

ABSTRACT
Functional in a tetrameric state, the protein product of the p53 tumor suppressor gene confers its tumor-suppressive activity by transactivating genes which promote cell-cycle arrest, senescence, or programmed cell death. How p53 distinguishes between these divergent outcomes is still a matter of considerable interest. Here we discuss the impact of 2 mutations in the tetramerization domain that confer unique properties onto p53. By changing lysines 351 and 357 to arginine, thereby blocking all post-translational modifications of these residues, DNA binding and transcriptional regulation by p53 remain virtually unchanged. On the other hand, by changing these lysines to glutamine (2KQ-p53), thereby neutralizing their positive charge and potentially mimicking acetylation, p53 is impaired in the induction of cell cycle arrest and yet can still effectively induce cell death. Surprisingly, when 2KQ-p53 is expressed at high levels in H1299 cells, it can bind to and transactivate numerous p53 target genes including p21, but not others such as miR-34a and cyclin G1 to the same extent as wild-type p53. Our findings show that strong induction of p21 is not sufficient to block H1299 cells in G1, and imply that modification of one or both of the lysines within the tetramerization domain may serve as a mechanism to shunt p53 from inducing cell cycle arrest.
KEYWORDS: acetylation, apoptosis, cell cycle, tumor suppressor p53, p21
Introduction
p53 is a DNA-binding transcription factor that carries out its tumor suppressive function by playing roles in cell cycle arrest, apoptosis, genomic stability and DNA repair, as well as other pathways (see refs.1-5). The p53 protein consists of 393 amino acid residues with a number of functional domains and isoforms of various lengths.6 The full-length wild-type (WT) protein contains a bipartite transactivation domain within the N-terminal region (residues 20–40 and 40–60), a proline-rich domain with a pro-apoptotic role (residues 60–90), a sequence-specific DNA binding domain (DBD) where most of the tumor-derived mutations reside (residues 100–300), an oligomerization domain which confers the tetrameric structure necessary for p53 function (residues 320–357), and a highly basic C-terminal domain (CTD) (residues 363–393) which possesses the ability to interact with DNA in a sequence non-specific manner.7,8
Well-studied as a sequence-specific DNA-binding transcription factor, p53 is most active in that regard in its tetrameric state. Maintenance of its proper conformation is controlled by the tetramerization domain.9,10 Although not nearly as frequently mutated in cancer as the DBD, the tetramerization domain of p53 has sustained certain tumor-derived mutations including L344 and R337. These are found mutated predominantly in Li-Fraumeni patients, affecting oligomerization and transactivation abilities.11 In particular, an inherited mutation in p53 (R337H) was found to be associated strongly with familial pediatric adrenocortical carcinoma.12 Most relevantly, it was shown that an ovarian carcinoma mutation, K351N, attenuates p53 function via a similar mechanism.13 This same mutation was shown to compromise p53 ubiquitination and subsequent mitochondrial localization, affecting the apoptotic response.14
The tetramerization domain contains a nuclear export signal (NES) (residues 340 to 351), which is masked upon tetramerization of p53, allowing it to accumulate in the nucleus.15 Two residues in the NES (L348 and L350) appear to be critical both for nuclear export and efficient tetramerization, suggesting an interplay between these 2 processes and optimal p53 function. Biochemical studies suggested phosphorylation of S392 could increase tetramer formation, while phosphorylation of S315 (within the linker region) might counteract this effect.16 Additionally, 2 reports conflictingly implicate CTD acetylation in either the promotion of tetramerization and maintenance of high acetyl-p53 levels by PTEN, or the disruption of oligomerization, followed by nuclear export.17-18
Following a wide range of cellular stresses, p53 becomes extensively modified at both the N-termini and C-termini by a number of phosphorylating, acetylating, ubiquitinating, sumoylating, methylating and neddylating enzymes.19-21 Acetylation and ubiquitination occur predominantly within the CTD of p53 and there is an important balance between ubiquitination and acetylation since acetylated lysines cannot simultaneously be ubiquitinated by Mdm2.22
Few modifications have been reported in the tetramerization domain of p53. One report implicated PRMT5-mediated arginine methylation of 3 residues, R333, R335 and R337 as being required for full induction of the GADD45, p21 and APAF1 genes.23 Another paper suggested K357 by mass spectrometry as undergoing acetylation in COS-1 cells,24 although no biological consequence of the modification was reported. Lysine residues 351 and 357 have been reported to be ubiquitinated by MSL2, a novel E3 ligase for p53 that promotes the cytoplasmic localization of the protein, but not its degradation.25 A large screen to identify ubiquitin-modified proteins confirmed the modification of lysine 357, but not lysine 351.26 However, mass spectrometry analysis of COS-1 p53 or etoposide-induced p53 from human foreskin fibroblasts indicates acetylation and methylation take place at lysines 351 and 357.27
From mining the TCGA database, we found various human cancers with alterations in K351, including one kidney carcinoma with a K351N mutation and a lung carcinoma with a mutation in 351 leading to a nonsense codon, as well as a malignant melanoma and an adrenal cortical carcinoma with K351E mutations (see Table 1).
Table 1.
Mutations in p53 tetramerization domain. Table showing selected mutations in p53 CTD from cancers across all TCGA datasets (Accessed from cBioPortal Sept 2015).
| Cancer | Alteration(s) | Mut. Type |
|---|---|---|
| Renal cell (RCC) | K351N | Missense |
| Lung | K35* | Nonsense |
| Melanoma | K351E | Missense |
| Adrenocortical (ACC) | K351E | Missense |
| Bladder | Δ349–351 | In-frame deletion |
Additionally, the modification of other residues within p53 requires an intact quaternary structure. For example, in a p53 protein where the tetramerization domain has been deleted, Chk1 can no longer phosphorylate p53.28 One study reported that oligomerization of p53 is essential for the acetylation of the protein's CTD lysines.29
Since the functional consequences of modifications at K351 and K357 are still being elucidated, and they are clearly of physiological interest, we generated cell lines expressing mutations to either glutamine or arginine for both residues. Our results indicate that these lysines are involved in differential regulation of p53 target genes and ensuing cellular outcomes, notably cell cycle arrest.
