
Fig. 3. Theoretical calculation and proposed mechanism of IRR.
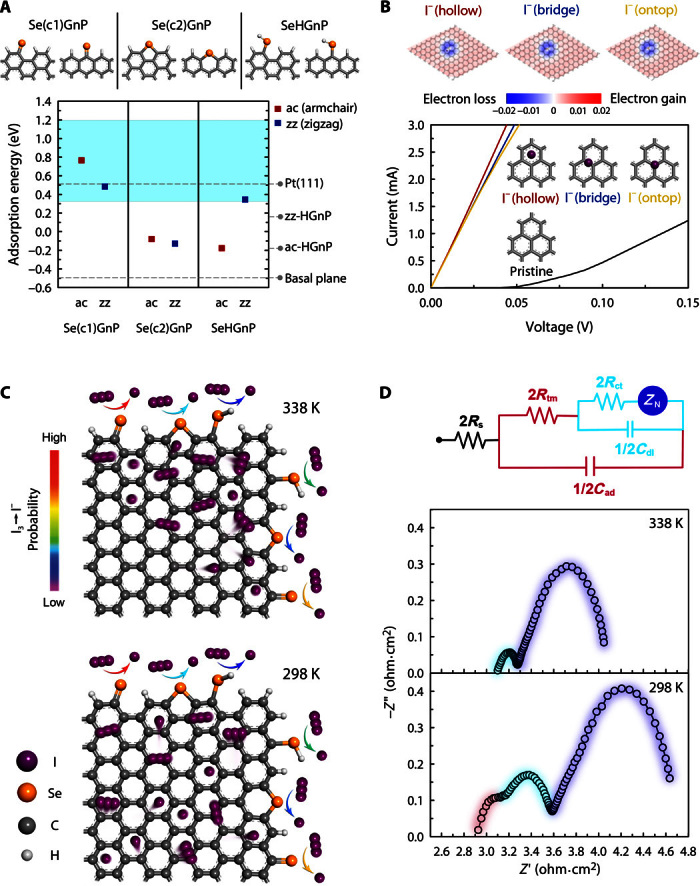
(A) For the representative single-coordinated, double-coordinated, and hydrogenated Se [Se(c1), Se(c2), and SeH, respectively]–doped armchair (ac) and zigzag (zz) graphene edges (top panel), the adsorption energies of the I atom explicitly solvated by acetonitrile molecules were evaluated and compared with the undoped edge and basal plane cases (bottom panel). The Pt(111) value of 0.52 eV has been taken from the study of Li et al. (24). In the bottom panel, the shaded region indicates the IRR activity criterion. (B) For various I−- and I3−-adsorbed graphene basal plane models (top panel and figs. S27 and S28), the current-voltage (I-V) curves were calculated and compared with those from pristine graphene (bottom panel). In the top panel, Mulliken charge populations were coded into atomic structures. (C) IRR mimetic diagram on the SeGnP surface. (D) Nyquist plots of SeGnP-CEs and their EC at room temperature.