

Abstract
Glucuronic acid (GlcAp) and/or methylglucuronic acid (MeGlcAp) decorate the major forms of xylan in hardwood and coniferous softwoods as well as many cereal grains. Accordingly, the complete utilization of glucuronoxylans or conversion to sugar precursors requires the action of main chain xylanases as well as α-glucuronidases that release the α- (1→2)-linked (Me)GlcAp side groups. Herein, a family GH115 enzymefrom the marine bacterium Saccharophagus degradans 2-40T, SdeAgu115A, demonstrated activity toward glucuronoxylan and oligomers thereof with preference toward MeGlcAp linked to internal xylopyranosyl residues. Unique biochemical characteristics of NaCl activation were also observed. The crystal structure of SdeAgu115A revealed a five-domain architecture, with an additional insertion C+ domain that had significant impact on the domain arrangement of SdeAgu115A monomer and its dimerization. The participation of domain C+ in substrate binding was supported by reduced substrate inhibition upon introducing W773A, W689A, and F696A substitutions within this domain. In addition to Asp-335, the catalytic essentiality of Glu-216 was revealed by site-specific mutagenesis. A primary sequence analysis suggested that the SdeAgu115A architecture is shared by more than half of GH115 members, thus defining a distinct archetype for GH115 enzymes.
Keywords: carbohydrate, crystal structure, enzyme catalysis, enzyme structure, protein domain, GH115 α-glucuronidase, carbohydrate degradation, catalytic apparatus, enzyme domain composition
Introduction
The xylan backbone consists of β-(1→4)-linked d-xylopyranosyl (Xylp)4 residues, which, depending on the source, can be partially substituted at O-2 positions and/or O-3 positions with α-l-arabinofuranosyl (Araf) and acetyl substituents and at O-2 positions with α-d-glucuronic acid (also called glucopyranosyluronic acid, GlcAp) or 4-O-methyl GlcAp (MeGlcAp) (1–3). Whereas glucuronoxylan is the predominant form of xylan in dicots, such as hardwoods, xylan from conifers (softwoods) contains both (Me)GlcAp and Araf substituents (4). Xylan from monocots (e.g. cereals and grasses) resembles softwood xylan, although with varying (Me)GlcAp and Araf contents. Consistent with the structural diversity of xylans, their complete enzymatic hydrolysis typically requires the concerted action of several enzymes, including endoxylanases and β-xylosidases, which target glycosidic linkages within the xylan backbone, along with α-glucuronidases, α-l-arabinofuranosidases, acetyl xylan esterases, feruloyl esterases, and glucuronoyl esterases, which target the branching substituents present in different xylan types (5–8). In addition to enabling the complete conversion of xylan to monosaccharides for digestion or utilization as biochemical precursors, debranching enzymes can be used to fine tune xylan solubility, rheology, and adherence to other biopolymers (9). In this way, α-glucuronidases may also be useful tools in the development of xylan-derived polymers with desired characteristics (10, 11).
To date, α-glucuronidases (EC 3.2.1.131/139) have been classified into three glycoside hydrolase (GH) families, namely GH4, GH67, and GH115, in the CAZy database (12). Whereas two family GH4 α-glucuronidases from Thermotoga maritime (TM0434 and TM0752) hydrolyze p-nitrophenyl-α-d-glucuronopyranoside (pNP-GlcAp), both lack activity toward 4-O-methyl-d-glucuronoxylan (13, 14); TM0752 also lacks activity toward aldouronic acids (13), whereas TM0434 activity toward aldouronic acids has not been tested. By contrast, α-glucuronidases from families GH67 and GH115 target the α-(1→2)-linkage between (Me)GlcAp and Xylp of xylan and/or corresponding oligosaccharides (15–17). However, GH67 activity is restricted to (Me)GlcAp substituents at the non-reducing end of the substrate, presumably due to the rather deep active site pocket adopted by this enzyme family (18–21). Most of the characterized α-glucuronidases from family GH115, on the other hand, are able to remove (Me)GlcAp from both terminal and internal Xylp units in xylan and xylooligomers (16, 17, 22, 23) and so may be particularly beneficial to efforts aimed at harnessing xylan as a polymer. Moreover, the recent characterization of BtGH115 from Bacteroides thetaiotaomicron (24) supports earlier predictions that GH115 is a polyspecific enzyme family (23), given the action of BtGH115 on terminal 4-O-methyl-glucuronic acid residues from internal positions of arabinogalactan but not glucuronoxylan (24).
At present, more than 300 GH115 sequences are recorded in the CAZy database; however, only five representatives have been characterized to date, including the first one from Schizophyllum commune (ScAgu115A) (16, 22) as well as PsAgu115A from Pichia stipitis (now Scheffersomyces stipitis) (17), SpAgu115A from Streptomyces pristinaespiralis (25), BoAgu115A from Bacteroides ovatus (23) and BtGH115A from B. thetaiotaomicron (24). 1H NMR analysis of two GH115 α-glucuronidases from P. stipitis and S. commune confirmed that, like GH67 enzymes, α-glucuronidases from GH115 adopt an inverting catalytic mechanism (26). The first crystal structure of a GH115 enzyme was reported in 2013. In their study, Bolam and co-workers (23) showed that BoAgu115A from B. ovatus forms a tight dimer with each monomer composed of four distinct domains (A–D). The dimerization interface comprises interactions between helical bundles of C domains in each protomer and reciprocally between the C-terminal β-sandwich (domain D) of protomer 1 and the TIM barrel of protomer 2 (domain B′) (23). The BoAgu115A-GlcAp complex structure allowed these authors to localize the enzyme's active site to the deep pocket within the TIM barrel domain (B/B′) of each protomer. Mutagenesis analysis demonstrated the important role of several residues, including Asp-206, Asp-332, or Glu-375, as candidates for catalytic residues and Arg-328 involved in substrate recognition. Notably, the overall structures of the first three domains of BoAgu115A bear distant evolutionary similarity with corresponding domains in GH67 enzymes, and although catalytic residues identified in each of these CAZy families do not directly align, they are all located at the C-terminal end of domain B. More recently, the BtGH115A structure revealed a four-domain architecture similar to that of BoAgu115A; however, the position of C-terminal domain D was altered and placed atop of domain C (24). Although representing the first two molecular images of GH115 family enzymes, the structural insights gained to date cannot be directly applied to more than 50% GH115 members due to a substantial sequence insertion, thus necessitating further studies of corresponding GH115 representatives.
Saccharophagus degradans 2-40T is an aerobic marine bacterium that can degrade several complex polysaccharides, including agar, chitin, alginic acid, carrageenan, cellulose, β-glucan, laminarin, pectin, pullulan, starch, and xylan (27–29). The S. degradans 2-40T genome sequence was reported in 2008 and was found to encode at least 128 carbohydrate active enzymes relevant to polysaccharide hydrolysis (30). In particular, for xylan utilization, the genome sequence revealed several genes encoding GH10 and GH11 endoxylanases as well as genes encoding xylan debranching enzymes, including α-l-arabinofuranosidases from families GH43 and GH51, acetyl xylan esterases from families CE1 to CE3, and α-glucuronidases from families GH67 and GH115 (31).
To further map the diversity of GH115 family enzymes, we undertook a detailed biochemical and structural characterization of the GH115 α-glucuronidase from S. degradans 2-40T (SdeAgu115A), which shares <40% sequence identity with previously characterized GH115 representatives (33% with BoAgu115A, 39% with PsAgu115A, 38% with ScAgu115A, and 23% with BtGH115A). The crystal structure of the SdeAgu115A monomer revealed five distinct domains, in contrast to the four-domain composition of BoAgu115A and BtGH115A (23, 24), resulting from the insertion of an additional 125-amino acid domain between domains C and D, herein referred to as C+. Detailed structural and mutagenesis studies of SdeAgu115A demonstrated the role of domain C+ in domain arrangement, enzyme dimerization, and substrate binding; Glu-216 in SdeAgu115A was also identified as an essential catalytic residue. Finally, biochemical analyses using defined xylo-oligosaccharides as well as multiple xylan sources confirmed SdeAgu115A action toward α-(1→2)-glycosidic linkages between (Me)GlcAp and singly substituted Xylp residues at both internal and terminal positions of the substrate.
Experimental Procedures
Materials
Pfx polymerase was purchased from Invitrogen. The QIAEX II gel extraction kit and QIAquick PCR purification kit were purchased from Qiagen. Phosphorylase b from rabbit muscle, beechwood xylan, and oat spelt xylan were purchased from Sigma-Aldrich; GH11 endoxylanase from Neocallimastix patriciarum and the d-glucuronic acid assay kit were purchased from Megazyme; GH10 xylanase (Shearzyme®) and GH11 xylanase (Pentopan® Mono BG) were obtained from Novozymes; pNP-GlcAp was purchased from TCI. Spruce arabinoglucuronoxylan was prepared as reported previously (32). Genomic DNA of S. degradans 2-40T (ATCC catalog no. 43961) was purchased from American Type Culture Collection. All other chemicals were analytical grade and obtained from Sigma-Aldrich or Fisher.