Results
Mutation of tetramerization domain lysines does not affect p53 localization or oligomerization.
Since conflicting data have come from studies in which p53 was overexpressed ectopically by transient transfection, which in some cases masked true in vivo function,30 we analyzed the effects of lysine mutations at residues 351 and 357 in the more physiological setting of inducible cell lines. Expression of p53 protein was regulated (by reducing or omitting tetracycline) to levels comparable with endogenous expression.31,32 When we undertook clonal selection of cells expressing lysine residues 351/357 mutations to arginine (2KR-p53) or glutamine (2KQ-p53), we obtained far fewer clones that expressed 2KR-p53 than their 2KQ-p53 counterparts. In fact, only 2 of the 2KR-p53 clones survived expansion and these expressed significant amounts of p53 protein (Table 2). This result suggests that, even though protein expression should be completely silent in the presence of tetracycline,33,34 there may be slight leakiness from the inducible promoter that expressed a hyperactive p53 that can block cell survival and clonal isolation. This phenomenon was previously observed when we attempted to clonally isolate an apoptotically hyperactive mutant of p53 (Table 2, ref. 30). We proceeded to characterize the p53 proteins with mutated tetramerization domain lysines (2KQ-p53 and 2KR-p53).
Table 2.
Isolation of inducible clones. Table showing the number and percent positive of isolated expression-positive tet-off inducible clones. Starred cell lines were isolated previously (30).
| Cell Line | # Positive | % Positive |
|---|---|---|
| WT-p53* | 13/37 | 35% |
| 2KR-p53 | 2/28 | 7% |
| 2KQ-p53 | 9/20 | 45% |
| T123A-p53* | 2/52 | 4% |
A diagram of p53 organization with the location of the 2 lysines in the tetramerization domain (denoted by asterisks) is shown in Figure 1A. As deduced from the solution structure of the tetramerization domain,35 both of these residues are solvent-accessible, and thus potentially amenable to modification (Figure S1A). Since mutations at other residues in the tetramerization region have previously been shown to disrupt p53 quaternary structure and expose a nuclear export signal,11,15 we first wanted to investigate the subcellular localization of our mutants. Immunofluorescence experiments with 2 clones each of 2KR-p53 and 2KQ-p53 expressing cells showed strong nuclear staining that was virtually identical to WT-p53 (Fig. 1B). The mutants further behaved like WT-p53 by showing nucleolar exclusion36 as evidenced both by DIC imaging and a lack of colocalization with nucleolin (data not shown).
Figure 1.
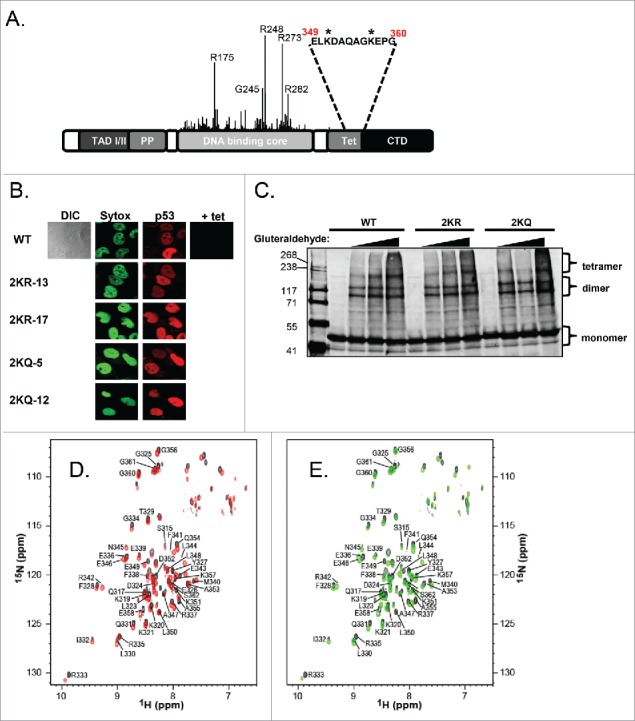
Mutation of the 2 lysine residues within the tetramerization domain of p53 does not alter subcellular localization or oligomerization. A) Schematic representation of the domains of p53. Location of tumor-derived mutations and relative frequency are indicated by the height of the line found at each amino acid position. The primary sequence encompassing lysine residues 351 and 357 in the tetramerization domain is listed and asterisks denote the positions of these residues within the sequence. (B) Subcellular localization of tetramerization domain mutants. Cell lines listed on left were induced to express p53 by removal of tetracycline for 24 hours. DNA was visualized by staining with Sytox Green (‘Sytox’) and p53 localization was detected by incubation with DO-1 monoclonal antibody followed by goat anti-mouse secondary antibody conjugated to Cy5 (‘p53’, red). The ‘+ tet’ image shows p53 staining without protein induction. DIC image of wild-type shows the whole cell including its nucleus and nucleoli. (C) H1299 cells were transfected with WT-p53, 2KR-p53, or 2KQ-p53 expression constructs. Twenty-four hours later, cells were harvested and crosslinked with 0.005%, 0.01%, or 0.02% gluteraldehyde. Complexes were separated by SDS-page and subjected to immunoblot analysis. (D) Superposition of the 1H-15N TROSY-HSQC spectra of WT-p53 (black) and the 2KQ-p53 mutant (red). (E) Superposition of the 1H-15N TROSY-HSQC spectra of WT-p53 (black) and the 2KR-p53 mutant (green).
