

Abstract
Background
Gibberella stalk rot caused by Fusarium graminearum is one of the most destructive soil-borne diseases of maize (Zea mays L.). Chemical means of controlling Gibberella stalk rot are not very effective; development of highly resistant hybrids is the best choice for disease control. Hence, understanding of the molecular basis underlying maize resistance against Gibberella stalk rot would undoubtedly facilitate the resistance breeding for stalk rot.
Results
Two quantitative trait loci (QTL), qRfg1 and qRfg2, conferring resistance to Gibberella stalk rot were detected in our previous study. Three near-isogenic lines (NILs) of maize with either qRfg1 (NIL1) or qRfg2 (NIL2), or neither (NIL3) were generated and subjected to RNA sequencing to study the transcriptional changes after F. graminearum inoculation at 0 (control), 6, and 18 h post-inoculation (hpi). In total, 536,184,652 clean reads were generated, and gene expression levels were calculated using FPKM (fragments per kilobase of exon model per million mapped reads). A total of 7252 differentially expressed genes (DEGs) were found in the three NILs after F. graminearum inoculation. As many as 2499 DEGs were detected between NIL1 and NIL3 at 0 hpi, of which 884 DEGs were more abundant in NIL1 and enriched in defense responses. After F. graminearum inoculation, 1070 and 751 genes were exclusively up- and downregulated, respectively, in NIL1 as compared to NIL3. The 1070 upregulated DEGs were enriched in growth/development, photosynthesis/biogenesis, and defense-related responses. Genes encoding putative auxin-induced proteins and GH3 family proteins in auxin signaling pathway were highly induced and lasted longer in NIL3. Genes involved in polar auxin transport (PAT) were more abundant in NIL3 as compared with NIL2.
Conclusions
The qRfg1 confers its resistance to Gibberella stalk rot through both constitutive and induced high expression of defense-related genes; while qRfg2 enhances maize resistance to the disease via relatively lower induction of auxin signaling and repression of PAT. The defense-related transcriptional changes underlying each QTL will undoubtedly facilitate our understanding of the resistance mechanism and resistance breeding for maize stalk rot.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-016-2780-5) contains supplementary material, which is available to authorized users.
Keywords: Zea mays, RNAseq, Gibberella stalk rot, Constitutive resistance, Tryptophan, Auxin signaling pathway, Polar auxin transport
Background
Plants live in complex environments in which they interact closely with a broad range of microorganisms, such as fungi, oomycetes, bacteria, viruses, and nematodes. Plant pathogens may gain access to a plant via a wound or natural opening such as stomata, or they may directly penetrate the plant surface. In response, plants have evolved sophisticated strategies to combat the invasion of different types of pathogens. Plants may use pathogen-associated molecular patterns (PAMPs) to elicit PAMP-triggered immunity (PTI) to combat pathogen infection [1, 2]. Induction of PTI is always accompanied by the induction of mitogen-activated protein kinases (MAPKs) and of calcium signaling, transcription of pathogen-responsive genes, production of reactive oxygen species (ROS), and deposition of callose to reinforce the cell wall at sites of infection [3]. For the pathogens escaped from PTI, effectors are employed to interfere with plant defenses. In turn, plants have evolved resistance (R) genes to recognize specific pathogen effectors, resulting in effector-triggered immunity (ETI) and a hypersensitive response (HR) at the infection site [2].
After a plant perceives a pathogen, conversion of the early pathogen-induced signals into defense responses depends largely on phytohormones [4]. Species of phytohormones induced in this process depends greatly on the lifestyle and infection strategy of the invader. The roles of salicylic acid (SA), jasmonic acid (JA), and ethylene (ET) in the regulation of plant immune responses have been established in many studies [4–6]. JA/ET primarily participates in deterrence of herbivores and resistance to necrotrophic pathogens, whereas the SA is primarily involved in resistance to biotrophic and hemibiotrophic pathogens [7, 8]. The roles of auxins in plant-pathogen interactions have also been described in recent years [9, 10]. Auxins can positively or negatively impact plant defense responses depending on the lifestyles of pathogens. Repression of the auxin response pathway in axr2-1 and axr1-1 mutants increases Arabidopsis susceptibility to the necrotrophic fungi Plectosphaerella cucumerina and Botrytis cinerea [11]. Nevertheless, disruption of auxin signaling in the Arabidopsis mutants axr1, axr2, and axr3 leads to enhanced resistance to the hemibiotrophic pathogen Fusarium oxysporum [12]. Auxin is synthesized in meristematic tissues like shoots, root tips, and lateral root initials, and transported within the plant either through phloem (known as non-polar auxin transport) or polar auxin transport (PAT). The inhibition of PAT with 2, 3, 5-triio-dobenzoic acid and 1-naphthylphthalamic acid increases resistance to F. oxysporum in Arabidopsis [12]. Flavonoids are endogenous inhibitors of PAT, and a tt4 mutant with a mutation in the CHALCONE SYNTHASE (CHS) increases the rate of auxin transport [13] and is more susceptible to F. oxysporum [12]. All these results suggest an important role for auxin signaling and PAT in plant defense.
Stalk rot is one of the most destructive soil-borne diseases of maize, and it causes substantial losses in yield and quality. Previous studies have clarified the genetic basis of stalk rot resistance, and several resistance quantitative trait loci (QTLs) and genes have been identified [14–20]. A transcriptome analysis conducted in a pair of resistant and susceptible near-isogenic lines (NILs) revealed that secondary metabolic pathways (e.g., biosynthesis of alkaloids and phenylpropanoids) and plant hormones may play important roles in maze resistance to F. graminearum-induced stalk rot [21]. Fusarium graminearum (teleomorph Gibberella zeae), the causal agent of Gibberella stalk rot, also causes Ear Rot [22, 23] and Crown Rot in maize [24] and Fusarium Head Blight (FHB) in wheat and barley [25]. F. graminearum, known as a hemibiotrophic pathogen, always uses cell-wall degrading enzymes (CWDEs) and trichothecenes to facilitate invasion [24]. Deoxynivalenol (DON), a type of trichothecene secreted by Fusarium species, usually serves as a virulence factor during the infection of host and is harmful to animal and human health [26, 27]. It can cause the HR and programmed cell death (PCD) during infection of host plants [28]. Although the virulence role of DON on plant cell is unknown, it can act as an eukaryotic protein synthesis inhibitor in vitro [29]. Hence, Fusarium species may utilize DON to suppress or delay the plant defense response against fungal attack by inhibiting the synthesis of resistance-related proteins [30].
In recent years, transcriptome analysis has been used to study plant-pathogen interactions. Defense responses towards Fusarium species and its secreted trichothecenes have been intensely investigated in maize [21, 31–33], wheat [34–37], and barley [38–40]. Defense responses in different hosts are similar to a certain degree, e.g., the activation of the JA/ET or SA signaling pathway, induction of the genes encoding pathogenesis-related (PR) proteins and proteins involved in mycotoxin transportation and degradation, increased expression of genes encoding enzymes involved in the phenylpropanoid-related pathway, and the expression of oxidative stress response genes.
Although much progress has been made in charactering the defense mechanisms against F. graminearum, the molecular mechanism of resistance to maize stalk rot still remains obscure. Therefore, understanding the responses of maize to F. graminearum infection is important for disease control and resistance breeding. In our previous studies, two QTLs, namely qRfg1 and qRfg2, increasing the resistance percentage of maize plants to Gibberella stalk rot by 32–43 % and ~12 %, were finally refined to an interval of ~500 and ~300 kb on chromosomes 10 and 1, respectively [15, 16]. In our present study, three NILs differing at qRfg1 and qRfg2 were used to study the defense mechanisms involved in stalk rot using RNA sequencing (RNAseq). Three different time points were included to gain comprehensive insight into the molecular mechanisms in the response of maize to F. graminearum infection.
Results
Phenotypic evaluation of three NILs
Two resistance QTLs, qRfg1 and qRfg2, were separately introducing from the donor resistant line 1145 into the susceptible line Y331 via marker-assisted backcrossing procedure. In the advanced backcross generation, two individuals with the shortest donor fragments harboring either qRfg1 or qRfg2 were chosen. These two individuals were intercrossed, followed by two-rounds of self-pollination, to generate three NILs harboring either qRfg1 (NIL1) or qRfg2 (NIL2), or neither (NIL3). The genetic background of each of three NILs was evaluated using a GoldenGate 3KSNP (Illumina, San Diego, CA, USA), and each NIL shared >99.9 % identical genetic background with the recurrent parent Y331. We assessed disease severity in the three NILs after inoculation with F. graminearum at the mature stage as described by Yang et al. [15] and seedling stage as described by Ye et al. [21]. As shown in Fig. 1a and b, both qRfg1 and qRfg2 significantly increased disease resistance at two stages. We also assessed morphological traits such as plant height, ear height, and node number. Consequently, qRfg1 significantly increased all three traits, whereas qRfg2 could only significantly increase two of the three traits, ear height and node number (Fig. 1c, d and e).