DNA Manipulation
The gene encoding the mature form of SdeAgu115A (GenBankTM accession number ABD81015) without a signal peptide (aa residues 1–32) was amplified from S. degradans genomic DNA with Pfx DNA polymerase (Invitrogen). The infusion cloning kit from Clontech was used to transfer purified PCR products to the p15Tv-L expression vector (GenBankTM accession number EF456736), generating p15Tv-L_SdeAgu115A. Site-specific mutagenesis was carried out according to a modified QuikChangeTM (Stratagene) method (33). All constructs were verified by DNA sequencing at the Center of Applied Genomics at the SickKids Hospital in Toronto.
GH115 Enzyme Sequence Analysis
Enzyme sequences were retrieved from the CAZy database and the JGI fungal genomes database (November 22, 2014). The sequences were curated by removing members that had sequences shorter than 200 amino acid residues or lacked the catalytic domain B. In total, 303 GH115 α-glucuronidase sequences were collected and then aligned using MUSCLE in Geneious version 6.0.6. Geneious Tree Builder version 6.0.6 was used to create phylogenetic tree with the neighbor-joining method.
Purification of Recombinant SdeAgu115A
Escherichia coli BL21(λDE3) codon plus strain harboring p15Tv-L_SdeAgu115A was propagated at 37 °C in 1 liter of Luria-Bertani medium supplemented with 0.5 m d-sorbitol, 2.5 mm glycine betaine, 34 μg ml−1 chloramphenicol, and 100 μg ml−1 ampicillin. At A600 nm of 0.6, the cultivation temperature was reduced to 15 °C, and recombinant protein expression was induced with 0.5 mm isopropyl β-d-thiogalactopyranoside overnight.
The induced cultures were harvested by centrifugation at 6000 × g for 15 min. Cell pellets (∼4.3 g fresh weight) were suspended in 30 ml of binding buffer (300 mm NaCl, 50 mm HEPES (pH 7.0), 5% glycerol, 5 mm imidazole) and disrupted by sonication. Cell debris was removed by centrifugation (17,500 × g, 20 min), and supernatants were passed through a 0.45-μm filter before being incubated for 45 min with 5.0 ml of pre-equilibrated nickel-nitrilotriacetic acid resin (Qiagen) at 4 °C. Resin was then washed with 300 ml of washing buffer (300 mm NaCl, 50 mm HEPES (pH 7.0), 5% (v/v) glycerol, 50 mm imidazole), and bound protein was eluted with ∼30 ml of elution buffer (300 mm NaCl, 50 mm HEPES (pH 7.0), 5% (v/v) glycerol, 250 mm imidazole) in a stepwise manner. Active fractions were pooled and adjusted to 1.0 m NaCl before being applied to a phenyl-SepharoseTM hydrophobic interaction chromatography column (1 × 18.5 cm) equilibrated with the same buffer (50 mm HEPES (pH 7.0) containing 1.0 m NaCl). Fractions (5 ml) were collected at a flow rate of 1.5 ml min−1 and linear gradient of 1.0 to 0 m NaCl in 50 mm HEPES (pH 7.0) over 5 column volumes. All chromatographic steps were performed using the BIOSHOP Duoflow system (Bio-Rad). Fractions containing SdeAgu115A were exchanged to 25 mm HEPES buffer (pH 7.0) containing 300 mm NaCl using a Bio-Gel P10 column. Amicon stirred ultrafiltration cells (Millipore) equipped with a polyethersulfone membrane (nominal molecular weight 100,000) were then used to concentrate purified protein samples before aliquots were flash-frozen in liquid nitrogen and then stored at −80 °C.
Protein concentrations were determined using the Bradford assay and bovine serum albumin as a standard (34). SDS-PAGE was performed and stained with Coomassie Blue according to established procedures (35).
Production of MeGlcAp-substituted xylooligosaccharides (U4m2XX and XU4m2XX)
Birchwood xylan 1% (w/v) prepared in 300 ml of 50 mm sodium acetate buffer (pH 5.0) was boiled for 5 min and then cooled before the addition of endoxylanases. GH10 xylanase (Shearzyme®) (20 microkatals/g xylan) and GH11 xylanase (Pentopan® Mono BG) (30 microkatals/g xylan) were separately added to generate U4m2XX and XU4m2XX, respectively. Enzymatic hydrolyses were performed at 40 °C for 72 h with a continuous mixing, and the extent of hydrolysis was regularly checked by TLC. The reaction was stopped by 20-min boiling, and the supernatant was collected after a centrifugation of 15,000 × g for 15 min. Acidic glucuronyl xylooligosaccharides (XOS) were isolated using Dowex 1 × 2 anion exchange resin (75 ml; Sigma), which had been regenerated with 10 volumes of degassed 2 m ammonium formate (pH 6.0) and washed with 2 volumes of water in an Äkta chromatography system (GE Healthcare). Unbound neutral XOS were washed with water (1 ml min−1). Bound acidic glucuronyl XOS were eluted with an 800-ml gradient of 0–400 mm ammonium formate (pH 6.0) and concentrated to 4 ml in a rotatory evaporator and further purified using BioGel-P2 size exclusion chromatography (8.9 × 135 cm; Bio-Rad) with Milli-Q water (Millipore) as the eluent. Pure sugar factions were concentrated using a rotary evaporator, and the total carbohydrates were determined by the phenol-sulfuric acid method using xylose as the standard (36). The purity was determined by TLC, high performance anion exchange chromatography with pulsed amperometric detection, and mass spectrometry using purified U4m2XX and XU4m2XX as the standard (37). Mass spectrometric analyses of purified XOS were performed using an Agilent XCT Plus model ion trap mass spectrometer (Agilent Technologies, Waldbronn, Germany) equipped with LC/MSD Trap Software version 5.2, as described by Vuong et al. (38). Hydrolysis products and chromatography fractions were analyzed with TLC on Silica Gel 60 aluminum plates (Merck, Darmstadt, Germany), which were developed in ethyl acetate/acetic acid/isopropyl alcohol/formic acid/water (25:10:5:1:15, v/v/v/v/v). The plate was sprayed with 0.2% orcinol solution in 80% ethanol (v/v) and 10% H2SO4 (v/v) and heated at 100 °C for 2–5 min to detect XOS.
Effect of Temperature, pH, Metal Ions, and NaCl
The thermostability of SdeAgu115A was evaluated by incubating the enzyme (300 μg/ml) at pH 6.5 in 0.2 ml of 200 mm MES for up to 4 h at 25, 35, 40, and 50 °C. Following incubation, residual activity was measured by transferring 6.0 μg of the heat-treated enzyme to a final 0.2-ml reaction mixture comprising 200 mm MES (pH 6.5) and 1.0% (w/v) beechwood xylan. After a 10-min incubation at 40 °C, reaction products were quantified using the Nelson-Somogyi assay for reducing sugars (39). The pH optimum of SdeAgu115A was determined by measuring activity as described above, this time in buffers with a pH range from 4.0 to 9.5. Buffers used were 200 mm CH3CO2Na (pH 4–5.5), 200 mm MES (pH 6.0–8.0), and 200 mm glycine-NaOH (pH 8.5–9.5). To determine pH stability, residual SdeAgu115A activity was measured after a 24-h incubation at 4 °C in buffers with a pH range from 4.0 to 9.5. The effect of metal ions was determined by adding 1.0 mm CaCl2, MgCl2, CoCl2, MnCl2, NiCl2, CdCl2, AgNO3, ZnCl2, CuSO4, or HgCl2 to the activity assay mixture.
To further investigate the impact of NaCl on SdeAgu115A activity, the enzyme was dialyzed against 100 mm MES buffer (pH 6.5) before activity measurements in 100 mm MES buffer (pH 6.5) containing 0–1.5 m NaCl.
Analytical Ultracentrifugation for Oligomeric State Analysis
Sedimentation equilibrium analytical ultracentrifugation was carried out using a Beckman Optima XL-A analytical ultracentrifuge equipped with an An-60 Ti rotor. Protein absorbance was monitored at 280 nm. Data were collected at speeds of 6000, 7000, and 8000 rpm using SdeAgu115A at varying enzyme concentrations (5.9, 3.0, and 1.5 μm) in 20 mm HEPES (pH 7.0, 300 mm NaCl) at 20 °C. Data analysis was performed using the Origin MicroCal XL-A/CL-I Data Analysis Software Package version 4.0. The density of buffer and the partial specific volumes of protein were calculated using SedNterp (40).
CD Spectra
CD was recorded with an Aviv model 202 circular dichroism spectrometer from 200 to 300 nm at 25 °C in a 0.5-cm path length cell. The spectral bandwidth was 1.0 nm, the step size was 1.0 nm, and the averaging time was 1.0 s. The protein sample was in 0.2 m sodium phosphate buffer (pH 7.5) with an adjusted concentration of 1.0 mg/ml.