In general, misfolded proteins may be quickly degraded by multiple degradation pathways via quality control mechanisms (as reviewed in ref.37). We performed half-life experiments to confirm that the tetramerization domain mutations were not causing hyperactive degradation of p53. Figure S1B shows that when transfected into H1299 cells, WT-p53, 2KQ-p53, and 2KR-p53 proteins each have a similar half-life (∼9 hours) as determined by cycloheximide chase. Although endogenous p53 has a half-life of about 20–30 minutes, ectopically p53 is generally known to be much more stable for as yet unknown reasons.38
Further, information about the tetramerization region of the wild-type and mutant protein was derived from NMR analysis. 1H-15N TROSY-HSQC experiments were performed with WT-p53, 2KR-p53 or 2KQ-p53 tetramerization domain proteins (310–362) to assess whether double mutations of the K351 and K357 residues perturbed the local structure of the protein. For both the 2KQ-p53 and 2KR-p53 mutants (310–362), the overall appearance of the HSQC spectra was similar to WT-p53 (Fig. 1D and E), indicating the mutations did not cause global unfolding and/or gross aggregation of the protein. The assignments of the 1H-15N TROSY-HSQC resonances of the mutants were straightforward because of close similarity of the chemical shifts to those of WT-p53 protein, except for a couple of resonances. Specifically, the 2KR-p53 spectrum, in contrast to the 2KQ-p53 spectrum, did not show a peak for the Leu350 resonance at the chemical shifts similar to the WT-p53 resonance (8.24, 123.8ppm). However, one new unassigned peak (8.51, 123.1ppm) was detected in the vicinity of the spectral region, allowing us to tentatively assign this peak as the L350 resonance. In summary, 1H-15N amide backbone resonances for all but 5 N-terminal residues (310–315) were assigned.
In Figure S1C, chemical shift differences between WT-p53 and the 2KR-p53 or 2KQ-p53 mutants are plotted versus residue number. The largest differences observed were spatially close to the mutated residues. The residues with great (>0.10 ppm) chemical shift deviations from WT-p53 were F341, A347, L350 and A355. However, the differences between 2KR and 2KQ at these residues are minor (≤0.05 ppm), with the exception of L350. Due to their similar spectra, and overall deviation from WT-p53 being minimal, we conclude that the quaternary structure of p53 is not significantly affected by these mutations.
To determine whether these mutations affected tetramerization of p53, we performed gluteraldehyde crosslinking gel electrophoresis comparing WT-p53, 2KR-p53 and 2KQ-p53 expressed in transfected H1299 cells and showed that variants formed equivalent populations of tetramers and dimers under denaturing conditions (Fig. 1C). Additionally, analytical size exclusion column chromatography analysis on Thioredoxin-fusion p53 (tetramerization domain only) showed that WT, 2KR and 2KQ have similar quaternary states at 150–500 mM NaCl (data not shown).
Together these results demonstrate that mutation of the lysines within p53's tetramerization domain to either arginine or glutamine may cause minor localized alterations, however these do not seem to affect the tetramerization status, localization or degradation of the protein itself.
2KQ-p53 is deficient in binding to and transactivation of p53 target genes
We examined the abilities of tetramerization domain lysine p53 mutants to function as transcription factors. By regulating the concentration of tetracycline in the medium, p53 protein expression was normalized for each cell line so that equivalent protein levels could be compared (Fig. 2A). Immediately a difference in activities between 2KR-p53 and 2KQ-p53 could be seen: 2KR-p53 was able to induce similar levels of p21 protein as WT-p53 while p21 resulting from 2KQ-p53 induction was barely increased above background basal levels. When looking at a direct readout of transcriptional activity by analyzing mRNA production via RT-PCR experiments, we saw similar results. Two clones of 2KR-p53 (2KR-13 and 2KR-17) induced equivalent amounts of p21, PIG3 and Mdm2 mRNA as WT-p53, yet 2 clones of 2KQ-p53 (2KQ-5 and 2KQ-12) were significantly impaired in this respect (Fig. 2B and C).
Figure 2.
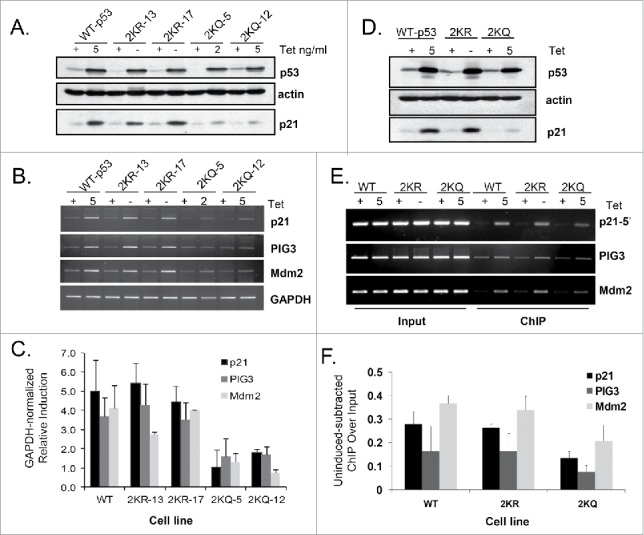
2KQ-p53 mutants show reduced ability to bind to and transactivate canonical p53 target genes, while 2KR-p53 mutants behave similarly to WT-p53. (A) Western Blot analysis. Tetracycline levels were regulated (as indicated) to express equivalent amounts of wild-type or mutant p53 protein, as determined by immunoblotting with 40 μg of whole cell extract. Levels of p21 protein induction were determined for each cell line and actin levels were assessed as a loading control. (B) mRNA induction by p53 variants. Total cellular RNA was prepared with cells from the same plate as (A) and cDNA was generated from Poly-A mRNA by oligo-dT priming. Two μl of each cDNA reaction was then used as a template in semi-quantitative RT-PCR to amplify the indicated endogenous target. Twenty μl PCR reactions were resolved by agarose gel electrophoresis, stained with ethidium bromide, and visualized on a Kodak gel imaging system. (C) Summary chart showing the average of 3 independent RT-PCR experiments, graphing arbitrary mRNA induction after normalizing to GAPDH and uninduced basal levels. Error bars indicate standard deviation. (D and E) Chromatin binding in vivo. Wild-type p53 (WT-p53), 2KR-17, or 2KQ-12 cell lines were induced with indicated tet amounts for 24 hours, crosslinked, lysed, sonicated, and processed for immunoblotting of p53, actin, or p21 (D) or ChIP (E). p53 immunoprecipitations were performed using a mixture of protein A and protein G beads preincubated with 1801 and DO-1 antibodies. In vivo DNA binding to p21 5′, PIG3 and Mdm2 response elements was determined by PCR using primers specific for regions within these genes. An aliquot of chromatin was taken before the immunoprecipitation and amplified by PCR to determine the relative number of cells in each ChIP sample (‘Input’). (F) Chart representing the average binding of each cell line to the indicated promoter, normalized to input and uninduced (+ tet) basal levels. Error bars indicate the standard deviation of at least 3 independent ChIP experiments.