Fig. 1.
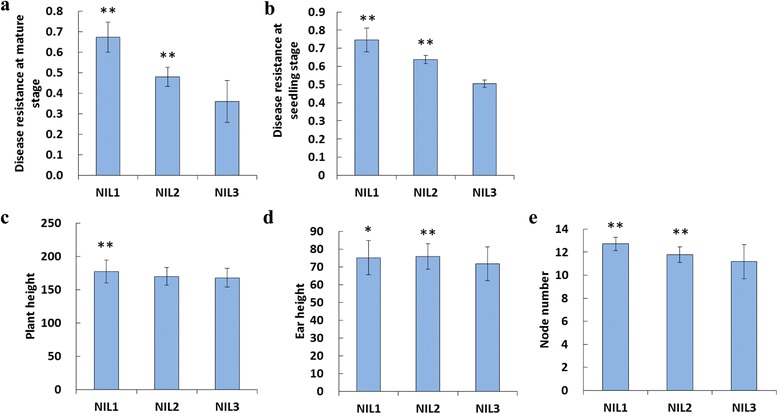
Phenotypic evaluation of the three NILs. Shown is disease resistance of the NILs after inoculation with F. graminearum at the mature (a) and seedling stages (b) and morphological traits evaluated in the field: c, plant height; d, ear height; e, nodes number. Cm was used for the measure of plant height and ear height; asterisks indicate significant differences for each NIL compared with NIL3 (two-tailed Student's t-test, *P < 0.05, **P < 0.01)
Profiling the defense transcriptome of the three NILs responsive to F. graminearum infection using RNAseq
To obtain a global gene expression profile of the three NILs during F. graminearum infection, roots of the NILs were sampled at 0 (control), 6, and 18 h post-inoculation (hpi) with two biological replicates, and the extracted RNA was sequenced using the Illumina HiSeq 2000 platform. Transcriptome sequence data for all samples can be found in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) under accession number PRJNA308408. After filtering and quality control of the raw reads, a total of 536,184,652 clean reads were yielded, and the average number of reads per sample ranged from 24,083,640 to 34,407,089 (average = 29,788,036; Table 1). The maize B73 genome was used as the reference for reads mapping (ZmB73_RefGen_v2; http://www.maizesequence.org), and TopHat was used to map the reads against the reference genome [41]. Finally, ~75 % (403,670,264/536,184,652) of the reads were mapped to the B73 genome, of which 86 % (347,503,158/403,670,264) were uniquely mapped (Table 1). All the uniquely mapped reads were transformed into FPKM (fragments per kilobase of exon model per million mapped reads) as implemented with Cufflinks [41], and DEseq passage was used to identify the differentially expressed genes (DEGs) between each comparison with a threshold of P-value/FDR < 0.05 and foldchange ≥ 2 [42]. The identified DEGs were annotated according to the maize genome; for those DEGs that did not have an unequivocal annotation in maize, the Arabidopsis description was chosen using a blast E-value cutoff of 10−10; otherwise, the gene were denominated as “Unknown”. Raw counts of expression data revealed a high Pearson’s correlation between each biological replicate (>0.90) for all the samples analyzed, indicating high reproducibility of the sequencing data (Additional file 1: Figure S1).
Table 1.
Read statistics for the three NILs after F. graminearum inoculation
| Summary | All Reads | Mapped Reads | Unmaped Reads | Unique Mapped Reads | Mapping Rates | Unique Mapping Rates | ||
|---|---|---|---|---|---|---|---|---|
| NIL1 | 0 hpi | repl_1 | 24,725,166 | 19,999,237 | 4,725,929 | 16,288,802 | 0.81 | 0.66 |
| repl_2 | 36,908,812 | 29,404,019 | 7,504,793 | 25,159,617 | 0.80 | 0.68 | ||
| 6 hpi | repl_1 | 32,929,308 | 23,508,743 | 9,420,565 | 21,025,580 | 0.71 | 0.64 | |
| repl_2 | 35,884,870 | 26,236,676 | 9,648,194 | 23,196,198 | 0.73 | 0.65 | ||
| 18 hpi | repl_1 | 37,364,076 | 28,211,864 | 9,152,212 | 25,150,279 | 0.76 | 0.67 | |
| repl_2 | 22,144,122 | 17,404,687 | 4,739,435 | 14,956,659 | 0.79 | 0.68 | ||
| NIL2 | 0 hpi | repl_1 | 24,034,692 | 17,845,686 | 6,189,006 | 15,475,706 | 0.74 | 0.64 |
| repl_2 | 24,132,588 | 17,996,916 | 6,135,672 | 16,093,485 | 0.75 | 0.67 | ||
| 6 hpi | repl_1 | 27,813,894 | 20,671,379 | 7,142,515 | 17,972,166 | 0.74 | 0.65 | |
| repl_2 | 26,652,486 | 19,881,940 | 6,770,546 | 16,687,925 | 0.75 | 0.63 | ||
| 18 hpi | repl_1 | 30,841,200 | 22,956,649 | 7,884,551 | 19,430,995 | 0.74 | 0.63 | |
| repl_2 | 25,759,186 | 20,212,586 | 5,546,600 | 14,044,578 | 0.78 | 0.55 | ||
| NIL3 | 0 hpi | repl_1 | 34,672,018 | 26,572,699 | 8,099,319 | 23,566,987 | 0.77 | 0.68 |
| repl_2 | 32,627,954 | 24,959,620 | 7,668,334 | 21,711,717 | 0.76 | 0.67 | ||
| 6 hpi | repl_1 | 30,388,570 | 22,870,029 | 7,518,541 | 20,120,065 | 0.75 | 0.66 | |
| repl_2 | 36,744,900 | 26,288,000 | 10,456,900 | 22,800,689 | 0.72 | 0.62 | ||
| 18 hpi | repl_1 | 25,082,644 | 18,406,593 | 6,676,051 | 16,339,773 | 0.73 | 0.65 | |
| repl_2 | 27,478,166 | 20,242,941 | 7,235,225 | 17,481,937 | 0.74 | 0.64 |
Identification of genes responsive to F. graminearum infection
To characterize transcriptome changes of the three NILs, we identified DEGs by comparing the gene expression profiles in the ranges of 0–6 hpi, 0–18 hpi, and 6–18 hpi for each NIL following F. graminearum inoculation. A total of 7252 DEGs were identified in the three NILs, and 4402, 3616, 5153 DEGs were identified in NIL1, NIL2, and NIL3, respectively. Three groups of genes could be discerned: i) 2956, 2665, and 2775 genes were upregulated, ii) 1809, 1242, and 2697 genes were downregulated, and iii) 363, 291, and 319 genes were upregulated or downregulated in NIL1, NIL2, and NIL3, respectively. For more details, 187, 137, and 103 genes were upregulated during 0–6 hpi and then downregulated during 6–18 hpi; while 176, 154, and 216 genes were downregulated during 0–6 hpi and then upregulated during 6–18 hpi in NIL1, NIL2, and NIL3, respectively (Fig. 2a).
Fig. 2.
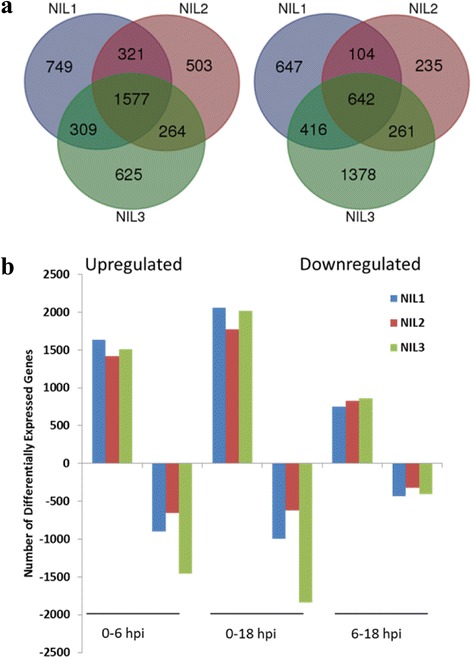
Differentially expressed genes in each NIL during the entire time course (a) and each post-inoculation interval (b) after inoculation with F. graminearum. Arrows indicate up- or downregulated after inoculation with F. graminearum in each NIL
During infection with F. graminearum, DEGs between any two of the three time points for each NIL were identified and shown in Fig. 2b. During each interval, there were much more upregulated genes than downregulated genes in each NIL, and NIL3 had the largest number of downregulated genes at any interval. As shown in Fig. 2b, the number of DEGs during the 6–18 hpi interval for each genotype was much smaller than that during the interval 0–6 hpi in each NIL, indicating a lesser transcriptome change from 6 to 18 hpi.