Affinity Gel Electrophoresis
The binding of SdeAgu115A and its mutants (E216A and D335A) to beechwood xylan was examined by native affinity gel electrophoresis (41) with minor modifications. Briefly, the native polyacrylamide gels contained 6.0% (w/v) acrylamide in 15 mm Tris, 150 mm glycine, 100 mm sodium phosphate buffer (pH 8.8), and 0.05 and 0.1% (w/v) of the test polysaccharide. Approximately 7.0 μg of SdeAgu115A and each mutant were loaded onto the gels and then run at 70 V for 6.0 h. Relative binding affinities were inferred from the migration distance of the wild type and mutant enzymes on gels with and without the test polysaccharides. BSA and phosphorylase b from rabbit muscle (7.0 μg) were used as reference proteins.
Protein Crystallization, Data Collection, and Structure Determination
For crystallographic purposes, the selenomethionine-substituted protein was overexpressed using M9 high yield growth medium (Medicilon Inc.) and purified according to the above-described procedure for recombinant SdeAgu115A.
SdeAgu115A was concentrated to 13.0 mg ml−1 in 10 mm HEPES (pH 7.5) containing 500 mm NaCl. The crystallization screen was performed by mixing 0.5 μl of protein with 0.5 μl of reservoir buffer using a Mosquito crystal (TTPlab) liquid-handling system. The SdeAgu115A crystals were obtained by a sitting drop vapor diffusion method at 23 °C in the presence of 200 mm sodium dihydrogen phosphate, 100 mm sodium acetate (pH 4.6), 200 mm ammonium acetate, and 30% PEG 4000. The crystals were flash-frozen in liquid nitrogen using paratone-N oil as cryoprotectant. Diffraction data for SdeAgu115A crystals were collected at the Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source, using beamline 19-ID with an ADSC Quantum 315R detector according to a previously described experimental set-up (42, 43). All diffraction data were processed using the HKL3000 suite of programs (44). The SdeAgu115A structure was determined by the single-wavelength anomalous diffraction method using phasing, density modification, and initial protein model building as implemented in the HKL3000 software package (45–51). Cycles of manual corrections of the model were carried out in COOT and with REFMAC of CCP4 and finalized with Phenix using the implemented TLS group refinement routine. The final model was refined against all reflections except for 5% randomly selected reflections, which were used for monitoring Rfree. Final refinement statistics are presented in Table 1. Structure figures were prepared using PyMOL v1.3 (Schrödinger LLC, New York).
TABLE 1.
Data and refinement statistics
| Data collection statistics for APS-SBC-19ID | |
| Space group | P212121 |
| Unit cell (Å) | |
| a | 116.4 |
| b | 124.3 |
| c | 180.0 |
| Resolution (Å) | 38.2–1.93 |
| Wavelength (Å) | 0.9794 |
| No. of observed reflections | 204,709 |
| No. of unique reflections | 48,738 |
| Rpima (%) | 4.0 (61.1)b |
| Completeness (%) | 97.7 (96.8) |
| I/δ | 19.1 (1.5) |
| Phasing | |
| Phasing method | SADc |
| Refinement statistics | |
| Rcryst (%) | 13.90 |
| Rfree (%) | 16.94 |
| Protein residues | 1866 |
| Sodium/phosphate/glycerol/acetate molecules | 2/4/2/2 |
| Solvent | 2185 |
| Root mean square deviation from target values | |
| Bond lengths (Å) | 0.011 |
| Bond angles (degrees) | 1.22 |
| Average B factors (Å2) | |
| Protein | 27.8 |
| Ions | 46.0 |
| Solvent | 37.8 |
| PDB code | 4ZMH |
| Ramachandran (%) F/A/Od | 97/3.0/0 |
a Rpim = Σhkl(1/(N − 1)½)Σi|Ii(hkl) − Īhkl|/ΣhklΣiIi(hkl).
b Numbers in parentheses are values for the highest resolution bin.
c Single-wavelength anomalous diffraction.
d As defined by MOLPROBITY (F (favored)/A (allowed)/O (outliers)).
Substrate Preference and Enzyme Kinetics
SdeAgu115A activity on various xylans and xylooligosaccharides was measured using the d-glucuronic acid assay kit from Megazyme. Reactions were performed for 10 min at 40 °C and were initiated by adding 5 μl of enzyme to 20 μl of 1.0% (w/v) substrate in 200 mm MES buffer (pH 6.5). Enzyme reactions were stopped by adding 2.5 μl of 2.0 m HCl, followed by incubation at 25 °C for 10 min. NaOH (2.5 μl of a 2.0 m solution) was used to neutralize reaction mixtures before product detection. In all cases, enzyme doses were optimized to obtain initial rates of reaction. d-(+)-Glucuronic acid (0.01–1.2 mg/ml) was used to generate a standard curve.
The activity on pNP-GlcAp was determined by measuring pNP release. The reaction mixture contained substrate in 0.15 ml of 200 mm MES buffer (pH 6.5). SdeAgu115A doses were adjusted to obtain initial rates of reaction at 40 °C over 10 min. Reactions were terminated by adding 0.15 ml of 4% (w/v) Na2CO3. The amount of pNP formed was measured by absorbance at 405 nm, where pNP (50–400 μm) was used to generate a standard curve.
Kinetic parameters were obtained using 0.1–25 mm pNP-GlcAp, 1.0–38.4 mm U4m2XX, and 1.0–31.9 mm XU4m2XX. Apparent kinetic parameters for beechwood xylan were also obtained at substrate concentrations from 0.5 to 40 mg/ml. In all cases, enzyme dose was adjusted to measure initial rates of reaction at 40 °C over 10 min (0.1, 0.7, 0.7, and 140 μg of SdeAgu115A for beechwood xylan, U4m2XX, XU4m2XX, and pNP-GlcAp, respectively). Kinetic parameters were calculated using GraphPad version 5.0 (GraphPad Software).
Finally, mass spectrometry was used to confirm the specific release of MeGlcAp from beechwood xylan by SdeAgu115A. Briefly, 160 μl of 1% (w/v) beechwood xylan was incubated with 40 μl of SdeAgu115A (54 μg) in water at 22 °C for 12 h. Electrospray ionization mass spectrometric analyses were performed using a Q-Exactive mass spectrometer (Thermo Scientific) in negative mode, and product spectra were compared with d-glucuronic acid.
Results and Discussion
Sequence Analysis and General Biochemical Properties of SdeAgu115A
Multiple-sequence alignment of 303 curated GH115 sequences exhibited two main groups of GH115 enzymes that were distinguished by the presence of an amino acid insertion motif corresponding to amino acid residues 667–792 in SdeAgu115A (Fig. 1A). Based on sequence alignment, nearly all (97%, 94 of 97 sequences) of the fungal GH115s appeared to comprise this conserved sequence insertion motif, whereas roughly 56% (109 of 195 sequences) of bacterial sequences, including previously characterized BoAgu115A and BtGH115A enzymes, lacked the insertion. In addition to these two major forms, 11 GH115 sequences featured additional C-terminal sequences, and six GH115 enzyme sequences corresponded to only the three N-terminal domains identified in BoAgu115A. Phylogenetic analysis of GH115 sequences showed that those with the insertion motif from both fungi and bacteria were grouped together (Fig. 1B), further suggesting a common yet uncharacterized structural arrangement shared by these enzymes. Notably, the N-terminal sequence of SdeAgu115A revealed a secretion motif (signal peptide/signal sequence) in line with the proposed extracellular function of these enzymes.
FIGURE 1.

Sequence analysis of curated GH115 α-glucuronidase sequences. A, the global view of multiple-sequence alignment to display the distribution of the inserted sequence region for domain C+ across 303 curated GH115 α-glucuronidase sequences. The amino acid sequences were retrieved from the CAZy database and the JGI fungal genomes database (November 22, 2014). All sequences were aligned using MUSCLE in Geneious version 6.0.6. B, phylogenetic analysis of GH115 α-glucuronidase sequences. The tree was constructed using Geneious Tree Builder version 6.0.6 and the neighbor-joining method. Enzymes predicted to comprise four domains are indicated by a blue dot; a red prism indicates biochemically characterized enzymes. In general, GH115 α-glucuronidases comprising four or five domains fall into two separate subgroups.
To characterize an α-glucuronidase from the GH115 subgroup containing an insertion motif, SdeAgu115A lacking the native signal peptide sequence (residues 1–32) was expressed in E. coli in fusion with an N-terminal His tag and purified to homogeneity. The weight-average molecular mass of native SdeAgu115A was determined to be 189 ± 3.9 kDa by sedimentation-equilibrium analytical ultracentrifugation (supplemental Fig. S1), indicating that SdeAgu115A forms homodimers under the used experimental conditions (25 mm HEPES buffer (pH 7.0) containing 0.3 m NaCl). By comparison, BoAgu115A was also shown to form a stable dimer in solution (23), whereas BtGH115A appears to remain in monomeric form (24).