We next evaluated DNA binding by these p53 variants in vivo in a ChIP assay. Again, immunoblot analysis of the lysates indicated similar levels of p53 expressed for each line along with weaker induction of p21 protein by 2KQ-p53 (Fig. 2D). In fact, binding by the 2KR and 2KQ mutants to 3 different p53 target gene promoters (p21-5′, PIG3 and Mdm2) in ChIP assays correlated well with the downstream induction of mRNA and protein; 2KR-p53 reproducibly bound as well as WT-p53, and 2KQ-p53 had decreased affinity for these sites (Fig. 2E and F).
Overall, our findings indicate that blocking lysine modification within the tetramerization domain does not impact DNA binding, whereas neutralizing charge at these residues decreases p53's overall affinity for DNA in vivo.
2KQ-p53 cannot arrest cells in G1 despite high levels of p21 or miR-34a
We investigated the ability of the p53 variants to affect 2 well studied downstream events in the p53 pathway, namely cell cycle arrest and apoptosis. As before, cells were induced to express equivalent amounts of p53 protein prior to determining their cell-cycle profile analyzed by FACS. A typical effect of WT-p53 expression in these cells is evidenced by a drop in S-phase and an increase in either G1 or the G2 population (Fig. 3A, top row). A mild increase in sub-G1 content was also seen, as reflected by the amount of fragmented DNA from apoptotic cells.39
Figure 3.

Even when overexpressed, 2KQ-p53 is unable to arrest but can induce cell death. (A) FACS analysis of wild-type and mutant p53 cell lines, expressing either no p53 or equivalent levels of protein. Percentage of cells in G1, S, or G2-phase and/or apoptotic sub-G1 is indicated. S-phase is highlighted to indicate arrest. (B) FACS analysis of WT-p53 and 2KQ-p53 cell lines, expressing either no p53 or equivalent levels of protein as indicated. Cells were treated for 48 hours with 0.5 mM 5-FU. Sub-G1 content is indicated to highlight apoptosis. (C) p21 localization in H1299 cell lines. Cell lines listed at top were grown in tetracycline-free media for 48 hours. DNA was visualized by staining with DAPI and p21 was detected by incubation with p21 antibody (Santa Cruz Biotechnology, F-5), then with Alexa Fluor 488 (sc-6246). (D and E) Western blot (D) and FACS analysis (E) at low WT-p53 and maximal 2KQ-p53 protein levels to determine relative p21 induction and cell cycle arrest respectively. (F) FACS analysis of other 2KQ-p53 clones induced to express maximal levels of p53 and p21.
Results with the tetramerization domain mutants were quite striking. Similar to cells expressing WT-p53, expression of 2KR-p53 caused a drop in S-phase and a strong arrest (Fig. 3A). 2KQ-p53, on the other hand, showed the opposite phenotype. Upon expression of this mutant, there was no discernable cell cycle arrest. Notably, the 2KQ-p53 mutant was able to induce cell death, both alone (weakly) and, more strongly in response to 5-FU (Fig. 3B) to a comparable degree as did WT-p53, indicating a selective deficiency in the ability of 2KQ-p53 to function within the cell cycle arrest pathway. We wondered if this phenotype was due to abnormal localization of the key cell cycle regulator, p21. However, immunofluorescence of this protein in the 3 cell lines in tetracycline-free conditions indicated that it is still localized in the nucleus (Fig. 3C).
It is interesting that 2KQ-p53, when regulated to express a similar amount of protein as WT-p53 was able to cause some form of cell death despite its deficiency in inducing apoptotic targets such as PIG3 (Fig. 2), Bax, Puma, Noxa, and PIDD (Figure S2). The ability of 2KQ-p53 to transactivate these genes was not significantly increased after treatment with either daunorubicin or 5-FU (data not shown). Under these conditions, then, there may be other anti-survival targets of p53 that can be induced by 2KQ-p53, or this mutant is competent in regulating a p53-mediated transcription-independent pathway.
The inability of 2KQ-p53 to arrest cells was an intriguing finding that called for further investigation. Since p21 is thought to be the major effector of p53-mediated cell-cycle arrest,40-42 it is possible that 2KQ-p53 was unable to cause a G1 arrest simply because it did not induce sufficient p21 mRNA and protein. To address this question we adjusted the amounts of tetracycline in the culture media of WT-p53- and 2KQ-p53-expressing cells to obtain points where the mutant induced markedly more p21 protein than did the WT-p53 cells (Fig. 3D). While WT-p53 was still capable of causing a robust arrest, surprisingly, 2KQ-p53 was completely inert in this regard even when expressed at higher levels than the wild-type protein (Fig. 3E). This effect was not unique to this clone of cells, as we observed this phenotype with 3 additional 2KQ-p53-expressing clones (Fig. 3F). In line with the FACS analysis, turning on WT-p53 caused a decrease in the levels of phospho-Rb, consistent with a G1 arrest, while overexpressing 2KQ-p53 did not lead to such a decrease (Fig. 3D). Furthermore, when WT-p53 was expressed in the background of p21 downregulation by siRNA (Figure S3A, B), cells were kept in G1 to a similar extent to those cells with control siRNA. Also, we expressed miR-34a in the 2KQ-p53 cells, to a level equivalent of that in the WT-p53 cells (Figure S3C). However, when 2KQ-p53 was induced, the addition of miR-34a did not rescue the ability of cells to undergo G1 arrest (Figure S3D). Thus, in this system, expression of p21 or miR-34a are not sufficient (and may not be necessary) to induce a p53-dependent cell cycle arrest.