The common defense responses among the different genotypes after F. graminearum infection
After F. graminearum inoculation, the transcriptome of the three NILs underwent dramatic adjustment, and thousands of infection-related genes were induced. There were 1577 and 642 genes commonly induced and repressed in the three NILs after F. graminearum inoculation (Fig. 2a).
Gene Ontology (GO) analysis was carried out, and the commonly induced genes were categorized into three functions: biological process (bp), cellular component (cc), and molecular function (mf). As shown in Fig. 3, only GO terms in biological process were listed. GO terms related to defense responses were significantly enriched in the commonly induced genes (FDR < 0.001), including jasmonic acid-mediated signaling pathway, oxidation-reduction process, response to chitin, MAPK cascade, response to oxidative stress, salicylic acid biosynthetic process, positive regulation of flavonoid biosynthetic process, response to salicylic acid stimulus, respiratory burst involved in defense response, response to ethylene stimulus, phenylpropanoid biosynthetic process, coumarin biosynthetic process, anthocyanin-containing compound biosynthetic process, response to cyclopentenone, hydrogen peroxide catabolic process. All other significantly enriched GO terms in each category were listed in Additional file 2: Table S1 (FDR < 0.05).
Fig. 3.
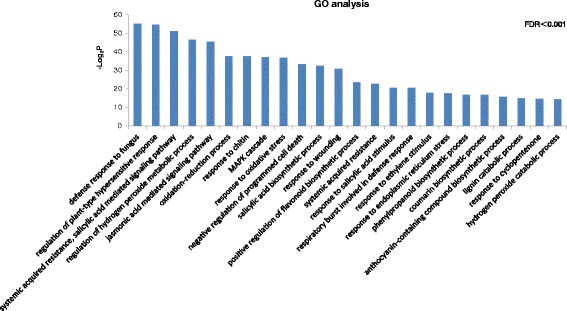
GO classification of genes commonly induced in three NILs. Genes were annotated in three main categories: biological process (bp), cellular component (cc), and molecular function (mf) and only bp were shown in this figure. All GO terms shown were significant at FDR ≤ 0.001
The importance of PRs for resistance to Fusarium species has been reported in many studies [31, 43, 44]. Among the commonly induced genes in three NILs, many PR genes were identified, including PR1, PR4, PR5 and PR10 (Fig. 4).
Fig. 4.
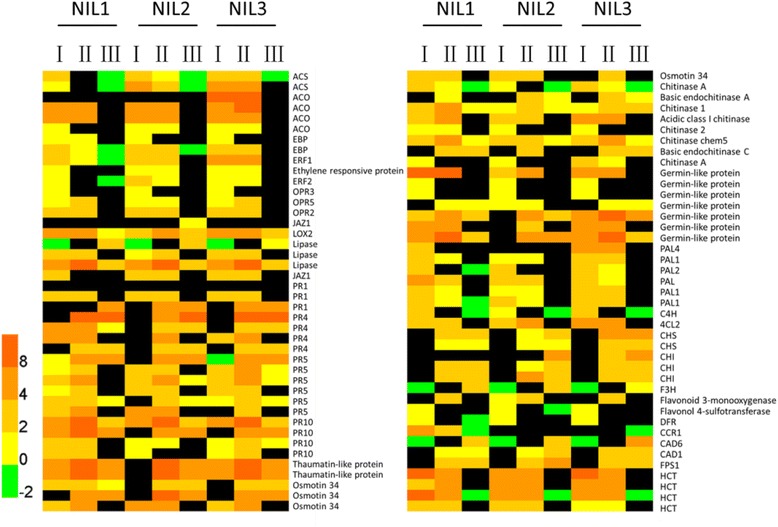
Samples of the resistance-related genes commonly upregulated in three NILs after inoculation of F. graminearum. The bottom colored scale represents the log2 of foldchange values for each gene. Black color indicates unchanged genes. The numerals I, II, and III indicated under each NIL denote a comparison between 0 and 6 hpi, 0 and 18 hpi, and 6 and 18 hpi, respectively. The function of each gene is listed to the right
After perception of a pathogen infection by the host, conversion of previous pathogen-induced signals into a defense response largely depends on phytohormones. Plant defense responses towards pathogen infection are regulated through a complex network of signaling that includes SA, JA, and ET [45, 46]. Genes encoding enzymes involved in the JA biosynthesis, like 12-oxo-phytodienoic acid reductase 5 (OPR5), Lipoxygenase 2 (LOX2), and JA signaling pathway, like jasmonate-zim-domain protein 1 (JAZ1), were commonly induced in three NILs (Fig. 6). As for ET, genes encoding enzymes involved in ET biosynthesis, like 1-aminocyclopropane-1-carboxylic acid oxidase (ACO), 1-aminocyclopropane-1-carboxylate synthase (ACS) and ET responsive proteins, like ethylene-responsive element binding protein (EBP), and ERF family proteins were commonly induced in three NILs (Fig. 4).
Fig. 6.
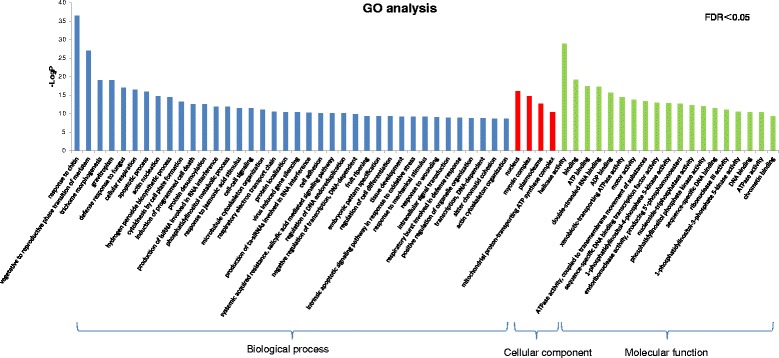
GO classification of genes more abundant in NIL1 than NIL3 at 0 hpi. Genes were annotated in three main categories: biological process (bp), cellular component (cc), and molecular function (mf). All the GO terms were significant at FDR ≤ 0.05
Genes involved in phenylpropanoid metabolism pathway, like phenylalanine ammonia-lyases (PALs), cinnamate-4-hydroxylase (C4H), and 4-coumarate-CoA ligase 2 (4CL2), were commonly induced in three NILs (Fig. 4).
F. graminearum-produced trichothecenes was reported to serve as virulence factor during infection of plants [24]. Detoxification genes, encoding glutathione S-transferases (GSTs), UDP-glucosyltransferases (UGTs), pleiotropic drug resistance family proteins (PDR), MATE efflux family proteins, heavy metal transport/detoxification superfamily proteins, and multidrug resistance-associated protein (MDR), were induced in three NILs in response to F. graminearum inoculation (Fig. 4).
In addition to the genes mentioned above, genes encoding pectin methylesterase inhibitor (PMEI), polygalacturonase inhibitor protein (PGIP), and xylanase inhibitor protein 1 involved in the inhibition of CWDE secretion by pathogens were commonly induced in three NILs (Fig. 4).
Validation via real-time quantitative reverse transcription-PCR (qRT-PCR)
A fraction of 19 genes that were commonly induced in the three NILs after F. graminearum inoculation were selected for the real-time quantitative reverse transcription-PCR (qRT-PCR) validation. The fold changes of each gene between any two of the three time points were calculated in each NIL. As shown in Additional file 3: Figure S2, the Pearson’s correlation coefficient between the data generated from the two platforms was high (R2 = 0.6859), indicating that the RNAseq analysis was well suited for analysis of F. graminearum infection-induced maize root transcriptome.
The qRfg1-dependent transcriptional profile in response to F. graminearum infection
To evaluate and characterize the effect of qRfg1 on the transcriptional profile, DEGs in and between NIL1 and NIL3 were analyzed. Basal differences were explored by comparing the NIL1 and NIL3 transcriptomes at 0 hpi. Differential expression analysis revealed 2499 DEGs, of which 884 were more abundant in NIL1 and 1615 were more so in NIL3 (Fig. 5a). Functional analysis of 884 DEGs in NIL1 revealed that biological processes related to defense responses, including response to chitin, defense response to fungus, induction of programmed cell death, response to JA stimulus, systemic acquired resistance, SA-mediated signaling, intrinsic apoptotic signaling in response to oxidative stress, response to wounding, intracellular signal transduction, and respiratory burst are significantly enriched (FDR ≤ 0.05) (Fig. 6). Additionally, biological processes related to growth/development (e.g., vegetative to reproductive phase transition of meristem, trichome morphogenesis, embryonic pattern specification, tissue development, regulation of cell differentiation) and cell division/organization (e.g., actin nucleation, microtubule cytoskeleton organization, actin cytoskeleton organization, positive regulation of organelle organization, regulation of DNA endoreduplication, cytokinesis by cell plate formation, sister chromatid cohesion) were also enriched. All these results indicate a superiority of resistance and growth condition of NIL1 compared with NIL3.