The SdeAgu115A activity on beechwood xylan was optimal at pH 6.5, and this recombinant enzyme retained more than 50% activity after 24-h incubation at 4 °C and pH 5.5–9.5 (Fig. 2A). In comparison, the optimal pH of BoAgu115A (23), ScAgu115A (16, 22), and PsAgu115 (17) was reported to be 7.0, 5.8, and 4.4, respectively. SdeAgu115A was stable for more than 3 h at 23 °C, whereas the half-life of SdeAgu115A at 35, 40, and 50 °C was ∼90, 60, and 2 min, respectively (Fig. 2B). Whereas SdeAgu115A lost all activity after incubation at 70 °C for 5 min, PsAgu115A lost 50% activity after 30 min at 60 °C and was stable for at least 3 h at 40 °C (17). ScAgu115A retained full activity after 24 h at 40 °C between pH 6 and 8 and 65% activity after 24 h of incubation at 45 °C (16, 22).
FIGURE 2.

The general properties of SdeAgu115A. A, pH activity profile (bar) and stability (dot) of purified SdeAgu115A; B, thermostability of purified SdeAgu115A; C, effect of metal ions on SdeAgu115A activity. The activity assay solution contained 1.0 mm selected metal ions or 10 mm sodium EDTA. Enzyme assays were performed using 1.0% (w/v) beechwood xylan and 0.03 μg/μl SdeAgu115A. n = 3; Error bars, S.D. The specific activity value corresponding to 100% was 2470 μmol of product/min/μmol of enzyme.
Of the 10 ions tested, the presence of 1.0 mm Ni2+, Cd2+, Ag+, Zn2+, Cu2+, and Hg2+ inhibited SdeAgu115A activity by ∼40–100%, whereas the rest did not have any significant affect on the enzyme's activity (Fig. 2C). Moreover, the addition of 10 mm EDTA did not significantly affect SdeAgu115A activity, indicating that this enzyme does not strictly depend on ion supplementation for its activity under the tested experimental conditions (Fig. 2C). Consistently, no metal ions were observed in the crystal structure of the enzyme. The effect of metal ions on the specific activity of other GH115 enzymes has not been tested. Altogether, these data will inform optimization efforts for future enzyme applications.
Crystal Structure of SdeAgu115A Revealed an Additional Domain
The SdeAgu115A crystal structure was determined using the single-wavelength anomalous diffraction approach and selenomethionine-labeled protein. The final model of SdeAgu115A consisted of two polypeptide chains in the asymmetric unit, spanning residues 40–974 (chain A) and 40–975 (chain B). Additional density was assigned as two sodium and two phosphate ions, one glycerol, and one acetate molecule. The final model exhibited excellent crystallographic and geometric statistics, which are summarized in Table 1.
In contrast to previously structurally characterized GH115 enzymes (23, 24), the crystal structure of the SdeAgu115A monomer revealed a five-consecutive domain architecture with N-terminal domains A (residues 40–205), B (residues 206–494), C (residues 495–670), and the unique C+ domain corresponding to the insertion (residues 671–790) conserved in one of two major groups within the GH115 family (Figs. 1 and 3); in SdeAgu115A, domain C+ is then followed by the C-terminal D domain (residues 791–975).
FIGURE 3.

The overall structure of SdeAgu115A, revealing a novel five-domain arrangement. I, the five-domain structure of SdeAgu115A (each domain is labeled and colored for clarity: blue for domain A, green for domain B, yellow for domain C, orange for domain C+, and red for domain D); II, SdeAgu115A overlaid with four-domain BoAgu115A shown in gray (helices are shown as cylinders); III, SdeAgu115A overlaid with four-domain BtGH115A shown in gray (helices are shown as cylinders); IV, the fold of domain C+ (residues 671–790), displaying a β-barrel mainly consisting of eight anti-parallel β-strands; V, schematic drawing of the domain organization for SdeAgu115A in comparison with BoAgu115A and BtGH115A.
Superimposition of SdeAgu115A with BoAgu115A and BtGH115A demonstrated a root mean square deviation of 2.4 and 2.1 Å, respectively (Fig. 3 and supplemental Table S1). Like BoAgu115A and BtGH115A, domain A of SdeAgu115A adopted a two-layer β-sandwich fold, composed of a central five-stranded β-sheet linked to a smaller two-stranded anti-parallel β-sheet between three α-helices (classified as Pfam 03648), whereas domain B displayed a distorted (β/α)8 TIM barrel fold featuring short third and fourth β-strands (classified as Pfam 15979). Domain C of SdeAgu115A comprised a five-helix bundle with a ∼35-residue long loop disrupted by a short β-strand. In the case of SdeAgu115A, the five-helix bundle is wedged between domains B and C+, whereas the loop and short strands are located at the side of this domain (Fig. 3A). In addition to close structural similarity to corresponding domains in BoAgu115A and BtGH115A, domains A–C in SdeAgu115A also share overall structural similarity with a GH67 α-glucuronidase from Geobacillus stearothermophilus (PDB code 1K9E) (20), GH84 O-GlcNAcase from Clostridium perfringens (PDB code 2VUR) (53), and GH20 lacto-N-biosidase from Bifidobacterium bifidum (PDB code 4H04) (54) (supplemental Table S1).
Also similar to BoAgu115A and BtGH115, the C-terminal domain D of SdeAgu115A adopted a “jelly-roll” β-sandwich fold consisting of two β-sheets, each having five anti-parallel β-strands. However, the presence of the C+ domain in SdeAgu115A significantly impacted the positioning of this domain D compared with BoAgu115A (Fig. 3) (23).
As mentioned above, domain C+ in the SdeAgu115A structure corresponds to the additional insertion sequence, which is present in more than half of GH115 members. The C+ domain in SdeAgu115A displayed a β-barrel composed of eight anti-parallel β-strands (Fig. 3IV). A structural similarity search using the Dali server (55) showed that it is most similar to the S pilus periplasmic chaperone protein SfaE (PDB code 1L4I) (56) and the chaperone protein FimC from E. coli (PDB code 3BWU) (57). Both proteins are involved in bacterial pilus biogenesis. More interestingly, domain C+ is also structurally similar to the Bacon domain (Bacteriodetes-associated carbohydrate-binding often N-terminal, Pfam 13004) in BoGH5A, a GH5 endoxyloglucanase from B. ovatus (supplemental Table S1) (58). Bacon domains have been identified in several other carbohydrate-active enzymes as well as proteases (2704 sequences deposited in the current Pfam database) (59), suggesting that the C+ domain in GH115 enzymes might play a role in carbohydrate binding as well as protein-protein interactions.
Distinct Domain Composition of SdeAgu115A Significantly Impacted Its Domain Arrangement and Dimerization
The overlay of SdeAgu115A (five domains) with BoAgu115A (four domains) clearly showed that insertion of the C+ domain in SdeAgu115A impacted the position of domain D, where domain C+ was placed at the position equivalent to the domain D in BoAgu115A and consequently shifted the domain D position to the top of domain C (Figs. 3 and 4). This change of domain composition and arrangement in SdeAgu115A prevented the enzyme from forming a butterfly-like dimer, as observed in the BoAgu115A structure (23). In BoAgu115A, the dimer is arranged by the crossing of complementary helical bundles of domain C from each promoter and is further strengthened by interactions of domain D of protomer A with domain B′ from protomer B and vice versa (Fig. 4II). In contrast, the interface of the SdeAgu115A dimer was contributed primarily through interactions between anti-parallel β-sheets and their flanking region (residues 665–670) from C domains of each protomer, which formed on the opposite side of domain C relative to domain D (Fig. 4I). Additional contacts between SdeAgu115A subunits were formed between the loops of the C+ domain in protomer A and loop regions from domains C′ and B′ in the protomer B. The overall interface between two SdeAgu115A promoters covers ∼1371 Å2 according to a PDBsum/PISA server estimate (60), which corresponds to only 3.9% of each promoter surface area and involves 42 residues in each subunit. In comparison, the overall interface between BoAgu115A promoters is ∼4000 Å2 and involves 158 residues, accounting for 12.4% of each subunit surface area.
FIGURE 4.
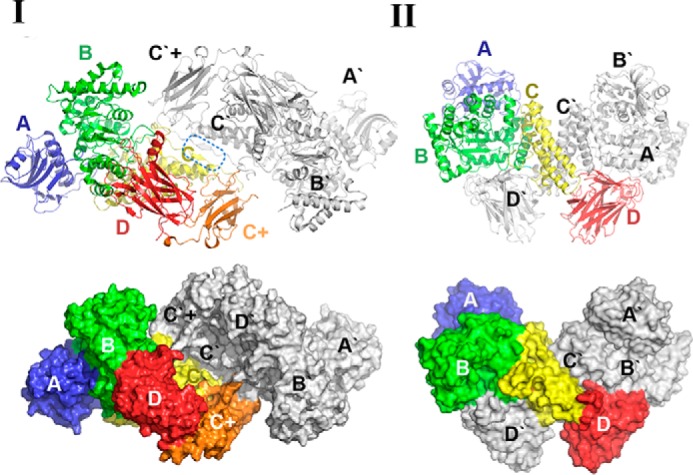
The dimerization of SdeAgu115A in comparison with BoAgu115A. I, dimerization pattern of SdeAgu115A. II, dimerization pattern of BoAgu115A. Side by side comparison between SdeAgu115A and BoAgu115A reveals different dimer formation schemes, which are presented in both schematic and surface forms. Each domain in protomer 1 is labeled and colored for clarity (blue for domain A, green for domain B, yellow for domain C, orange for domain C+, and red for domain D), whereas all domains from protomer 2 are colored in gray.
