 100 * (cocultured cells – T cells alone – media alone)/(tumor cells maximum – tumor cell minimum – media alone).
Acknowledgments
We would like to thank Perry Hackett (University of Minnesota) and Simon Olivares (M.D. Anderson Cancer Center) for their assistance with the Sleeping Beauty system. This research was supported by the Intramural Research Program of the NIH at the National Cancer Institute. On 7 May 2015, L.J.N.C. was appointed as the Chief Executive Officer at ZIOPHARM Oncology and remains at MD Anderson Cancer Center as a Visiting Scientist. The authors declare no other competing financial interests.
References
- Rosenberg, SA and Restifo, NP (2015). Adoptive cell transfer as personalized immunotherapy for human cancer. Science 348: 62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran, E, Ahmadzadeh, M, Lu, YC, Gros, A, Turcotte, S, Robbins, PF et al. (2015). Immunogenicity of somatic mutations in human gastrointestinal cancers. Science 350: 1387–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klebanoff, CA, Rosenberg, SA and Restifo, NP (2016). Prospects for gene-engineered T cell immunotherapy for solid cancers. Nat Med 22: 26–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg, SA, Yang, JC, Sherry, RM, Kammula, US, Hughes, MS, Phan, GQ et al. (2011). Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res 17: 4550–4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinrichs, CS and Rosenberg, SA (2014). Exploiting the curative potential of adoptive T-cell therapy for cancer. Immunol Rev 257: 56–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins, PF, Lu, YC, El-Gamil, M, Li, YF, Gross, C, Gartner, J et al. (2013). Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med 19: 747–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, YC, Yao, X, Crystal, JS, Li, YF, El-Gamil, M, Gross, C et al. (2014). Efficient identification of mutated cancer antigens recognized by T cells associated with durable tumor regressions. Clin Cancer Res 20: 3401–3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, YC, Yao, X, Li, YF, El-Gamil, M, Dudley, ME, Yang, JC et al. (2013). Mutated PPP1R3B is recognized by T cells used to treat a melanoma patient who experienced a durable complete tumor regression. J Immunol 190: 6034–6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran, E, Turcotte, S, Gros, A, Robbins, PF, Lu, YC, Dudley, ME et al. (2014). Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 344: 641–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran, E and Rosenberg, SA (2014). T-cell therapy against cancer mutations. Oncotarget 5: 4579–4580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence, MS, Stojanov, P, Polak, P, Kryukov, GV, Cibulskis, K, Sivachenko, A et al. (2013). Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499: 214–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein, B, Papadopoulos, N, Velculescu, VE, Zhou, S, Diaz, LA Jr and Kinzler, KW (2013). Cancer genome landscapes. Science 339: 1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crompton, JG, Sukumar, M, Roychoudhuri, R, Clever, D, Gros, A, Eil, RL et al. (2015). Akt inhibition enhances expansion of potent tumor-specific lymphocytes with memory cell characteristics. Cancer Res 75: 296–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baitsch, L, Baumgaertner, P, Devêvre, E, Raghav, SK, Legat, A, Barba, L et al. (2011). Exhaustion of tumor-specific CD8⁺ T cells in metastases from melanoma patients. J Clin Invest 121: 2350–2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gros, A, Robbins, PF, Yao, X, Li, YF, Turcotte, S, Tran, E et al. (2014). PD-1 identifies the patient-specific CD8⁺ tumor-reactive repertoire infiltrating human tumors. J Clin Invest 124: 2246–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiti, SN, Huls, H, Singh, H, Dawson, M, Figliola, M, Olivares, S et al. (2013). Sleeping beauty system to redirect T-cell specificity for human applications. J Immunother 36: 112–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deniger, DC, Switzer, K, Mi, T, Maiti, S, Hurton, L, Singh, H et al. (2013). Bispecific T-cells expressing polyclonal repertoire of endogenous γδ T-cell receptors and introduced CD19-specific chimeric antigen receptor. Mol Ther 21: 638–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivics, Z, Hackett, PB, Plasterk, RH and Izsvák, Z (1997). Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell 91: 501–510. [DOI] [PubMed] [Google Scholar]