Fig. 5.
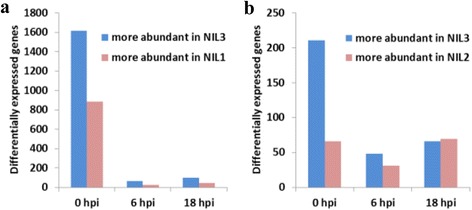
DEGs between each pair of NILs. DEGs between NIL1 and NIL3 (a) and between NIL2 and NIL3 (b) after inoculation with F. graminearum
Transcriptional changes induced by F. graminearum infection in resistant and susceptible genotypes were identified during the time course. A total of 2956 and 1809 genes were up- and downregulated in NIL1; while 2775 and 2697 were up- and downregulated in NIL3 (Fig. 2a). The majority of these genes were co-regulated in both lines (1886 genes commonly upregulated and 1058 genes commonly downregulated); the numbers of upregulated genes in both lines were almost the same, but there were more downregulated genes in NIL3 compared with NIL1 (Fig. 2a). As shown in Fig. 2a, 1070 and 889 genes were exclusively upregulated and 751 and 1639 genes exclusively downregulated in NIL1 and NIL3 respectively after F. graminearum inoculation. There were more genes exclusively upregulated and fewer genes exclusively downregulated in NIL1 compared with NIL3. Among the 1070 genes in NIL1, genes assigned to the biological process category were enriched in plant growth/development, plant photosynthesis/biogenesis, and defense-related responses (Additional file 4: Table S2). For the 751 exclusively downregulated genes in NIL1, biological process related to defense responses were enriched (Additional file 5: Table S3).
The qRfg2-dependent transcriptional profile in response to F. graminearum infection
Inoculation with F. graminearum altered the expression of genes involved in tryptophan (Trp) biosynthesis in NIL2 and NIL3, including genes encoding Anthranilate synthase component II, Orange pericarp1 (ORP1; also called Tryptophan synthase beta chain 1 Fragment), and Orange pericarp2 (ORP2; also called Tryptophan synthase beta chain 2) (Fig. 7). Anthranilate synthase component II is a homolog of Anthranilate synthase beta subunit 1 (ASB1) in Arabidopsis, which catalyzes the first step of Trp biosynthesis as a heterocomplex with anthranilate synthase alpha subunit (ASA1 or ASA2). The Trp pathway leads to the biosynthesis of indole-glucosinolate and indole 3-indolacetic acid (IAA) [12]. After F. graminearum inoculation, genes involved in glucosinolate metabolism were induced in NIL2 (Fig. 7a), including cytochrome P450, family 81, subfamily F, polypeptide 2 (CYP81F2), 3-isopropylmalate dehydratase large subunit 2, and UDP-glucose:thiohydroximate S-glucosyltransferase (UGT74B1). 3-isopropylmalate dehydratase large subunit is a homology to isopropyl malate isomerase large subunit 1 (IIL1) in Arabidopsis, and UDP-glucose:thiohydroximate S-glucosyltransferase (UGT74B1) is involved in the conversion of indole-3-acetaldoxime (IAOx) to indole-3-methyl-glucosinolate (IG). Both of them were involved in glucosinolate biosynthetic process [47]. In this study, IIL1 and CYP81F2 genes were induced in NIL3 (Fig. 7b). All these results suggested that F. graminearum infection altered the expression of genes in Trp biosynthesis.
Fig. 7.
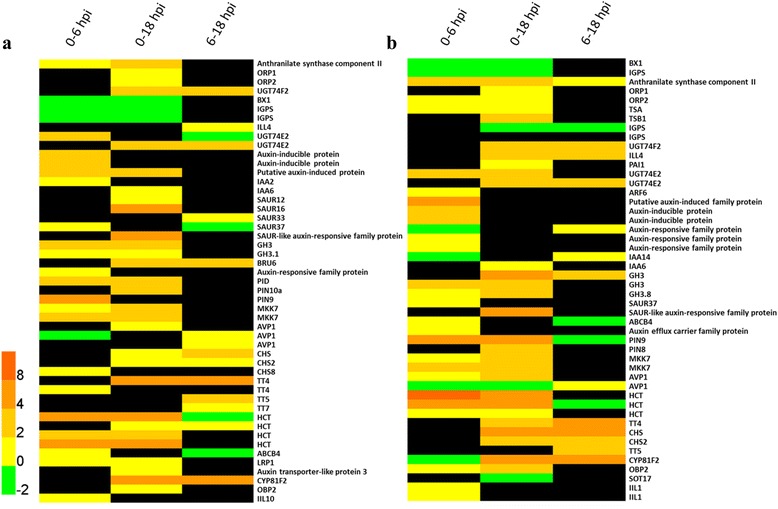
DEGs involved in Trp biosynthesis, auxin signaling and PAT. Genes involved in Trp biosynthesis, auxin signaling and PAT were induced in NIL2 (a) and NIL3 (b) after inoculation with F. graminearum. The bottom colored scale represents the log2 of foldchange values for each gene. Black color indicates unchanged genes
The inoculation of F. graminearum also induced the expression of genes encoding proteins involved in auxin signaling pathway in NIL2 and NIL3, such as auxin-inducible proteins, auxin-induced protein, AUX/IAA family proteins, SAUR family proteins, GH3 family proteins, auxin responsive family protein, and auxin response factor 6 (ARF6) (Fig. 7); However, foldchanges for several co-upregulated genes encoding one putative auxin-induced protein and two GH3 family proteins were higher in NIL3 than NIL2 (Fig. 8a); The gene encoding auxin-repressed 12.5 kDa protein was more repressed in NIL2 (foldchange = 0.49) than in NIL3 (foldchange = 0.20) (Fig. 8a). Several genes in auxin signaling pathway exhibited different expression patterns in NIL2 and NIL3. Two genes encoding auxin-inducible proteins kept increasing in gene expression during 0-18 hpi in NIL3, but only 0-6 hpi in NIL2. Gene encoding an AUX/IAA family protein was repressed in NIL2 and NIL3 at 6 hpi, whereas induced at 18 hpi in NIL3. Genes encoding proteins involved in auxin signaling pathway like SAUR37 and nucleoside diphosphate kinase 2 (NDPK2), and two auxin-inducible proteins, were more abundant in NIL3 than NIL2 (Fig. 8b).
Fig. 8.
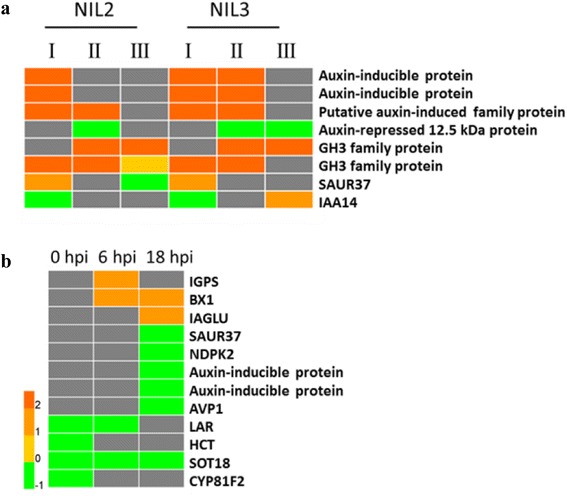
DEGs involved Trp biosynthesis and auxin-related processes between NIL2 and NIL3. Coregulated genes involved in auxin signaling in NIL2 and NIL3 (a) and DEGs involved in Trp biosynthesis, auxin signaling pathway and PAT between NIL2 and NIL3 (b) after inoculation of F. graminearum. The bottom colored scale represents the log2 of foldchange values for each gene Gray color indicates unchanged genes. The numerals I, II, and III indicated under each NIL denote a comparison between 0 and 6 hpi, 0 and 18 hpi, and 6 and 18 hpi, respectively
Genes involved in PAT were also found differentially expressed after F. graminearum inoculation. Genes that were induced in both NIL2 and NIL3 include PIN9 encoding an auxin efflux carrier family protein, MKK7 encoding a MAP kinase kinase 7, AVP1 encoding an inorganic H pyrophosphatase family protein, and ABCB4 encoding an ATP binding cassette subfamily B4 (Fig. 7). Genes encoding PID (a positive regulator of cellular auxin efflux), PIN10a, auxin transporter-like protein 3, and LRP1 were exclusively induced in NIL2 (Fig. 7a). Genes encoding PIN8, LPR1, and auxin efflux carrier family protein were induced in NIL3 (Fig. 7b). Flavonoids are endogenous inhibitors of PAT. In our study, genes involved in flavonoid biosynthesis like CHS, TT4, TT5, and TT7 were induced in both NIL2 and NIL3. P-coumaroyl CoA is a common substrate of two enzymes: CHS, which catalyzes the formation of flavonoids, and HCT, which is involved in the biosynthesis of lignin. Induction of HCT decreased the accumulation of flavonoids and relieved the inhibition of PAT. HCT were induced in NIL2 and NIL3 after inoculation with F. graminearum (Fig. 7).