Even when overexpressed in vivo, 2KQ-p53 is deficient in binding and inducing miR-34a and CCNG1
Since levels of p21 were clearly not the sole determinant of G1 arrest in this system, we next wanted to investigate how 2KQ-p53 regulates other p53 target genes when expressed at high levels. As before, cells were plated and conditions were set to express more 2KQ-p53 protein than WT-p53 protein, and the induction of a panel of p53 targets was assessed by qRT-PCR (Fig. 4A). We found that when overexpressed, 2KQ-p53 was as competent as WT p53 in inducing several targets, with the notable exceptions of CCNG1 and miR-34a. Intriguingly, both of these targets have been implicated in the G1 arrest pathway.43-45 We saw similar if not more dramatic results with another clone of 2KQ-p53 (Figure S4A).
Figure 4.
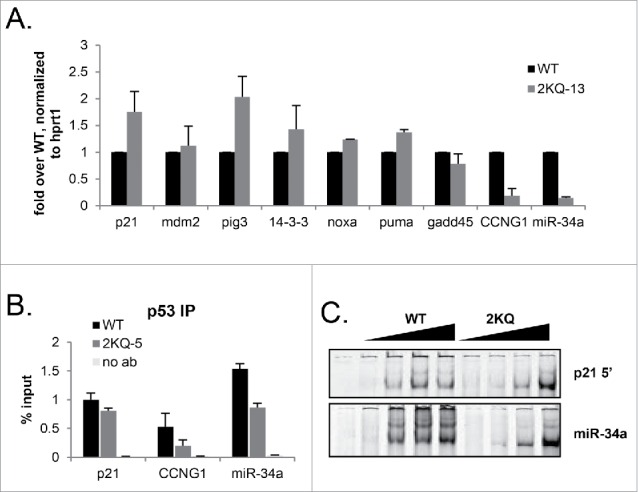
When overexpressed in vivo, 2KQ-p53 is deficient in binding and inducing miR-34a and CCNG1. (A) mRNA induction at low WT-p53 and maximal 2KQ-p53 p53 protein levels (as in Fig 3C) to analyze transactivation potential of the 2KQ-p53 mutant. Cells were induced or not with tet for 24 hours, harvested, and RNA was extracted. After cDNA was synthesized, samples were amplified for indicated p53 target genes by qRT-PCR. (B) ChIP analysis at low WT-p53 and maximal 2KQ-p53 protein levels to assess in vivo DNA binding. Samples were processed for ChIP as in Figure 2D, except that immunoprecipitated DNA was analyzed by qRT-PCR. For (A) and (B), the average of 3 experiments is shown, and error bars show standard deviation of 3 experiments. (C) Purified p53 WT-p53 or 2KQ-p53 protein was incubated with 10 ng of p21 5′ RE or miR34a RE fluorescently labeled 44mer probe, in the presence of excess 44mer mutant p21 competitor. P53:DNA complexes were separated by electrophoresis and visualized by the Licor Odyssey system.
p53 DNA binding can directly correlate with mRNA induction32 and so we next asked whether the marked deficiency of 2KQ-p53 in inducing CCNG1 and miR-34a mRNA was due to an impaired ability to bind the p53 canonical response elements at these loci. This question was especially important because many alterations to the C-terminus of p53 have been shown to impact binding by the core domain in vitro (reviewed in ref.46). As before, cells were plated and induced with tetracycline for 24 hours to express more 2KQ-p53 protein than WT-p53 protein, and a ChIP assay was performed to assay for p53 binding to its distal p21, miR34-a, and CCNG1 response elements. Nicely mimicking our RNA data, when 2KQ-p53 was overexpressed, it was capable of binding the p21 distal response element to a comparable degree to WT-p53. However, even when it was expressed at these high levels, 2KQ-p53 was still markedly deficient in binding the miR-34a and CCNG1 response elements in a ChIP assay (Fig. 4B). Similar results were observed in a transient transfection/ChIP assay (Figure S4B, C).
We examined whether the 2KQ-p53 binding deficiency observed in vivo was due to impaired direct binding to DNA by performing an electrophoretic mobility shift assay (EMSA) with purified WT-p53 and 2KQ-p53 proteins. WT-p53 was capable of strong binding to a probe derived from the p21 5′ response element, as well as a probe derived from the miR-34a response element, as evidenced by a shift of IRDye-labeled DNA (Fig. 4C). Excess short oligonucleotide was used as a carrier to compete away any nonspecific interactions in these reactions since long carrier can engage the p53 C-terminus and has led to confusing interpretations of EMSA data in the past.47,48 Surprisingly, given our ChIP data, at high concentrations, 2KQ-p53 protein bound probes derived from p21 and miR-34a response elements somewhat better than WT-p53 (Fig. 4C). The respective WT-p53 and 2KQ-p53 binding to p21 and miR-34a probes was nearly identical across a concentration curve, suggesting that whatever is responsible for the differential binding profiles of WT-p53 and 2KQ-p53 to these sites in a ChIP assay is not recapitulated in vitro. Even though EMSA results do not always reflect cellular DNA binding,32,49 it was nonetheless surprising that at high concentrations 2KQ-p53 appeared to bind DNA better than WT-p53, however it transactivated p53 targets to a lesser degree in vivo. It is possible that the differential binding abilities of WT-p53 and 2KQ-p53 were related to the slight structural perturbations seen in our NMR spectra, as were the somewhat different display of p53:DNA complexes formed by WT and 2KQ p53 proteins on the miR-34a probe. Nonetheless, since the overall binding deficiency of 2KQ-p53 at high concentrations was not observed on naked DNA spanning the miR-34a binding site in vitro, these results suggest that the binding impairment of 2KQ-p53 is not inherent to the sequence of the p53 RE at this locus.