- Hackett, PB, Largaespada, DA and Cooper, LJ (2010). A transposon and transposase system for human application. Mol Ther 18: 674–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jena, B, Dotti, G and Cooper, LJ (2010). Redirecting T-cell specificity by introducing a tumor-specific chimeric antigen receptor. Blood 116: 1035–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kebriaei, P, Huls, H, Jena, B, Munsell, M, Jackson, R, Lee, DA et al. (2012). Infusing CD19-directed T cells to augment disease control in patients undergoing autologous hematopoietic stem-cell transplantation for advanced B-lymphoid malignancies. Hum Gene Ther 23: 444–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh, H, Huls, H, Kebriaei, P and Cooper, LJ (2014). A new approach to gene therapy using Sleeping Beauty to genetically modify clinical-grade T cells to target CD19. Immunol Rev 257: 181–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng, PD, Cohen, CJ, Yang, S, Hsu, C, Jones, S, Zhao, Y et al. (2009). Efficient nonviral Sleeping Beauty transposon-based TCR gene transfer to peripheral blood lymphocytes confers antigen-specific antitumor reactivity. Gene Ther 16: 1042–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field, AC, Vink, C, Gabriel, R, Al-Subki, R, Schmidt, M, Goulden, N et al. (2013). Comparison of lentiviral and sleeping beauty mediated αβ T cell receptor gene transfer. PLoS One 8: e68201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen, CJ, Gartner, JJ, Horovitz-Fried, M, Shamalov, K, Trebska-McGowan, K, Bliskovsky, VV et al. (2015). Isolation of neoantigen-specific T cells from tumor and peripheral lymphocytes. J Clin Invest 125: 3981–3991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haga-Friedman, A, Horovitz-Fried, M and Cohen, CJ (2012). Incorporation of transmembrane hydrophobic mutations in the TCR enhance its surface expression and T cell functional avidity. J Immunol 188: 5538–5546. [DOI] [PubMed] [Google Scholar]
- Szymczak, AL, Workman, CJ, Wang, Y, Vignali, KM, Dilioglou, S, Vanin, EF et al. (2004). Correction of multi-gene deficiency in vivo using a single ‘self-cleaving' 2A peptide-based retroviral vector. Nat Biotechnol 22: 589–594. [DOI] [PubMed] [Google Scholar]
- Singh, H, Figliola, MJ, Dawson, MJ, Olivares, S, Zhang, L, Yang, G et al. (2013). Manufacture of clinical-grade CD19-specific T cells stably expressing chimeric antigen receptor using Sleeping Beauty system and artificial antigen presenting cells. PLoS One 8: e64138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deniger, DC, Yu, J, Huls, MH, Figliola, MJ, Mi, T, Maiti, SN et al. (2015). Sleeping beauty transposition of chimeric antigen receptors targeting receptor tyrosine kinase-like orphan receptor-1 (ROR1) into diverse memory T-cell populations. PLoS One 10: e0128151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geurts, AM, Yang, Y, Clark, KJ, Liu, G, Cui, Z, Dupuy, AJ et al. (2003). Gene transfer into genomes of human cells by the sleeping beauty transposon system. Mol Ther 8: 108–117. [DOI] [PubMed] [Google Scholar]
- Singh, H, Figliola, MJ, Dawson, MJ, Huls, H, Olivares, S, Switzer, K et al. (2011). Reprogramming CD19-specific T cells with IL-21 signaling can improve adoptive immunotherapy of B-lineage malignancies. Cancer Res 71: 3516–3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gattinoni, L, Lugli, E, Ji, Y, Pos, Z, Paulos, CM, Quigley, MF et al. (2011). A human memory T cell subset with stem cell-like properties. Nat Med 17: 1290–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alves, NL, van Leeuwen, EM, Remmerswaal, EB, Vrisekoop, N, Tesselaar, K, Roosnek, E et al. (2007). A new subset of human naive CD8+ T cells defined by low expression of IL-7R alpha. J Immunol 179: 221–228. [DOI] [PubMed] [Google Scholar]