Genes influencing auxin transport were also differentially expressed between NIL2 and NIL3. Gene encoding a leucoanthocyanidin reductase (LAR) that is involved in the biosynthesis of proanthocyanidins, a type of plant flavonoid [48], was more abundant in NIL3 at 0 and 6 hpi; The gene AVP1 was more abundant in NIL3 at 18 hpi; one HCT and CYP81F2 were more abundant in NIL3 at 0 hpi; one SOT18 was more abundant in NIL3 at 0, 6 and 18 hpi (Fig. 8b).
Discussion
The inhibition of gene expression and the constitutive resistance to F. graminearum conferred by qRfg1
The qRfg1 can significantly increase the maize resistance to F. graminearum at mature and seedling stages. Morphological traits such as plant height, ear height, and node number were all significantly increased by qRfg1 in NIL1 (Fig. 1c, d, and e). All these results indicate that qRfg1 play important roles in maize growth/development, as well as resistance against F. graminearum.
It has been reported that genes conferring bio/abiotic stress resistance may also be involved in the regulation of plant growth/development. Ghd7 was reported to be involved in the regulation of multiple processes including flowering time, hormone metabolism, and response to biotic and abiotic stresses [49]. In the young panicles of OX-Ghd7HJ19 transgenic rice, fewer genes were upregulated and more genes were downregulated compared with wild type, which indicates an inhibitory role for Ghd7 in transcription [49]. In our study, the basal difference was obtained by comparing NIL1 with NIL3 at 0 hpi; more genes (1615) were highly expressed in NIL3 compared with NIL1 (884), which corresponded well with the role of Ghd7 in the inhibition of gene expression.
Plant responses to pathogen infection may be quite diverse. Some plants may resist pathogen infection by inducing defense-related genes and pathways, while other plants may be well prepared for oncoming stress via the constitutive high expression of resistance genes specific for a biotic or abiotic stress, i.e., the plants create a resistance barrier before the full force of the stress occurs. In this study, lots of defense-related biological processes were enriched in the relevant genes more abundantly expressed in NIL1 (Fig. 6). All these results suggest that qRfg1 can counterattack the infection of F. graminearum via constitutive resistance. A previous study showed that OX-Ghd7HJ19 transgenic rice had altered expression of genes involved in hormone signaling [49]. In our study, genes more abundant in NIL1 were significantly enriched in biological processes, such as response to JA stimulus and SA-mediated signaling, suggesting the role of qRfg1 in the constitutive resistance to infection via the orchestration of phytohormones. Of the genes more abundant in NIL1, in addition to GO terms related to defense responses, biological processes related to growth/development, and cell division/organization were also significantly represented (Fig. 6). These results are consistent with the documented phenotype of NIL1 and suggest that in addition to the defense resistance, qRfg1 also participates in the regulation of growth/development.
After inoculation with F. graminearum, biological processes related to plant growth/development, plant photosynthesis/biogenesis, and defense-related responses were significantly represented in the exclusively upregulated genes in NIL1 (Additional file 4: Table S2). All these results demonstrated that qRfg1 provides maize resistance to F. graminearum through two approaches: the constitutive high expression of genes related to the resistance and induced defense responses after F. graminearum infection.
qRfg2 increase the resistance to F. graminearum via the relatively repression of auxin signaling
The qRfg2 can significantly increase the maize resistance to F. graminearum at both mature and seedling stages (Fig. 1a and b). The candidate gene for qRfg2 was predicated to encode an auxin-regulated protein [16]. It has been reported that auxin signaling contributes to the plant resistance. Repression of auxin signaling either through mutation of auxin signaling components or inhibition of auxin transport compromises Arabidopsis resistance to the necrotrophic fungi P. cucumerina and B. cinerea [11]. However, auxin signaling is required for susceptibility of Arabidopsis to F. oxysporum [12]. AXR2 and AXR3 encode the IAA/AUX proteins IAA7 and IAA17, respectively, which repress the expression of auxin-inducible genes. Arabidopsis mutants axr2 and axr3 increased F. oxysporum resistance by delaying symptom development relative to wild-type plants, whereas no significant difference in disease resistance between tir1 and wild-type plants is evident [12]. These results suggest that auxin signaling downstream of the auxin receptor contribute to the susceptibility of Arabidopsis to F. oxysporum. In our study, many genes involved in auxin signaling were upregulated in NIL2 and NIL3 (Fig. 7). All these results indicate that auxin signaling is required for susceptibility of both NIL2 and NIL3 to F. graminearum. As shown in Fig. 8a, several genes encoding proteins involved in auxin signaling were induced more in NIL3 than NIL2, including auxin-inducible proteins, putative auxin induced protein, GH3 family proteins, and SAUR family protein, suggesting that qRfg2 may increase the resistance to F. graminearum as a consequence of the relatively repression of auxin signaling genes.
PAT is required for the susceptibility of maize to F. graminearum infection
Auxin is synthesized in meristematic tissues like shoots, root tips, and lateral root initials, and it is transported within the plant either through phloem or PAT. Compared with auxin synthesized in roots, a relatively large proportion of auxin is transported to roots from aerial parts. Therefore, pathogens may target PAT components as a more effective way of modulating auxin levels to cause disease. It has been reported that inhibition of PAT through chemical or genetic means results in increased susceptibility to foliar fungal pathogens of Arabidopsis [11]. Arabidopsis treated with 2, 3, 5-triiodobenzoic acid, a chemical inhibitor of PAT, reduces disease development after F. oxysporum infection, but this inhibitor can also inhibit spore germination in vitro [12]. BIG, also called LPR1, is required for PAT [50]. BIG responds to F. oxysporum infection [12], and a resistance phenotype was observed in the big mutant [51]. In our study, LPR1 was induced in both NIL2 and NIL3 (Fig. 7). PAT is controlled by the location of auxin influx and efflux carriers to the plasma membrane. Auxin efflux is regulated by PIN proteins together with the multidrug resistance/phosphoglycoprotein (MDR/PGP) ATP binding cassette transporters [52]. In our study, PIN8, PIN9, PIN10a, and ATP binding cassette subfamily B4 (ABCB4) genes were induced in NIL2 and NIL3 (Fig. 7). It has been reported that F. oxysporum inoculation increases the expression of genes involved in flavonoids. Flavonoids are the endogenous inhibitors of PAT, and a tt4 mutant with a mutation in the CHS is more susceptible to F. oxysporum [12, 13]. HCT silencing lines has a severe growth inhibition owing to the inhibition of auxin transport and increased flavonoid accumulation [53]. In our study, several genes involved in flavonoid synthesis were differentially expressed between NIL2 and NIL3. One LAR, homolog of BAN in Arabidopsis, which encoding a NAD(P)-binding Rossmann-fold superfamily protein negatively regulates flavonoid biosynthesis, was more abundant in NIL3 at 0 and 6 hpi. Genes playing important roles in PAT were also more abundantly expressed in NIL3 compared with NIL2, including AVP1, LAR, HCT (Fig. 8b). All these results suggest a role for PAT in the susceptibility of plants to F. graminearum infection, and disruptions of PAT would undoubtedly result in altered resistance of maize to F. graminearum. The qRfg2 may increase maize resistance to F. graminearum by inhibiting PAT.
Conclusions
The qRfg1 increases maize resistance to F. graminearum through both the constitutive and induced high expression of defense-related genes, and after F. graminearum inoculation it can elegantly fine-tune the metabolic processes between defense and growth. The qRfg2 mediated resistance via the differential expression or induction of genes involved in auxin signaling and PAT, implying again the linking of growth to resistance. The findings of this study will undoubtedly facilitate the future analyses of both the resistance mechanism and resistance breeding for maize stalk rot.
Methods
Plant materials and growth conditions
To evaluate the phenotype at plant maturity, seeds of three NILs were sown at the experimental farm with regular watering and fertilizer application. For the seedling-stage test, germination was conducted as described by Ye et al.[21]. Germination was carried out at 27 ± 1 °C with 16 h of light and 8 h of darkness and watered daily (200 μl of ddH2O) until roots were 6–8 cm long.
Inoculation with F. graminearum and disease assay
For the maturity-stage test, the inoculation of maize roots with F. graminearum and disease assess at 1 month post-inoculation were carried out as described by Yang et al. [15]. For the seedling-stage test, the macrospore suspension used for inoculation was prepared using the liquid Mung bean medium as described by Buerstmayr et al. [54]. The inoculation of seedlings and disease assay was carried out as described by Ye et al. [21]. Disease was evaluated 2 days after inoculation, and resistance percentage was calculated as a measure of host resistance.