Lysine 357 can be acetylated by p300/CBP
Given our results with acetyl-mimicking mutants and previously reported findings24,26,27 we sought to confirm that lysine residues 351 or 357 could be acetylated in vivo. We first transfected constructs expressing WT-p53 alone or with either HA-tagged p300 or CBP, or Flag-tagged pCAF into H1299 cells. The cells were harvested and subjected to immunoblot analysis with a rabbit polyclonal antibody that recognizes doubly acetylated p53 at K351 and K357 (anti-Ac-351/7). Both p300 and CBP were able to acetylate these residues (Fig. 5A). While pCAF was not apparently able to do so when expressed in these cells, it is acknowledged that a firm conclusion cannot be drawn since the version of pCAF we had available has a different epitope tag and so we cannot compare its levels to those of p300 and CBP. Tip60, a HAT implicated in the acetylation of p53 at Lys120,50 was also apparently unable to acetylate these residues (data not shown). Although our p300 construct expressed poorly, on a per mole basis, it is possible that it could acetylate K351 and K357 to a similar extent as did CBP.
Figure 5.
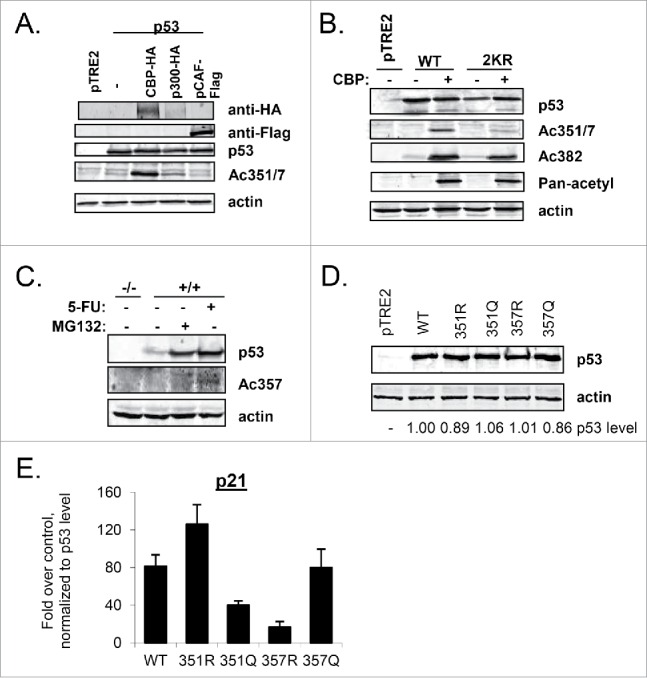
Lysine 357 can be acetylated in vivo. (A) H1299 cells were transfected with WT-p53 and either p300-HA, CBP-HA, or pCAF-Flag expression plasmids. Cells were lysed and proteins were separated by SDS-PAGE and transferred to a nitrocellulose membrane. Immunoblot analysis with indicated antibodies was performed. (B) H1299 cells were transfected with CBP-HA and either WT-p53 or 2KR-p53. After 24 hours, cells were lysed and analyzed as in A. (C) HCT116 cells expressing p53 (+/+) were treated with 5FU (0.5 mM ) for 24 hours or MG132 (25 uM) for 6 hours, lysed, and subjected to immunoblot analysis with antibodies recognizing p53, p53-Ac357, or actin. HCT116 p53-null cells (−/−) were run alongside as a control. (D) H1299 cells were transfected with constructs expressing single tetramerization domain lysine p53 mutants as indicated. Cells were lysed and immunoblot analysis to detect p53 level and actin as indicated. (E) Parallel cultures of H1299 cells were treated as in D. RNA was extracted and analyzed by qPCR for p21 expression, which was normalized to hprt1 levels. Error bars are the standard deviation of 3 experiments.
To determine if the antibody was specific to these residues, we performed a similar transfection experiment in which either WT-p53 or 2KR-p53 was transfected with or without CBP. The signal from the Ac351/7 antibody was markedly reduced when 2KR-p53 cotransfected with CBP was compared with WT-p53 cotransfected with CBP (Fig. 5B). Mutations to arginine within these residues of the tetramerization domain did not appear to compromise p53 reactivity with a pan-acetyl antibody, while reactivity with an Ac-K382-specific antibody was decreased as compared to WT-p53 (Fig. 5C).
While the anti-Ac351/357 antibody could be used to detect ectopically overexpressed p53, unfortunately, it was not strong enough to detect endogenously expressed p53. A rabbit polyclonal antibody was generated that specifically recognized p53 acetylated at K357 (Ac357; Figure S5). HCT116 cells, which contain wild-type p53, were treated with either MG132, a proteasome inhibitor that stabilizes p53 in the absence of DNA damage, or with 5-FU, to induce damage-stabilized p53. The cells were harvested and subjected to immunoblot analysis with anti-Ac357 (Fig. 5C). Acetylation of K357 was induced in response to damage by 5-FU, but was not visible when p53 was stabilized in the absence of damage by MG132, indicating that 5-FU initiates a signaling pathway that results in acetylation of p53 at K357.
Our results extend previous findings from mass spectrometric analysis that p53 can be acetylated at K357.26-27 Taken together, these data indicate that CBP (and likely p300), at least when they are overexpressed, are capable of acetylating p53 at K351 and/or K357 in mammalian cells, and that acetylation of K357 occurs endogenously in response to genotoxic stress, unlocking the exciting possibility of a physiological importance for these modifications.
Given that our data thus far demonstrated that 2KQ-p53 was largely defective in binding and transactivating p53 target genes (Fig. 2), it was perhaps counterintuitive that acetylation of p53 at K357 was increased after DNA damage, when p53 function is amplified. We therefore constructed single mutants of p53-K351 (R or Q) and p53-K357 (R or Q). To determine whether the individual p53 mutants of these lysines gave rise to the same phenotype as the double mutant, their transactivation ability was assessed in a transient transfection assay (Fig. 5D). Interestingly, at comparable levels of p53 protein (Fig 5D), both K351Q-p53 and K357R-p53 showed reduced transcription of p21.
mRNA when compared with WT-p53, while K357Q-p53 and K351R-p53 could transactivate p21 as well as, or better than, WT-p53 (Fig. 5E). To date, we have not yet been able to confirm whether K351 is also acetylated. These data indicate that if K351 is in fact acetylated such modification may be the reason for the defects in 2KQ binding and transactivation of select p53 targets. In that case, modification of K351 and K357 would play opposing roles in regulating p53 function, and the negative role of K351 modification would be dominant over a positive role of K357 modification.