- Nkwanyana, NN, Gumbi, PP, Roberts, L, Denny, L, Hanekom, W, Soares, A et al. (2009). Impact of human immunodeficiency virus 1 infection and inflammation on the composition and yield of cervical mononuclear cells in the female genital tract. Immunology 128(1 Suppl): e746–e757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamann, D, Kostense, S, Wolthers, KC, Otto, SA, Baars, PA, Miedema, F et al. (1999). Evidence that human CD8+CD45RA+CD27- cells are induced by antigen and evolve through extensive rounds of division. Int Immunol 11: 1027–1033. [DOI] [PubMed] [Google Scholar]
- Luft, T, Rizkalla, M, Tai, TY, Chen, Q, MacFarlan, RI, Davis, ID et al. (2001). Exogenous peptides presented by transporter associated with antigen processing (TAP)-deficient and TAP-competent cells: intracellular loading and kinetics of presentation. J Immunol 167: 2529–2537. [DOI] [PubMed] [Google Scholar]
- Chinnasamy, N, Wargo, JA, Yu, Z, Rao, M, Frankel, TL, Riley, JP et al. (2011). A TCR targeting the HLA-A*0201-restricted epitope of MAGE-A3 recognizes multiple epitopes of the MAGE-A antigen superfamily in several types of cancer. J Immunol 186: 685–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draper, LM, Kwong, ML, Gros, A, Stevanović, S, Tran, E, Kerkar, S et al. (2015). Targeting of HPV-16+ epithelial cancer cells by TCR gene engineered T cells directed against E6. Clin Cancer Res 21: 4431–4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanitis, E, Smith, JB, Dangaj, D, Flingai, S, Poussin, M, Xu, S et al. (2014). A human ErbB2-specific T-cell receptor confers potent antitumor effector functions in genetically engineered primary cytotoxic lymphocytes. Hum Gene Ther 25: 730–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leisegang, M, Engels, B, Schreiber, K, Yew, PY, Kiyotani, K, Idel, C et al. (2015). Eradication of large solid tumors by gene therapy with a T cell receptor targeting a single cancer-specific point mutation. Clin Cancer Res (epub ahead of print). [DOI] [PMC free article] [PubMed]
- Rosati, SF, Parkhurst, MR, Hong, Y, Zheng, Z, Feldman, SA, Rao, M et al. (2014). A novel murine T-cell receptor targeting NY-ESO-1. J Immunother 37: 135–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolint, P, Betts, MR, Koup, RA and Oxenius, A (2004). Immediate cytotoxicity but not degranulation distinguishes effector and memory subsets of CD8+ T cells. J Exp Med 199: 925–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidmann, E, Brieger, J, Jahn, B, Hoelzer, D, Bergmann, L and Mitrou, PS (1995). Lactate dehydrogenase-release assay: a reliable, nonradioactive technique for analysis of cytotoxic lymphocyte-mediated lytic activity against blasts from acute myelocytic leukemia. Ann Hematol 70: 153–158. [DOI] [PubMed] [Google Scholar]
- Hackett, PB, Ekker, SC, Largaespada, DA and McIvor, RS (2005). Sleeping beauty transposon-mediated gene therapy for prolonged expression. Adv Genet 54: 189–232. [DOI] [PubMed] [Google Scholar]
- Walisko, O, Schorn, A, Rolfs, F, Devaraj, A, Miskey, C, Izsvák, Z et al. (2008). Transcriptional activities of the Sleeping Beauty transposon and shielding its genetic cargo with insulators. Mol Ther 16: 359–369. [DOI] [PubMed] [Google Scholar]
- Orchard, PJ, Blazar, BR, Burger, S, Levine, B, Basso, L, Nelson, DM et al. (2002). Clinical-scale selection of anti-CD3/CD28-activated T cells after transduction with a retroviral vector expressing herpes simplex virus thymidine kinase and truncated nerve growth factor receptor. Hum Gene Ther 13: 979–988. [DOI] [PubMed] [Google Scholar]
- Graef, P, Buchholz, VR, Stemberger, C, Flossdorf, M, Henkel, L, Schiemann, M et al. (2014). Serial transfer of single-cell-derived immunocompetence reveals stemness of CD8(+) central memory T cells. Immunity 41: 116–126. [DOI] [PubMed] [Google Scholar]
- Gattinoni, L, Klebanoff, CA, Palmer, DC, Wrzesinski, C, Kerstann, K, Yu, Z et al. (2005). Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J Clin Invest 115: 1616–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casati, A, Varghaei-Nahvi, A, Feldman, SA, Assenmacher, M, Rosenberg, SA, Dudley, ME et al. (2013). Clinical-scale selection and viral transduction of human naïve and central memory CD8+ T cells for adoptive cell therapy of cancer patients. Cancer Immunol Immunother 62: 1563–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willoughby, JE, Kerr, JP, Rogel, A, Taraban, VY, Buchan, SL, Johnson, PW et al. (2014). Differential impact of CD27 and 4-1BB costimulation on effector and memory CD8 T cell generation following peptide immunization. J Immunol 193: 244–251. [DOI] [PubMed] [Google Scholar]