Sample preparation, RNA isolation and real-time qRT-PCR
After inoculation, young maize seedling roots were sampled at 0 (control: dipped into the spore suspension and then sampled immediately), 6, and 18 hpi. Each sample consisted of 10 roots pooled together with two biological replicates in parallel. All the samples were immediately frozen in liquid nitrogen and stored at −70 °C. Total RNA from roots was extracted using TRIzol reagent (Invitrogen). The concentration and quality of total RNA were determined with a Nanodrop spectrophotometer and 1 % agarose gel electrophoresis. For real-time qRT-PCR, cDNA was synthesized using the M-MLV First Strand kit (Invitrogen). DEGs were validated with a Roter-Gene™ 6000 (Corbett Research, Sydney, Australia) using SYBR Green II (Takara). Expression levels of genes in samples were normalized using endogenous maize GAPDH, the relative expression levels were calculated using the 2−ΔΔCt method. Primer sequences used in this step were designed using Primer 3.0 and listed in Additional file 6: Table S4.
Illumina sequencing and data analysis
Totally 9 samples with two replicates were sequenced using the Illumina HiSeq 2000 platform. All the clean reads in each sample were mapped to the maize B73 genome using Tophat 2.0.7 [7]. Default settings were used during mapping, and only the unique hits were kept for further analysis. FPKM was used to estimate transcript expression levels in all samples. DEseq was applied to detect DEGs between each chosen sample pairs [42]. Significantly differentially expressed transcripts were identified using a cutoff P-value/FDR < 0.05 and foldchange ≥2. Genes were annotated according to the maize genome; for genes without an unequivocal annotation in maize, the Arabidopsis description was chosen using the blast E-value of 10−10. Those identified DEGs were subjected to GO enrichment analyses as described [55]. For GO analysis, the GO descriptions for each gene in maize and Arabidopsis were edited by removing the duplicate GO term if the gene had the same GO annotation in maize and Arabidopsis, but all GO terms were retained if the gene had different GO annotations.
Acknowledgements
The study was financially supported by 948 project of the Ministry of Agriculture of The People’s Republic of China (2011-G15). We thank Prof. Han Zhao and Yuanda Lv from Jiangsu Academy of Agricultural Sciences for their help with data analysis. We thank Novelbio Co.,Ltd for data analysis.
Funding
The study was financially supported by 948 project of the Ministry of Agriculture of The People’s Republic of China (2011-G15).
Availability of data and materials
The data sets were deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) under accession number PRJNA308408.
Authors’ contributions
YL and YG contributed equally to this research. YL wrote the manuscript and analyzed data. MX and YG participated in the design of the work and contributed to the writing of the manuscript. YL and CM conducted the experiments. DZ, CW, YG, and QY contributed the plant materials. All authors have read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Additional files
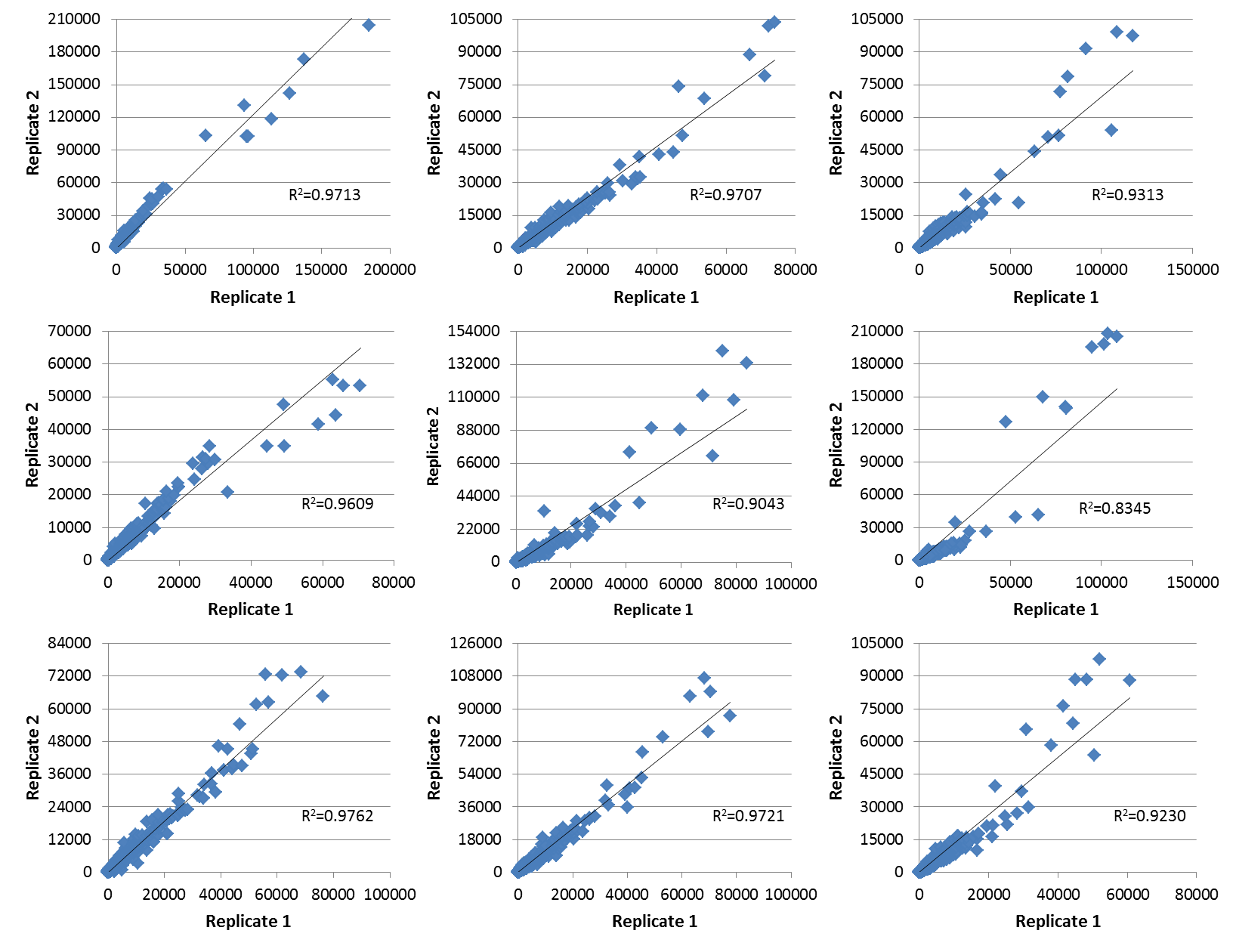{kind=link}
Correlation tests for the replicates. The x and y axis means the reads number in each replicates. R 2 is the square of Pearson’s correlation coefficient. (PNG 96 kb)
GO classification of genes commonly induced in the three NILs after inoculation with F. graminearum. Genes were annotated in three main categories: biological process (bp), cellular component (cc), and molecular function (mf). All GO terms shown were significant at FDR < 0.05. (XLSX 32 kb)
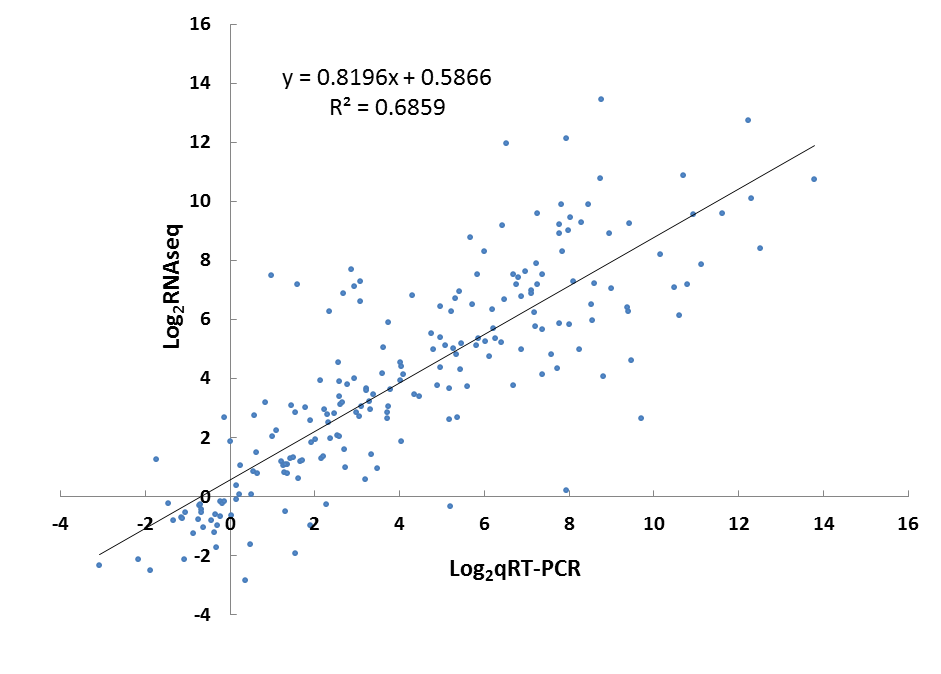{kind=link}
Validation of the RNAseq results via real-time qRT-PCR. qRT-PCR was performed for 19 genes commonly induced in three NILs after inoculation with F. graminearum. The log2 transformed qRT-PCR expression data are plotted against log2 transformed RNAseq data and fit to a linear regression. (PNG 18 kb)
GO classification of genes exclusively upregulated in NIL1 compared with NIL3 after inoculation with F. graminearum. Genes were annotated in three main categories: biological process (bp), cellular component (cc), and molecular function (mf). All GO terms shown were significant at FDR < 0.05. (XLSX 13 kb)
GO classification of genes exclusively downregulated in NIL1 compared with NIL3 after inoculation with F. graminearum. Genes were annotated in three main categories: biological process (bp), cellular component (cc), and molecular function (mf). All GO terms shown were significant at P ≤ 0.01. (XLSX 14 kb)
Primers used for qRT-PCR to validate the selected DEGs in RNAseq. (XLSX 9 kb)
Footnotes
Yongjie Liu and Yanling Guo are co-first authors.