Discussion
While the extreme CTD of p53 is highly modified, few modifications have been described within p53's tetramerization domain.19,51 We sought to investigate the potential role of modification within this region by mutating the only 2 lysines in this domain of p53 (351 and 357, Fig. 1A) to either block all post-translational modification (lysine to arginine, 2KR-p53) or to neutralize basic charge (lysine to glutamine, 2KQ-p53).
Although mutation of other residues within this region can drastically impact p53 tetramer formation and function,52,53 we found that neither 2KR-p53 nor 2KQ-p53 possessed unusual localization or tetramerization ability. Our data strongly suggest that alteration of the lysines in the tetramerization domain does not have a deleterious effect on the correct folding of p53. To that end, the cellular phenotypes we observe seem to be a direct effect of the lysine mutations and not caused by perturbations in overall structure.
When examining the effects of lysines 351 and 357 mutation on p53s role as a DNA-binding transcription factor, some interesting results were observed. Mutation to arginine did not have any observable impact on transactivation or in vivo DNA binding (Fig. 2). On the other hand, mutation to glutamine had a significant impact on both of these functions, reducing p53's ability to bind response elements in vivo and induce mRNA production by about half. Although acetylation on lysines in regions flanking the tetramerization domain have been shown to enhance p53 transcriptional activity, our results point to a requirement of the positively charged residues in this region for proper recognition of DNA response elements by the core domain in the context of chromatin and efficient transcription. It is possible that even though neutralization of these lysines does not disrupt tetramerization and localization they could impart some subtle effects on the quaternary structure of p53 which impairs its access to DNA, as suggested by our NMR data. However, since at higher concentrations, the 2KQ-p53 mutant was not deficient in binding naked p21 or miR-34a response elements in an EMSA assay (Fig. 5C), it is also possible that the altered promoter specificity in vivo was due to the ability of 2KQ-p53 to interact in an altered fashion with p53 binding partners. Many proteins have been shown to bind to p53 and to affect its propensity to bind select response elements,5,20 and further experiments are underway to explore the differential association of WT-p53 and 2KQ-p53 with such proteins. Alternately, the chromatin landscape at p53 target promoters may account for different interactions of WT and 2KQ-p53.
How p53 discriminates between inducing cell cycle arrest or cell death, the so-called “molecular switch,” has been attributed to a number of players, yet none of them are sufficient to drive this process in all cases. In fact, mouse studies have conflictingly suggested that p53's arrest or apoptotic functions are individually dispensable for tumor suppression.54,55 Mechanisms underlying this decision making process could be as simple as levels of p53 protein, presumably dictated by the extent of cellular stress,32 or as complex as a coordination of modifications, interactions and conformational changes to the protein in a cell-type specific context. A growing list of mutations within various regions of p53 has been shown to have an impact on the downstream function of p53. Some of these mutations exhibit increased apoptotic activity (S121F, S46F),56,57 while others have been shown to drastically decrease the cell-destructive potential of p53 (R213Q, A175P, A143P, del300-308).58-61 In these cases, the combined effect of the mutation on target gene activation, cell cycle arrest and apoptosis has been variable, supporting the idea that p53 serves as a “master gene” in which a single change can lead to simultaneous downstream changes.62 Even naturally-occurring polymorphisms within p53 can influence serine-46 phosphorylation (P47S)63 or cause a propensity for apoptosis by increasing p53's export from the nucleus (P72R) and binding to the mitochondria which activates a transcription-independent death pathway (reviewed in ref.64). Other interacting proteins such as the ASPP family of proteins which bind to the core and proline-rich regions of p53,65,66 or kinases such as PKCdelta and HIPK2 which are recruited to p53 by p53DINP1 can selectively upregulate genes involved in cell death.67-69
With 2KQ-p53, a global defect in transcriptional ability could certainly explain the lack of arrest. However, since p21 is thought to be the major effector of p53-mediated G1 arrest, our findings that 2KQ-p53 still failed to arrest cells even after inducing wild-type (or greater) levels of p21 induction were surprising. Furthermore, expressing miR-34a did not appreciably halt cells from proliferating. That 2KQ-p53 was still capable of killing cells after 5-FU treatment was also unexpected, considering that apoptotic targets like PIG3 are known to possess generally weaker binding sites70 and that in our hands 2KQ-p53 was less capable of inducing a number of apoptotic targets. This is a rare instance of a version of p53 incapable of arrest, yet still functional for programmed cell death – although which type of cell death is currently unknown, and will be explored in the future. The 2KQ mutant p53 that we describe here functions to the contrary of several previously described p53 mutants, including K120R, R175P and E177R, which are defective in apoptotic induction but are still able to regulate cell cycle arrest.50,71-73 It is also unknown whether our mutant would be incapable of senescence, since it cannot arrest, and future work will look into these capabilities.74,75
Data with the 2KQ-p53 mutants point to some exciting possibilities. First, there may be additional p53 transcriptional targets required for G1 arrest along with, or completely separate to p21. Lending support to this notion, p21-null mice develop normally, but curiously only partially fail to arrest in response to irradiation, indicating that a parallel, p21-independent pathway for G1 arrest exists.41 Based on data described herein and elsewhere, it is possible that this parallel pathway involves CCNG1 as well as other targets that we have not identified. CCNG1 was one of the earliest p53 targets to be discovered,76 yet its function remains controversial. It is known that CCNG1 can either induce a G1 arrest or apoptosis when expressed at high levels, in a fashion partially dependent on the Rb protein.45 However, at low levels of expression, CCNG1 appears to promote proliferation.47 Also of note, a p21-independent, p53-mediated repression of c-Myc was shown to be necessary for human and mouse cells to arrest in G1.77 C-Myc is an oncogene that drives cellular proliferation, and it can overcome p53-mediated activation of p21 and GADD45.78-80 It is possible that while WT-p53 can effect this repression of c-Myc, 2KQ-p53 cannot, thus preventing an efficient arrest of cells in G1. Another possibility is the existence of a pathway activated by 2KQ-p53 which can selectively block p21's inhibition of cyclin/CDK complexes. Next-generation sequencing studies would undoubtedly be worthwhile to look for additional targets induced or repressed by these mutants and to find novel targets directly involved in these phenotypes.