The recently determined structure of BtGH115A unveiled a second four-domain architecture for the GH115 family (24). This structure closely resembled the domain organization of SdeAgu115A, with its domain D positioned adjacent to domain C and a 15-residue loop to link domains C and D (Fig. 3III); however, BtGH115A was reported to be a monomer (24). Comparison of the BoAgu115A and BtGH115A structures suggests that the placement of domain D atop domain C blocks access to one side of domain C, thereby limiting the interface available for oligomer formation in BtGH115A. In SdeAgu115A, the insertion of domain C+ along with the anti-parallel β-sheets formed by residues 667–670 in domain C appeared to reconstitute the interface for stable dimerization of the enzyme.
Site-specific Mutagenesis Highlighted the Role of Individual Residues in Catalysis
In line with previously structurally characterized GH115 enzymes, the active site of SdeAgu115A localized in the large cavity at the C-terminal end of the β/α TIM barrel of the domain B. To probe the essential residues for catalysis, four conserved acidic amino acid residues from domain B (Asp-215, Glu-216, Asp-335, and Glu-381) and one basic arginine residue (Arg-331), were individually replaced by alanine (Fig. 5). In addition to the D335A variant, no activity was detected for the E216A variant of SdeAgu115A (Table 2). CD spectra of SdeAgu115A and its mutants (D335A and E216A) confirmed similar overall structures of the proteins (Fig. 6A). Affinity gel electrophoresis was also used to evaluate the impact of mutating Asp-335 and Glu-216 on substrate binding. The slightly slower migration and band smearing observed for the wild-type enzyme and D335A and E216A variants in gels containing beechwood xylan indicated weak binding to the substrate (Fig. 6B). Nevertheless, replacing Asp-335 or Glu-216 with alanine appeared to reduce this binding (Fig. 6B), suggesting that an activity-associated conformational change may enhance substrate binding by this enzyme. By contrast, D215A and E381A variants remained active, albeit with 15 and 330 times lower catalytic efficiencies, respectively, on beechwood xylan (Table 2). Similar to the R328A mutant of BoAgu115A, the R331A substitution in SdeAgu115A led to a 26,000-fold decrease in catalytic efficiency, further confirming the importance of this residue for GH115 enzyme activity, probably due to its involvement in positioning and stabilizing the Asp-335 side chain and likely interactions with the carboxylate group in (Me)GlcAp as well.
FIGURE 5.
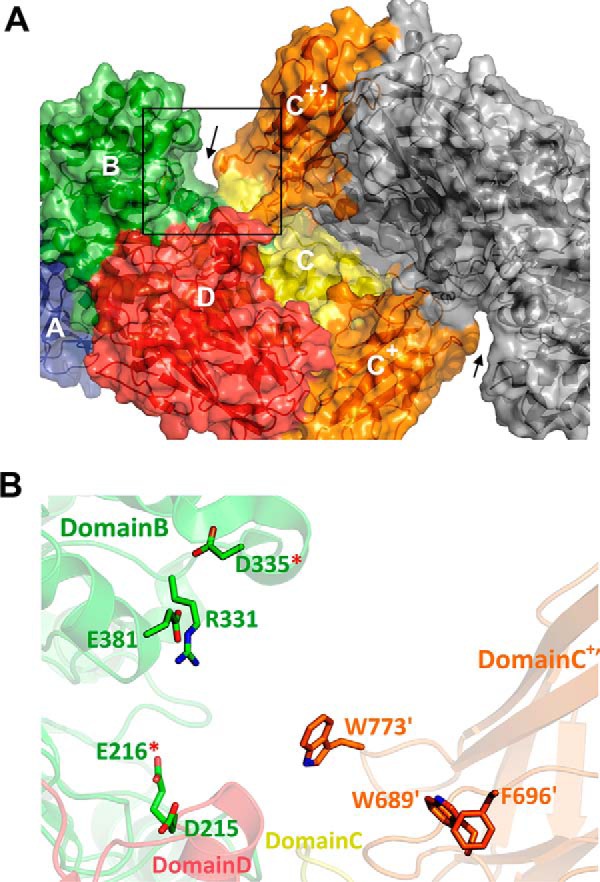
The active site region in SdeAgu115A. A, surface representation of the overall structure of SdeAgu115A dimer, with one of the active site regions highlighted by the rectangular box. B, close-up of the highlighted area that is formed by domains B (green), C (yellow), and D (red) from protomer A and domain C+′ (orange) from protomer B. The side chains of the candidate residues tested for their role in catalysis and substrate binding are displayed as sticks and labeled.
TABLE 2.
Kinetic analysis of SdeAgu115A mutant enzymes on beechwood xylan
Kinetic parameters were calculated based on the conventional substrate inhibition equation except for R331A. Values are mean ± S.D. (n = 3). ND, no detectable activity.
| Enzyme | Residue location | kcat(app) | Km(app) | Ki(app) | kcat(app)/Km(app) |
|---|---|---|---|---|---|
| s−1 | mg/ml | mg/ml | s−1 mg−1 ml | ||
| Wild type | 289 ± 110 | 43 ± 18 | 3 ± 1.5 | 6.7 | |
| D215A | Domain B | 1 ± 0.1 | 2 ± 0.5 | 119 ± 32 | 0.45 |
| E216A | Domain B | ND | ND | ND | ND |
| E381A | Domain B | 0.3 ± 0.15 | 12 ± 8 | 5 ± 3 | 0.02 |
| D335A | Domain B | ND | ND | ND | ND |
| R331A | Domain B | 0.008 ± 0.001 | 31 ± 8 | —a | 2.5 × 10−4 |
| W773A | Domain C+ | 129 ± 32 | 26.4 ± 8.3 | 19.6 ± 7.5 | 4.9 |
| W689A | Domain C+ | 116 ± 20 | 12 ± 3 | 49 ± 20 | 9.3 |
| F696A | Domain C+ | 86 ± 13 | 13 ± 3 | 24 ± 6 | 6.5 |
a Substrate inhibition was not observed for R331A at concentrations of beechwood xylan up to 40 mg/ml; therefore, its data were fitted to the Michaelis-Menten equation.
FIGURE 6.

CD spectra and affinity gel electrophoresis analysis of SdeAgu115A and E216A and D335A variants. A, CD spectra of SdeAgu115A along with E216A and D335A variants displayed similar overall structure. B, affinity gel electrophoresis analysis with beechwood xylan. The protein band in the BSA lane represents the BSA dimer.
Asp-206, Asp-332, or Glu-375 has been proposed to act as the catalytic acid/base in BoAgu115A (23), whereas these roles were assigned to residues Asp-299 and Glu-346 in BtGH115A (24). However, both E375A and D206A variants of BoAgu115A remained active on beechwood xylan, which is consistent with observations for E381A and D215A variants of SdeAgu115A (Table 2). To date, the catalytic essentiality was biochemically confirmed only for the equivalent residue of Asp-335 in SdeAgu115A (Asp-332 in BoAgu115A). Herein, complete activity loss was also observed for E216A in SdeAgu115A, suggesting its essential role in catalysis. Despite the high conservation of the Glu-216 residue among GH115 enzymes, this residue has not previously been reported as a catalytically important residue (23, 24). Whereas Glu-216 and Asp-335 may represent a catalytic acid/base pair in SdeAgu115A, the distance between Oϵ1 of Glu-216 and Oδ2 of Asp-335 is 22.6 Å (Fig. 5B), which is considerably higher than the typical distance of 8–12 Å observed between catalytic residues in other glycosidases that use an inverting mechanism (61). Because these residues are localized to a flexible region, it is possible that their position can change upon substrate binding, as was also proposed for BoAgu115A (23). An overlay of B domains from three available GH115 structures revealed a significant positional shift for Cδ in equivalent residues of Asp-335 in SdeAgu115A (12.6 Å relative to Cδ in Asp-299 of BtGH115, 13.6 Å relative to Asp-332 in BoAgu115A). Comparatively, shifts of equivalent residues to Arg-331 in SdeAgu115A are low, with a distance of 2.5 Å for Cζ relative to Arg-295 in BtGH115A and 3.0 Å relative to Arg-328 in BoAgu115A. These shifts can be explained by movement in the loop formed by residues 330–340 in SdeAgu115A (Fig. 7). Unfortunately, attempts to obtain the SdeAgu115A-GlcAp complex structure to further clarify involvement of specific residues in catalysis and substrate binding were unsuccessful (see “Experimental Procedures” for details).
FIGURE 7.

Superimposition of catalytic sites in domain B of SdeAgu115A, BoAgu115A, and BtGH115A. SdeAgu115A is shown in green, BoAgu115A in orange, and BtGH115A in gray. The identified catalytic essential residues in this study are indicated by red stars.