Contributor Information
Yongjie Liu, Email: liu_yongj@126.com.
Yanling Guo, Email: yanglingguo1981@163.com.
Chuanyu Ma, Email: machuanyu071@126.com.
Dongfeng Zhang, Email: zhangdongfeng@cau.edu.cn.
Chao Wang, Email: wangchao19871001@163.com.
Qin Yang, Email: yq_smile_745@163.com.
Mingliang Xu, Email: mxu@cau.edu.cn.
References
- 1.Dodds PN, Rathjen JP. Plant immunity: towards an integrated view of plant-pathogen interactions. Nat Rev Genet. 2010;11:539–48. doi: 10.1038/nrg2812. [DOI] [PubMed] [Google Scholar]
- 2.Jones JD, Dangl JL. The plant immune system. Nature. 2006;444:323–9. doi: 10.1038/nature05286. [DOI] [PubMed] [Google Scholar]
- 3.Nürnberger T, Brunner F, Kemmerling B, Piater L. Innate immunity in plants and animals: striking similarities and obvious differences. Immunol Rev. 2004;198:249–66. doi: 10.1111/j.0105-2896.2004.0119.x. [DOI] [PubMed] [Google Scholar]
- 4.Pieterse CM, Van der Does D, Zamioudis C, Leon-Reyes A, Van Wees SC. Hormonal modulation of plant immunity. Annu Rev Cell Dev Biol. 2012;28:489–521. doi: 10.1146/annurev-cellbio-092910-154055. [DOI] [PubMed] [Google Scholar]
- 5.Loake G, Grant M. Salicylic acid in plant defence-the players and protagonists. Curr Opin Plant Biol. 2007;10(5):466–72. doi: 10.1016/j.pbi.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 6.Robert-Seilaniantz A, Grant M, Jones JD. Hormone crosstalk in plant disease and defense: more than just jasmonate-salicylate antagonism. Ann Rev Phytopathol. 2011;49:317–43. doi: 10.1146/annurev-phyto-073009-114447. [DOI] [PubMed] [Google Scholar]
- 7.Gimenez-Ibanez S, Solano R. Nuclear jasmonate and salicylate signaling and crosstalk in defense against pathogens. Front Plant Sci. 2013;4:72. doi: 10.3389/fpls.2013.00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grant M, Lamb C. Systemic immunity. Curr Opin Plant Biol. 2006;9(4):414–20. doi: 10.1016/j.pbi.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 9.Bari R, Jones JD. Role of plant hormones in plant defence responses. Plant Mol Biol. 2009;69:473–88. doi: 10.1007/s11103-008-9435-0. [DOI] [PubMed] [Google Scholar]
- 10.Denancé N, Sánchez-Vallet A, Goffner D, Molina A. Disease resistance or growth: the role of plant hormones in balancing immune responses and fitness costs. Front Plant Sci. 2013;4:155. doi: 10.3389/fpls.2013.00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Llorente F, Muskett P, Sanchez-Vallet A, Lopez G, Ramos B, Sanchez-Rodriguez C, et al. Repression of the auxin response pathway increases Arabidopsis susceptibility to necrotrophic fungi. Mol Plant. 2008;1:496–509. doi: 10.1093/mp/ssn025. [DOI] [PubMed] [Google Scholar]
- 12.Kidd BN, Kadoo NY, Dombrecht B, Tekeoglu M, Gardiner DM, Thatcher LF, et al. Auxin signaling and transport promote susceptibility to the root-infecting fungal pathogen Fusarium oxysporum in Arabidopsis. Mol Plant Microbe Interact. 2011;24:733–48. doi: 10.1094/MPMI-08-10-0194. [DOI] [PubMed] [Google Scholar]
- 13.Peer WA, Murphy AS. Flavonoids and auxin transport: modulators or regulators? Trends Plant Sci. 2007;12:556–63. doi: 10.1016/j.tplants.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 14.Pè ME, Gianfranceschi L, Taramino G, Tarchini R, Angelini P, Dani M, et al. Mapping quantitative trait loci (QTLs) for resistance to Gibberella zeae infection in maize. Mol Gen Genet. 1993;241:11–6. doi: 10.1007/BF00280195. [DOI] [PubMed] [Google Scholar]
- 15.Yang Q, Yin GM, Guo YL, Zhang DF, Chen SJ, Xu ML. A major QTL for resistance to Gibberella stalk rot in maize. Theor Appl Genet. 2010;121:673–87. doi: 10.1007/s00122-010-1339-0. [DOI] [PubMed] [Google Scholar]
- 16.Zhang DF, Liu YJ, Guo YL, Yang Q, Ye JR, Chen SJ, et al. Fine-mapping of qRfg2, a QTL for resistance to Gibberella stalk rot in maize. Theor Appl Genet. 2012;124:585–96. doi: 10.1007/s00122-011-1731-4. [DOI] [PubMed] [Google Scholar]
- 17.Yang DE, Zhang CL, Zhang DS, Jin DM, Weng ML, Chen SJ, et al. Genetic analysis and molecular mapping of maize (Zea mays L.) stalk rot resistant gene Rfg1. Theor Appl Genet. 2004;108:706–11. doi: 10.1007/s00122-003-1466-y. [DOI] [PubMed] [Google Scholar]
- 18.Yang DE, Jin DM, Wang B, Zhang DS, Nguyen H-T, Zhang CL, et al. Characterization and mapping of Rpi1, a gene that confers dominant resistance to stalk rot in maize. Mol Genet Genomics. 2005;274:229–34. doi: 10.1007/s00438-005-0016-5. [DOI] [PubMed] [Google Scholar]
- 19.Jung M, Weldekidan T, Schaff D, Paterson A, Tingey S, Hawk J. Generation-means analysis and quantitative trait locus mapping of anthracnose stalk rot genes in maize. Theor Appl Genet. 1994;89:413–8. doi: 10.1007/BF00225375. [DOI] [PubMed] [Google Scholar]
- 20.Song FJ, Xiao MG, Duan CX, Li HJ, Zhu ZD, Liu BT, et al. Two genes conferring resistance to Pythium stalk rot in maize inbred line Qi319. Mol Genet Genomics. 2015;290:1–7. [DOI] [PubMed]
- 21.Ye JR, Guo YL, Zhang DF, Zhang N, Wang C, Xu ML. Cytological and molecular characterization of quantitative trait locus qRfg1, which confers resistance to Gibberella stalk rot in maize. Mol Plant Microbe Interact. 2013;26:1417–28. doi: 10.1094/MPMI-06-13-0161-R. [DOI] [PubMed] [Google Scholar]
- 22.Robertson-Hoyt LA, Jines MP, Balint-Kurti PJ, Kleinschmidt CE, White DG, Payne GA, et al. QTL mapping for Fusarium ear rot and Fumonisin contamination resistance in two maize populations. Crop Sci. 2006;46:1734. doi: 10.2135/cropsci2005.12-0450. [DOI] [Google Scholar]
- 23.Robertson LA, Kleinschmidt CE, White DG, Payne GA, Maragos CM, Holland JB. Heritabilities and correlations of Fusarium ear rot resistance and Fumonisin contamination resistance in two maize populations. Crop Sci. 2006;46:353. doi: 10.2135/cropsci2005.0139. [DOI] [Google Scholar]
- 24.Stephens AE, Gardiner DM, White RG, Munn AL, Manners JM. Phases of infection and gene expression of Fusarium graminearum during crown rot disease of wheat. Mol Plant Microbe Interact. 2008;21:1571–81. doi: 10.1094/MPMI-21-12-1571. [DOI] [PubMed] [Google Scholar]
- 25.Goswami RS, Kistler HC. Heading for disaster: Fusarium graminearum on cereal crops. Mol Plant Pathol. 2004;5:515–25. doi: 10.1111/j.1364-3703.2004.00252.x. [DOI] [PubMed] [Google Scholar]
- 26.Marasas WFO, Van Rensburg SJ, Mirocha CJ. Incidence of Fusarium species and the mycotoxins, deoxynivalenol and zearalenone, in corn produced in esophageal cancer areas in Transkei. J Agri Food Chem. 1979;27:1108–12. doi: 10.1021/jf60225a013. [DOI] [PubMed] [Google Scholar]
- 27.Marasas WFO, Nelson PE, Toussoun TA. Toxigenic Fusarium species. Identity and mycotoxicology. Toxigenic Species Identity & Mycotoxicology. 1984. [DOI] [PubMed]