- Borowski, AB, Boesteanu, AC, Mueller, YM, Carafides, C, Topham, DJ, Altman, JD et al. (2007). Memory CD8+ T cells require CD28 costimulation. J Immunol 179: 6494–6503. [DOI] [PubMed] [Google Scholar]
- Suzuki, T, Ogawa, S, Tanabe, K, Tahara, H, Abe, R and Kishimoto, H (2008). Induction of antitumor immune response by homeostatic proliferation and CD28 signaling. J Immunol 180: 4596–4605. [DOI] [PubMed] [Google Scholar]
- Beane, JD, Lee, G, Zheng, Z, Mendel, M, Abate-Daga, D, Bharathan, M et al. (2015). Clinical scale zinc finger nuclease-mediated gene editing of PD-1 in tumor infiltrating lymphocytes for the treatment of metastatic melanoma. Mol Ther 23: 1380–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacon, JA, Pilon-Thomas, S, Sarnaik, AA and Radvanyi, LG (2013). Continuous 4-1BB co-stimulatory signals for the optimal expansion of tumor-infiltrating lymphocytes for adoptive T-cell therapy. Oncoimmunology 2: e25581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacon, JA, Sarnaik, AA, Chen, JQ, Creasy, C, Kale, C, Robinson, J et al. (2015). Manipulating the tumor microenvironment ex vivo for enhanced expansion of tumor-infiltrating lymphocytes for adoptive cell therapy. Clin Cancer Res 21: 611–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maus, MV, Thomas, AK, Leonard, DG, Allman, D, Addya, K, Schlienger, K et al. (2002). Ex vivo expansion of polyclonal and antigen-specific cytotoxic T lymphocytes by artificial APCs expressing ligands for the T-cell receptor, CD28 and 4-1BB. Nat Biotechnol 20: 143–148. [DOI] [PubMed] [Google Scholar]
- Forget, MA, Malu, S, Liu, H, Toth, C, Maiti, S, Kale, C et al. (2014). Activation and propagation of tumor-infiltrating lymphocytes on clinical-grade designer artificial antigen-presenting cells for adoptive immunotherapy of melanoma. J Immunother 37: 448–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackett, PB Jr, Aronovich, EL, Hunter, D, Urness, M, Bell, JB, Kass, SJ et al. (2011). Efficacy and safety of Sleeping Beauty transposon-mediated gene transfer in preclinical animal studies. Curr Gene Ther 11: 341–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, QJ, Yu, Z, Griffith, K, Hanada, K, Restifo, NP and Yang, JC (2016). Identification of T-cell Receptors Targeting KRAS-Mutated Human Tumors. Cancer Immunol Res 4: 204–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenstein, T, Leisegang, M, Uckert, W and Schreiber, H (2015). Targeting cancer-specific mutations by T cell receptor gene therapy. Curr Opin Immunol 33: 112–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodenko, B, Toebes, M, Hadrup, SR, van Esch, WJ, Molenaar, AM, Schumacher, TN et al. (2006). Generation of peptide-MHC class I complexes through UV-mediated ligand exchange. Nat Protoc 1: 1120–1132. [DOI] [PubMed] [Google Scholar]
- Chacon, JA, Wu, RC, Sukhumalchandra, P, Molldrem, JJ, Sarnaik, A, Pilon-Thomas, S et al. (2013). Co-stimulation through 4-1BB/CD137 improves the expansion and function of CD8(+) melanoma tumor-infiltrating lymphocytes for adoptive T-cell therapy. PLoS One 8: e60031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howie, B, Sherwood, AM, Berkebile, AD, Berka, J, Emerson, RO, Williamson, DW et al. (2015). High-throughput pairing of T cell receptor α and β sequences. Sci Transl Med 7: 301ra131. [DOI] [PubMed] [Google Scholar]
- Han, A, Glanville, J, Hansmann, L and Davis, MM (2015). Corrigendum: Linking T-cell receptor sequence to functional phenotype at the single-cell level. Nat Biotechnol 33: 210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goff, SL, Johnson, LA, Black, MA, Xu, H, Zheng, Z, Cohen, CJ et al. (2010). Enhanced receptor expression and in vitro effector function of a murine-human hybrid MART-1-reactive T cell receptor following a rapid expansion. Cancer Immunol Immunother 59: 1551–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasetto, A, Frelin, L, Aleman, S, Holmström, F, Brass, A, Ahlén, G et al. (2012). TCR-redirected human T cells inhibit hepatitis C virus replication: hepatotoxic potential is linked to antigen specificity and functional avidity. J Immunol 189: 4510–4519. [DOI] [PubMed] [Google Scholar]