Domain C+ Participated in Xylan Substrate Binding
Further analysis of domain B in SdeAgu115A revealed a long and deep cleft above and flanking the active site cavity, which could accommodate the backbone of xylan substrates. Notably, this cleft further extended into domain D of the same SdeAgu115A monomer (Fig. 5A). By contrast, the predicted xylan-binding cleft of BoAgu115A was formed by domain B and the C-terminal domain D′ of the neighboring protomer (23).
Two loop regions spanning residues 689–696 and 770–775 in the additional C+′ domain of SdeAgu115A extended the sides of the xylan binding cleft, increasing its depth (Fig. 5A). To assess the potential role of these two loop regions in substrate binding, three aromatic residues (Trp-773, Trp-689, and Phe-696) within these loops were individually mutated to alanine, and their activities were evaluated. Although kcat values of all three mutants (W773A, W689A, and F696A) was decreased by 3–5 times compared with the wild type, these mutants displayed catalytic efficiency on beechwood xylan similar to that of the wild type enzyme due to slightly decreased Km(app) values (Table 2). All three mutants also displayed reduced substrate inhibition with increased Ki(app) values, particularly for W689A, where the Ki(app) was increased by 15 times relative to wild type. The reduction in Km(app) values observed for each of the W773A, W689A, and F696A mutants was at first surprising given the presumed role of these aromatic residues in substrate stacking. However, the relative stability of the α-(1→2) linkage between MeGlcAp and Xylp compared with β-(1→4)-d-Xylp backbone linkages is well recognized (62). It is conceivable, then, that the aromatic residues present in loop regions of the additional domain C+ increase torsional strain within the bound substrate, thus decreasing the stability of the targeted glycosidic linkage.
Implications of SdeAgu115A Structure on Its Substrate Profile
The glucurono-xylooligosaccharides were generated with a yield >10% (w/w); their purity (over 98%) was confirmed by TLC, high performance anion exchange chromatography with pulsed amperometric detection, and electrospray ionization-MS. The activity (kcat) of SdeAgu115A on XU4m2XX and U4m2XX was 173.3 ± 3.0 and 65.7 ± 3.0 s−1, respectively (Fig. 8), indicating that the enzyme was active on both terminal and internal MeGlcAp substitutions. Furthermore, SdeAgu115A revealed a catalytic efficiency (kcat/Km) of 16.6 s−1 mm−1 for XU4m2XX, which is 5.5 times higher than that of U4m2XX (3.0 s−1 mm−1) (Fig. 8). The comparatively high catalytic efficiency measured using XU4m2XX was explained by both a 2.6-fold increase in kcat and a 2.1-fold decrease in Km compared with the value obtained using U4m2XX. A similar pattern was reported for BoAgu115A (23) and suggests that, as in BoAgu115A, at least one subsite (+2NR) beyond the +1 subsite of SdeAgu115A participates in xylan backbone binding.
FIGURE 8.

Kinetic parameters of SdeAgu115A on selected substrates: XU4m2XX (A), U4m2XX (B), beechwood xylan (C), and pNP-GlcAp (D). Data corresponding to A, B, and D were fitted to the Michaelis-Menten equation. Activity data collected using beechwood xylan were fitted to a conventional substrate inhibition equation. The normalized concentration of the MeGlcAp in the glucuronoxylan (mm) is presented as km(app). Error bars, S.D.
Among the xylan substrates, SdeAgu115A showed the highest activity toward beechwood glucuronoxylan, followed by spruce arabinoglucuronoxylan and oat spelt xylan (Table 3). This is the first report directly comparing GH115 specific activity on hardwood, softwood, and cereal xylans. Beechwood glucuronoxylan comprises a β-(1→4)-Xylp backbone substituted with α-(1→2)-MeGlcAp, where the ratio of Xylp to MeGlcAp is ∼8:1 (3) (Fig. 9). Similarly, the β-(1→4)-Xylp backbone of spruce arabinoglucuronoxylan and oat spelt xylan is decorated by α-(1→2)-MeGlcAp; however, these xylans are further substituted by Araf, where the ratio of MeGlcAp to Araf:Xylp in spruce arabinoglucuronoxylan and oat spelt xylan is ∼2:1:11 and 1:3:24, respectively (32, 64). The lower activity of SdeAgu115A toward oat spelt xylan could be explained by the comparatively low water solubility of this substrate preparation, which would lower the effective MeGlcAp content available to the enzyme. Lower activity toward spruce arabinoglucuronoxylan was more difficult to explain, given the comparatively high water solubility of this substrate and the 1.5-fold higher MeGlcAp content compared with beechwood xylan. Previous studies of GH115 α-glucuronidase using acetylated glucurono-xylooligosaccharides demonstrate that although GH115 enzymes can access (Me)GlcAp substituents from internal regions of xylan, they may be restricted to singly substituted Xylp residues (16). Analogously, it could be that lower activity of SdeAgu115A on spruce arabinoglucuronoxylan is due to partial obstruction by Araf residues despite its position on other Xylp units, which would further support the formation of an extended substrate binding site by this and potentially other GH115 enzymes.
TABLE 3.
Activity profile of SdeAgu115A
The final concentration of substrates in activity assay solution was 1% (w/v). No activity was detected toward alginate, rhamnogalacturonan, apple pectin, and gum arabic. Values shown are mean ± S.D. (n = 3). Infusion negative ion spectra confirmed that only MeGlcAp (m/z 207) was released from beechwood xylan by SdeAgu115A (data not shown).
| Polymeric substrates | Activity | Relative activitya |
|---|---|---|
| μmol product/min/μmol enzyme | % | |
| Beechwood xylan | 2470 ± 72 | 100 ± 2.9 |
| Spruce arabinoglucuronoxylan | 917 ± 6 | 37 ± 0.2 |
| Oat spelt xylan | 24 ± 1 | 1.0 ± 0.04 |
a Relative activity was calculated based on maximum activity values for each substrate category.
FIGURE 9.

Schematic structures of xylan substrates used in activity studies of SdeAgu115A.
SdeAgu115A displayed low but detectable activity toward pNP-GlcAp (Fig. 8), which has not been tested for GH67 and other GH115 enzymes characterized to date. Still, kcat values obtained using pNP-GlcAp were 8200–36,000 times lower than apparent kcat values obtained using glucurono-xylooligosaccharides and beechwood xylan (Fig. 8), underscoring the critical importance of including non-synthetic substrates in screens for new enzyme activity. Kinetic analyses also revealed substrate inhibition of SdeAgu115A by beechwood xylan (Fig. 8C), suggesting the non-catalytic interactions between SdeAgu115A and this substrate. Substrate inhibition can be caused by the non-productive binding of substrate molecules to different subsites within the enzyme substrate binding site (65). Thus, the substrate inhibition observed for SdeAgu115A implies a multisubsite binding cleft at the surface of this enzyme. Notably, substrate inhibition has not been reported for other GH115 enzymes characterized to date.
Impact of NaCl on the Aggregation and Activity of SdeAgu115A
SdeAgu115A was observed to aggregate in 20 mm HEPES buffer (pH 7.0). The aggregation was reversed by adding NaCl up to 40 mm in 20 mm HEPES buffer (pH 7.5) (Fig. 10A). Interestingly, the presence of NaCl (up to 300 mm) enhanced SdeAgu115A activity toward beechwood xylan up to 6-fold. Moreover, 80% of SdeAgu115A activity was retained in 1.5 m NaCl (Fig. 10B). NaCl tolerance and activation have been reported for several enzymes from other marine bacteria (52, 63). Altogether, it was consistent with the marine habitat of the enzyme source organism.
FIGURE 10.

The effect of NaCl to SdeAgu115A reversible aggregation and its activity on beechwood xylan. A, dialysis of enzyme solution against 20 mm Hepes buffer (pH 7.0) led to protein aggregation; turbidity at A450 nm decreased through titration with NaCl. B, the Nelson-Somogyi method was used to measure SdeAgu115A activity in 100 mm MES buffer (pH 6.5) containing different NaCl concentrations. Full relative activity (100%) was obtained at 300 mm NaCl. Error bars, S.D.
Conclusion
Only a few α-glucuronidases from family GH115have been characterized to date. Two recently published GH115 structures both exhibited four-domain architectures. Herein, we present a GH115 α-glucuronidase (SdeAgu115A) from the marine bacterium S. degradans 2-40T with a novel five-domain architecture, which will be shared by more than half of GH115 members. The domain C+ insertion in SdeAgu115A significantly impacted domain arrangement and its dimerization, which is the predominant functional oligomeric state of this enzyme. In addition, an essential catalytic residue, Glu-216 in SdeAgu115A, was identified for the first time in a GH115 enzyme. Furthermore, site-specific mutagenesis revealed the participation of the C+ domain in xylan substrate binding and the benefit of altering the C+ region to modulate substrate inhibition. These results may benefit efforts to minimize detrimental impacts of Araf and acetyl substitutions on GH115 action through protein engineering. Determination of high resolution complex structures of SdeAgu115A in complex with ligands will be key to further evaluating these possibilities.