- 28.Kazan K, Gardiner DM, Manners JM. On the trail of a cereal killer: recent advances in Fusarium graminearum pathogenomics and host resistance. Mol Plant Pathol. 2012;13:399–413. doi: 10.1111/j.1364-3703.2011.00762.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pestka JJ. Deoxynivalenol-induced proinflammatory gene expression: mechanisms and pathological sequelae. Toxins. 2010;2:1300–17. doi: 10.3390/toxins2061300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harris L, Desjardins AE, Plattner R, Nicholson P, Butler G, Young J, et al. Possible role of trichothecene mycotoxins in virulence of Fusarium graminearum on maize. Plant Dis. 1999;83:954–60. doi: 10.1094/PDIS.1999.83.10.954. [DOI] [PubMed] [Google Scholar]
- 31.Lanubile A, Ferrarini A, Maschietto V, Delledonne M, Marocco A, Bellin D. Functional genomic analysis of constitutive and inducible defense responses to Fusarium verticillioides infection in maize genotypes with contrasting ear rot resistance. BMC Genomics. 2014;15:710. doi: 10.1186/1471-2164-15-710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reinprecht Y, Wu XG, Yan S, Labey L, Dasilva E, Martin J, et al. A microarray-based approach for identifying genes for resistance to Fusarium graminearum in maize (Zea Mays L.) Cereal Res Commun. 2008;36(Suppl 6):253–9. doi: 10.1556/CRC.36.2008.Suppl.B.23. [DOI] [Google Scholar]
- 33.Campos-Bermudez VA, Fauguel CM, Tronconi MA, Casati P, Presello DA, Andreo CS. Transcriptional and metabolic changes associated to the infection by Fusarium verticillioides in maize inbreds with contrasting ear rot resistance. PLoS One. 2013;8:e61580. doi: 10.1371/journal.pone.0061580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Steiner B, Kurz H, Lemmens M, Buerstmayr H. Differential gene expression of related wheat lines with contrasting levels of head blight resistance after Fusarium graminearum inoculation. Theor Appl Genet. 2009;118:753–64. doi: 10.1007/s00122-008-0935-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Golkari S, Gilbert J, Ban T, Procunier JD. QTL-specific microarray gene expression analysis of wheat resistance to Fusarium head blight in Sumai-3 and two susceptible NILs. Genome. 2009;52:409–18. doi: 10.1139/G09-018. [DOI] [PubMed] [Google Scholar]
- 36.Jia HY, Cho S, Muehlbauer GJ. Transcriptome analysis of a wheat near-isogenic line pair carrying Fusarium head blight-resistant and-susceptible alleles. Mol Plant Microbe Interact. 2009;22:1366–78. doi: 10.1094/MPMI-22-11-1366. [DOI] [PubMed] [Google Scholar]
- 37.Schweiger W, Steiner B, Ametz C, Siegwart G, Wiesenberger G, Berthiller F, et al. Transcriptomic characterization of two major Fusarium resistance quantitative trait loci (QTLs), Fhb1 and Qfhs. ifa‐5A, identifies novel candidate genes. Mol Plant Pathol. 2013;14:772–85. doi: 10.1111/mpp.12048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boddu J, Cho S, Kruger WM, Muehlbauer GJ. Transcriptome analysis of the barley-Fusarium graminearum interaction. Mol Plant Microbe Interact. 2006;19:407–17. doi: 10.1094/MPMI-19-0407. [DOI] [PubMed] [Google Scholar]
- 39.Gardiner SA, Boddu J, Berthiller F, Hametner C, Stupar RM, Adam G, et al. Transcriptome analysis of the barley-deoxynivalenol interaction: evidence for a role of glutathione in deoxynivalenol detoxification. Mol Plant Microbe Interact. 2010;23:962–76. doi: 10.1094/MPMI-23-7-0962. [DOI] [PubMed] [Google Scholar]
- 40.Boddu J, Cho S, Muehlbauer GJ. Transcriptome analysis of trichothecene-induced gene expression in barley. Mol Plant Microbe Interact. 2007;20:1364–75. doi: 10.1094/MPMI-20-11-1364. [DOI] [PubMed] [Google Scholar]
- 41.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protocols. 2012;7:562–78. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xiao J, Jin XH, Jia XP, Wang HY, Cao AZ, Zhao WP, et al. Transcriptome-based discovery of pathways and genes related to resistance against Fusarium head blight in wheat landrace Wangshuibai. BMC Genomics. 2013;14:197. doi: 10.1186/1471-2164-14-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kruger WM, Pritsch C, Chao S, Muehlbauer GJ. Functional and comparative bioinformatic analysis of expressed genes from wheat spikes infected with Fusarium graminearum. Mol Plant Microbe Interact. 2002;15:445–55. doi: 10.1094/MPMI.2002.15.5.445. [DOI] [PubMed] [Google Scholar]
- 45.Axtell MJ, Chisholm ST, Dahlbeck D, Staskawicz BJ. Genetic and molecular evidence that the Pseudomonas syringae type III effector protein AvrRpt2 is a cysteine protease. Mol Microbiol. 2003;49:1537–46. doi: 10.1046/j.1365-2958.2003.03666.x. [DOI] [PubMed] [Google Scholar]
- 46.Dong X. SA, JA, ethylene, and disease resistance in plants. Curr Opin Plant Biol. 1998;1:316–23. doi: 10.1016/1369-5266(88)80053-0. [DOI] [PubMed] [Google Scholar]
- 47.Skirycz A, Reichelt M, Burow M, Birkemeyer C, Rolcik J, Kopka J, et al. DOF transcription factor AtDof1. 1 (OBP2) is part of a regulatory network controlling glucosinolate biosynthesis in Arabidopsis. Plant J. 2006;47:10–24. doi: 10.1111/j.1365-313X.2006.02767.x. [DOI] [PubMed] [Google Scholar]
- 48.Koes R, Verweij W, Quattrocchio F. Flavonoids: a colorful model for the regulation and evolution of biochemical pathways. Trends Plant Sci. 2005;10:236–42. doi: 10.1016/j.tplants.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 49.Weng XY, Wang L, Wang J, Hu Y, Du H, Xu CG, et al. Grain number, plant height, and heading Date7 is a central regulator of growth, development, and stress response. Plant Physiol. 2014;164:735–47. doi: 10.1104/pp.113.231308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gil P, Dewey E, Friml J, Zhao Y, Snowden KC, Putterill J, et al. BIG: a calossin-like protein required for polar auxin transport in Arabidopsis. Genes Dev. 2001;15:1985–97. doi: 10.1101/gad.905201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kazan K, Manners JM. Jasmonate signaling: toward an integrated view. Plant Physiol. 2008;146:1459–68. doi: 10.1104/pp.107.115717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Titapiwatanakun B, Murphy AS. Post-transcriptional regulation of auxin transport proteins: cellular trafficking, protein phosphorylation, protein maturation, ubiquitination, and membrane composition. J Exp Bot. 2009;60:1093–107. doi: 10.1093/jxb/ern240. [DOI] [PubMed] [Google Scholar]
- 53.Besseau S, Hoffmann L, Geoffroy P, Lapierre C, Pollet B, Legrand M. Flavonoid accumulation in Arabidopsis repressed in lignin synthesis affects auxin transport and plant growth. Plant Cell. 2007;19:148–62. doi: 10.1105/tpc.106.044495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Buerstmayr H, Steiner B, Lemmens M, Ruckenbauer P. Resistance to Fusarium head blight in winter wheat: heritability and trait associations. Crop Sci. 2000;40:1012–8. doi: 10.2135/cropsci2000.4041012x. [DOI] [Google Scholar]
- 55.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene Ontology: tool for the unification of biology. Nat Genet. 2000;25:25–9. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data sets were deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) under accession number PRJNA308408.