Author Contributions
E. R. M. and A. S. conceived and coordinated the study; W. W., R. Y., B. P. N., T. V. V., R. D. L., X. X., H. C., M. T., P. G., and G. T. conducted the experiments; P. G. and G. T. prepared the spruce arabinoglucuronoxylan used in the study; and W. W., R. Y., B. P. N., A. S., and E. R. M. wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank G. Brown and Professor A. Yakunin (University of Toronto) for technical support and Dr. Masoud Vedadi and Dr. Guillermo Senisterra (Structural Genomics Consortium, Toronto, Canada) for circular dichroism spectra acquisition. We also thank K. Parikka for preparing figures of xylan structures. Structural results shown in this report are derived from work performed at Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source. Argonne National Laboratory is operated by UChicago Argonne, LLC, for the United States Department of Energy, Office of Biological and Environmental Research, under contract DE-AC02-06CH11357.
This work was supported by the Government of Ontario through the project “Forest FAB: Applied Genomics for Functionalized Fibre and Biochemicals” (Grant ORF-RE-05-005 to E. R. M.). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Table S1 and Fig. S1.
The atomic coordinates and structure factors (code 4ZMH) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- Xylp
- d-xylopyranosyl
- GlcAp
- glucuronic acid (glucopyranosyluronic acid)
- MeGlcAp
- methylglucuronic acid (4-O-methyl glucopyranosyluronic acid)
- pNP-GlcAp
- p-nitrophenyl-α-d-glucuronopyranoside
- pNP
- p-nitrophenyl
- XOS
- xylooligosaccharides
- Araf
- α-l-arabinofuranosyl
- PDB
- Protein Data Bank.
References
- 1. Busse-Wicher M., Gomes T. C., Tryfona T., Nikolovski N., Stott K., Grantham N. J., Bolam D. N., Skaf M. S., and Dupree P. (2014) The pattern of xylan acetylation suggests xylan may interact with cellulose microfibrils as a twofold felical screw in the secondary plant cell wall of Arabidopsis thaliana. Plant J. 79, 492–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Scheller H. V., and Ulvskov P. (2010) Hemicelluloses. Annu. Rev. Plant Biol. 61, 263–289 [DOI] [PubMed] [Google Scholar]
- 3. Teleman A., Lundqvist J., Tjerneld F., Stålbrand H., and Dahlman O. (2000) Characterization of acetylated 4-O-methylglucuronoxylan isolated from aspen employing 1H and 13C NMR spectroscopy. Carbohydr. Res. 329, 807–815 [DOI] [PubMed] [Google Scholar]
- 4. Timell T. (1967) Recent progress in the chemistry of wood hemicelluloses. Wood Sci. Technol. 1, 45–70 [Google Scholar]
- 5. Shallom D., and Shoham Y. (2003) Microbial hemicellulases. Curr. Opin. Microbiol. 6, 219–228 [DOI] [PubMed] [Google Scholar]
- 6. de Vries R. P., Kester H. C., Poulsen C. H., Benen J. A., and Visser J. (2000) Synergy between enzymes from Aspergillus involved in the degradation of plant cell wall polysaccharides. Carbohydr. Res. 327, 401–410 [DOI] [PubMed] [Google Scholar]
- 7. Coughlan M. P., and Hazlewood G. P. (1993) β-1,4-d-Xylan-degrading enzyme systems: biochemistry, molecular biology and applications. Biotechnol. Appl. Biochem. 17, 259–289 [PubMed] [Google Scholar]
- 8. Gilbert H. J., Stålbrand H., and Brumer H. (2008) How the walls come crumbling down: recent structural biochemistry of plant polysaccharide degradation. Curr. Opin. Plant Biol. 11, 338–348 [DOI] [PubMed] [Google Scholar]
- 9. Bosmans T. J., Stépán A. M., Toriz G., Renneckar S., Karabulut E., Wågberg L., and Gatenholm P. (2014) Assembly of debranched xylan from solution and on nanocellulosic surfaces. Biomacromolecules 15, 924–930 [DOI] [PubMed] [Google Scholar]
- 10. Mikkonen K. S., and Tenkanen M. (2012) Sustainable food-packaging materials based on future biorefinery products: xylans and mannans. Trends Food Sci. Technol. 28, 90–102 [Google Scholar]
- 11. Sedlmeyer F. B. (2011) Xylan as by-product of biorefineries: characteristics and potential use for food applications. Food Hydrocoll. 25, 1891–1898 [Google Scholar]
- 12. Henrissat B. (1991) A classification of glycosyl hydrolases based sequence similarities amino acid. Biochem. J. 280, 309–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Suresh C., Kitaoka M., and Hayashi K. (2003) A thermostable non-xylanolytic α-glucuronidase of Thermotoga maritima MSB8. Biosci. Biotechnol. Biochem. 67, 2359–2364 [DOI] [PubMed] [Google Scholar]
- 14. Suresh C., Rus'd A. A., Kitaoka M., and Hayashi K. (2002) Evidence that the putative α-glucosidase of Thermotoga maritima MSB8 is a pNP α-d-glucuronopyranoside hydrolyzing α-glucuronidase. FEBS Lett. 517, 159–162 [DOI] [PubMed] [Google Scholar]
- 15. Bronnenmeier K., Meissner H., Stocker S., and Staudenbauer W. L. (1995) α-d-Glucuronidases from the xylanolytic thermophiles Clostridium stercorarium and Thermoanaerobacterium saccharolyticum. Microbiology 141, 2033–2040 [DOI] [PubMed] [Google Scholar]
- 16. Tenkanen M., and Siika-aho M. (2000) An α-glucuronidase of Schizophyllum commune acting on polymeric xylan. J. Biotechnol. 78, 149–161 [DOI] [PubMed] [Google Scholar]
- 17. Ryabova O., Vrsanská M., Kaneko S., van Zyl W. H., and Biely P. (2009) A novel family of hemicellulolytic α-glucuronidase. FEBS Lett. 583, 1457–1462 [DOI] [PubMed] [Google Scholar]
- 18. Biely P., de Vries R. P., Vrsanská M., and Visser J. (2000) Inverting character of α-glucuronidase A from Aspergillus tubingensis. Biochim. Biophys. Acta 1474, 360–364 [DOI] [PubMed] [Google Scholar]
- 19. Rosa L., Ravanal M. C., Mardones W., and Eyzaguirre J. (2013) Characterization of a recombinant α-glucuronidase from Aspergillus fumigatus. Fungal Biol. 117, 380–387 [DOI] [PubMed] [Google Scholar]
- 20. Golan G., Shallom D., Teplitsky A., Zaide G., Shulami S., Baasov T., Stojanoff V., Thompson A., Shoham Y., and Shoham G. (2004) Crystal structures of Geobacillus stearothermophilus α-glucuronidase complexed with its substrate and products: mechanistic implications. J. Biol. Chem. 279, 3014–3024 [DOI] [PubMed] [Google Scholar]
- 21. Nurizzo D., Nagy T., Gilbert H. J., and Davies G. J. (2002) The structural basis for catalysis and specificity of the Pseudomonas cellulosa α-glucuronidase, GlcA67A. Structure 10, 547–556 [DOI] [PubMed] [Google Scholar]
- 22. Chong S.-L., Battaglia E., Coutinho P. M., Henrissat B., Tenkanen M., and de Vries R. P. (2011) The α-glucuronidase Agu1 from Schizophyllum commune is a member of a novel glycoside hydrolase family (GH115). Appl. Microbiol. Biotechnol. 90, 1323–1332 [DOI] [PubMed] [Google Scholar]
- 23. Rogowski A., Baslé A., Farinas C. S., Solovyova A., Mortimer J. C., Dupree P., Gilbert H. J., and Bolam D. N. (2014) Evidence that GH115 α-glucuronidase activity, which is required to degrade plant biomass, is dependent on conformational flexibility. J. Biol. Chem. 289, 53–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Aalbers F., Turkenburg J. P., Davies G. J., Dijkhuizen L., and Lammerts van Bueren A. (2015) Structural and functional characterization of a novel family GH115 4-O-methyl-α-glucuronidase with specificity for decorated arabinogalactans. J. Mol. Biol. 427, 3935–3946 [DOI] [PubMed] [Google Scholar]
- 25. Fujimoto Z., Ichinose H., Biely P., and Kaneko S. (2011) Crystallization and preliminary crystallographic analysis of the glycoside hydrolase family 115 α-glucuronidase from Streptomyces pristinaespiralis. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 67, 68–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kolenová K., Ryabova O., Vrsanská M., and Biely P. (2010) Inverting character of family GH115 α-glucuronidases. FEBS Lett. 584, 4063–4068 [DOI] [PubMed] [Google Scholar]
- 27. Andrykovitch G., and Marx I. (1988) Isolation of a new polysaccharide-digesting bacterium from a salt marsh. Appl. Environ. Microbiol. 54, 1061–1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ekborg N. A., Taylor L. E., Longmire A. G., Henrissat B., Weiner R. M., and Hutcheson S. W. (2006) Genomic and proteomic analyses of the agarolytic system expressed by Saccharophagus degradans 2-40. Appl. Environ. Microbiol. 72, 3396–3405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Taylor L. E. 2nd, Henrissat B., Coutinho P. M., Ekborg N. A., Hutcheson S. W., and Weiner R. M. (2006) Complete cellulase system in the marine bacterium Saccharophagus degradans strain 2-40T. J. Bacteriol. 188, 3849–3861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Weiner R. M., Taylor L. E. 2nd, Henrissat B., Hauser L., Land M., Coutinho P. M., Rancurel C., Saunders E. H., Longmire A. G., Zhang H., Bayer E. A., Gilbert H. J., Larimer F., Zhulin I. B., Ekborg N. A., et al. (2008) Complete genome sequence of the complex carbohydrate-degrading marine bacterium, Saccharophagus degradans strain 2-40 T. PLoS Genet. 4, e1000087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hutcheson S. W., Zhang H., and Suvorov M. (2011) Carbohydrase systems of Saccharophagus degradans degrading marine complex polysaccharides. Mar. Drugs 9, 645–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Escalante A., Gonçalves A., Bodin A., Stepan A., Sandström C., Toriz G., and Gatenholm P. (2012) Flexible oxygen barrier films from spruce xylan. Carbohydr. Polym. 87, 2381–2387 [Google Scholar]
- 33. Liu H., and Naismith J. H. (2008) An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol. 8, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 35. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 36. DuBois M., Gilles K. A., Hamilton J. K., Rebers P. A., and Smith F. (1956) Colorimetric method for determination of sugars and related substances. Anal. Chem. 28, 350–356 [Google Scholar]
- 37. Koutaniemi S., Guillon F., Tranquet O., Bouchet B., Tuomainen P., Virkki L., Petersen H. L., Willats W. G. T., Saulnier L., and Tenkanen M. (2012) Substituent-specific antibody against glucuronoxylan reveals close association of glucuronic acid and acetyl substituents and distinct labeling patterns in tree species. Planta 236, 739–751 [DOI] [PubMed] [Google Scholar]
- 38. Vuong T. V., Vesterinen A.-H., Foumani M., Juvonen M., Seppälä J., Tenkanen M., and Master E. R. (2013) Xylo- and cello-oligosaccharide oxidation by gluco-oligosaccharide oxidase from Sarocladium strictum and variants with reduced substrate inhibition. Biotechnol. Biofuels. 6, 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Smogyi M. (1952) Notes on sugar determination. J. Biol. Chem. 195, 19–23 [PubMed] [Google Scholar]
- 40. Laue T. M., Shah B. D., Ridgeway T. M., and Pelletier S. M. (1992) Computer-aided interpretation of analytical sedimentation data for proteins. in Analytical Ultracentrifugation in Biochemistry and Polymer Science (Harding S. E., Rowe A. J., and Horton J. C., eds) pp. 90–125, Royal Society of Chemistry, London [Google Scholar]
- 41. Freelove A. C. J., Bolam D. N., White P., Hazlewood G. P., and Gilbert H. J. (2001) A novel carbohydrate-binding protein is a component of the plant cell wall-degrading complex of Piromyces equi. J. Biol. Chem. 276, 43010–43017 [DOI] [PubMed] [Google Scholar]
- 42. Nocek B., Mulligan R., Bargassa M., Collart F., and Joachimiak A. (2008) Crystal structure of aminopeptidase N from human pathogen Neisseria meningitidis. Proteins 70, 273–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rosenbaum G., Alkire R. W., Evans G., Rotella F. J., Lazarski K., Zhang R. G., Ginell S. L., Duke N., Naday I., Lazarz J., Molitsky M. J., Keefe L., Gonczy J., Rock L., Sanishvili R., et al. (2006) The Structural Biology Center 19ID undulator beamline: facility specifications and protein crystallographic results. J. Synchrotron Radiat. 13, 30–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Minor W., Cymborowski M., Otwinowski Z., and Chruszcz M. (2006) HKL-3000: The integration of data reduction and structure solution: from diffraction images to an initial model in minutes. Acta Crystallogr. D Biol. Crystallogr. 62, 859–866 [DOI] [PubMed] [Google Scholar]
- 45. Collaborative Computational Project, Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 46. Terwilliger T. C., and Berendzen J. (1999) Automated MAD and MIR structure solution. Acta Crystallogr. D Biol. Crystallogr. 55, 849–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Terwilliger T. (2004) SOLVE and RESOLVE: automated structure solution, density modification, and model building. J. Synchrotron Radiat. 11, 49–52 [DOI] [PubMed] [Google Scholar]
- 48. Cowtan K. (2001) Fast Fourier feature recognition. Acta Crystallogr. D Biol. Crystallogr. 57, 1435–1444 [DOI] [PubMed] [Google Scholar]
- 49. Sheldrick G. M. (2002) Macromolecular phasing with SHELXE. Z. Kristallogr. 217, 644–650 [Google Scholar]
- 50. Cowtan K., and Main P. (1998) Miscellaneous algorithms for density modification. Acta Crystallogr. D Biol. Crystallogr. 54, 487–493 [DOI] [PubMed] [Google Scholar]
- 51. Cowtan K. D., and Zhang K. Y. (1999) Density modification for macromolecular phase improvement. Prog. Biophys. Mol. Biol. 72, 245–270 [DOI] [PubMed] [Google Scholar]
- 52. Uchimura K., Miyazaki M., Nogi Y., Kobayashi T., and Horikoshi K. (2010) Cloning and sequencing of alginate lyase genes from deep-sea strains of Vibrio and Agarivorans and characterization of a new Vibrio enzyme. Mar. Biotechnol. 12, 526–533 [DOI] [PubMed] [Google Scholar]
- 53. Pathak S., Dorfmueller H. C., Borodkin V. S., and van Aalten D. M. F. (2008) Chemical dissection of the link between streptozotocin, O-GlcNAc, and pancreatic cell death. Chem. Biol. 15, 799–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ito T., Katayama T., Hattie M., Sakurama H., Wada J., Suzuki R., Ashida H., Wakagi T., Yamamoto K., Stubbs K. A., and Fushinobu S. (2013) Crystal structures of a glycoside hydrolase family 20 lacto-N-biosidase from Bifidobacterium bifidum. J. Biol. Chem. 288, 11795–11806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Holm L., and Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Knight S. D., Choudhury D., Hultgren S., Pinkner J., Stojanoff V., and Thompson A. (2002) Structure of the S pilus periplasmic chaperone SfaE at 2.2 Å resolution. Acta Crystallogr. D Biol. Crystallogr. 58, 1016–1022 [DOI] [PubMed] [Google Scholar]
- 57. Eidam O., Dworkowski F. S. N., Glockshuber R., Grütter M. G., and Capitani G. (2008) Crystal structure of the ternary FimC-FimFt-FimDN complex indicates conserved pilus chaperone-subunit complex recognition by the usher FimD. FEBS Lett. 582, 651–655 [DOI] [PubMed] [Google Scholar]
- 58. Larsbrink J., Rogers T. E., Hemsworth G. R., McKee L. S., Tauzin A. S., Spadiut O., Klinter S., Pudlo N. A., Urs K., Koropatkin N. M., Creagh A. L., Haynes C. A., Kelly A. G., Cederholm S. N., Davies G. J., et al. (2014) A discrete genetic locus confers xyloglucan metabolism in select human gut Bacteroidetes. Nature 506, 498–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mello L. V., Chen X., and Rigden D. J. (2010) Mining metagenomic data for novel domains: BACON, a new carbohydrate-binding module. FEBS Lett. 584, 2421–2426 [DOI] [PubMed] [Google Scholar]
- 60. de Beer T. A. P., Berka K., Thornton J. M., and Laskowski R. A. (2014) PDBsum additions. Nucleic Acids Res. 42, D292–D296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rye C. S., and Withers S. G. (2000) Glycosidase mechanisms. Curr. Opin. Chem. Biol. 4, 573–580 [DOI] [PubMed] [Google Scholar]
- 62. De Ruiter G. A., Schols H. A., Voragen A. G. J., and Rombouts F. M. (1992) Carbohydrate analysis of water-soluble uronic acid-containing polysaccharides with high-performance anion-exchange chromatography using methanolysis combined with TFA hydrolysis is superior to four other methods. Anal. Biochem. 207, 176–185 [DOI] [PubMed] [Google Scholar]
- 63. Lemak S., Tchigvintsev A., Petit P., Flick R., Singer A. U., Brown G., Evdokimova E., Egorova O., Gonzalez C. F., Chernikova T. N., Yakimov M. M., Kube M., Reinhardt R., Golyshin P. N., Savchenko A., and Yakunin A. F. (2012) Structure and activity of the cold-active and anion-activated carboxyl esterase OLEI01171 from the oil-degrading marine bacterium Oleispira antarctica. Biochem. J. 445, 193–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kormelink F. J. M., and Voragen A. G. J. (1993) Degradation of different [(glucurono)arabino]xylans by a combination of purified xylan-degrading enzymes. Appl. Microbiol. Biotechnol. 38, 688–695 [Google Scholar]
- 65. Copeland R. A. (2000) Enzymes: A Practical Introduction to Structure, Mechanism, and Data Analysis, pp. 109–145, Wiley-VCH, Weinheim, Germany [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.