An impetus for this study was the discovery that p53 can be modified on lysines 351 and 357, however except for in the case of ubiquitination, the implications of this are largely unknown.24,26,27 Here, we show that p53 can be acetylated in vivo on K357, although the functional significance of this modification alone is not yet clear. While our finding that acetylation of K357 increases after 5-FU treatment might seem at odds with our results that 2KQ is impaired in transactivating p53 target genes, we cannot exclude the possibility that modification of K351 negatively regulates p53 target gene expression and is dominant over K357. Indeed, given our transfection data with the tetramerization domain single mutants (Fig. 5D, E), this appears to be the case, at least for p21. It is also possible that there are differential kinetics and outcomes of modification of these residues. Such possibilities await future experimentation and further work is required to elucidate how these and potentially other modifications on lysines 351 and 357 can essentially “flip the molecular switch” of p53 to drive either cell cycle arrest or apoptosis.
Materials & methods
Cell line creation and cell culture
H1299 cells expressing tetracycline (tet) regulated (“tet-off”) WT-p53 were previously described.30,31 Mammalian expression constructs in the pTRE2 backbone (Invitrogen) expressing p53-(K351R/K357R; 2KR) and p53-(K351Q/K351Q; 2KQ) were mutated from the wild-type sequence using the QuikChange protocol (Qiagen) according to the manufacturer's specifications. H1299-derived inducible cell lines expressing 2KR-p53 and 2KQ-p53 were then created using a 2-step tetracycline-regulated system and clonally selected with 400 µg/ml Hygromycin B (Invitrogen) as previously described.31 The cells were grown in Dulbecco's Modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 5 µg/ml tetracycline (Sigma), 100 µg/ml Hygromycin B (BD Biosciences), and 200 µg/ml G418 (Gibco). At least 25 clones were picked and screened for each line and all lines used in this study had their p53 sequence confirmed. Transfection experiments were performed with Lipofectamine 2000 (Invitrogen), according to the manufacturer's protocols.
Immunofluorescence (IF)
Cells were plated without drugs to 90% confluency in a 35-mm dish with a glass coverslip, washed twice 22 hours after seeding, and induced to express p53 (with tetracycline amounts indicated in the figure legends). Twenty four hours after induction, cells were washed twice with PBS followed by incubation with 4% paraformaldehyde (Sigma-Aldrich) for 15 minutes then washed 3 times with PBS, treated with PBS/0.5% Triton X-100 for one and a half minutes and blocked with 0.5% BSA (Sigma-Aldrich) in PBS for 30 minutes prior to the incubation with the antibodies. Samples were incubated with monoclonal antibodies PAb 1801 and PAb DO-1 (50 μL) for one hour at room temperature. The coverslips were then washed 3 times with PBS followed by incubation for another hour with 50 µl of diluted (1:100) secondary Cy5 goat anti-mouse IgG antibody (Jackson ImmunoResearch) and washed 3 times with PBS. DNA staining was performed using 10 nM SYTOX Green nucleic acid dye (Molecular Probes) for 10 minutes. The coverslips were then mounted with 10 µl cold 50% glycerol. The images were collected using confocal laser scanning microscopy (Olympus Model 1X70) with Fluoview software. In order to directly visualize cells and nuclei, Differential Interference Contrast (DIC) images were taken in parallel. For p21 IF, the indicated cell lines were grown in tetracycline-free media for 48 hours before fixation, same as above. They were incubated with PAb p21 (F-5, Santa Cruz Biotechnology) for one hour and then with Alexa Fluor 488 (sc-6246) for one hour. Cover slips were mounted with 20 μl Vectashield Mounting Medium with DAPI (Vector Laboratories, H-1200, Burlingame, CA) and visualized as above.
Creation of acetyl-specific antibodies
Polyclonal antibodies were generated by Cell Signaling Technologies and produced by immunizing animals with synthetic acetylated peptides (KLH-coupled) corresponding to residues surrounding lysine 351 and lysine 357 of human p53. Antibodies were purified using protein A and peptide affinity chromatography.
Protein purification and NMR spectroscopy
N-terminally Flag-tagged WT-p53 and 2KQ-p53 mutant proteins that contain an inserted site at their N-termini were affinity purified as previously described from Sf-9 cells infected with the corresponding baculovirus for p53.81 Constructs coding for p53 residues 310–362 were amplified from WT-p53, 2KQ-p53, and 2KR-p53 mutants respectively. The cDNAs were subcloned into pET32a (EMD Chemicals, Inc.) using EcoRI and XhoI sites. A TEV protease recognition sequence (ENLYFQS) was created between the BamHI and EcoRI sites in pET32, at the C-terminus of Thioredoxin. After TEV protease cleavage, p53 proteins contained Ser-Glu-Phe at their N-termini. For isotopic labeling, proteins were expressed in E. coli Rosetta 2 (DE3), using 0.4 mM IPTG for induction and growth at 23°C for 16 h in modified minimal media using 15NH4Cl and U-13C6-Glucose or U-[13C6,2H7]-Glucose as sole nitrogen and/or carbon sources. Soluble forms of His-tagged proteins were first purified using 5 mL Ni-NTA columns and followed by gel-filtration column chromatography using Hi-Load Superdex200 16/60 (GE Healthcare) with a buffer containing 25 mM sodium phosphate, pH 7.5, 50 mM NaCl, 1 mM DTT, and 0.02 % sodium azide. After removal of Thioredoxin, final purifications of p53 proteins were performed over a Hi-Trap QP column (GE Healthcare) at pH 7.5 using a 0–1 M NaCl gradient. Buffer exchange was carried out using Amicon concentrators (Millipore).
All NMR experiments were conducted at 17°C using protein samples in 25 mM sodium phosphate buffer, pH 7.5, 150 mM NaCl, 2 mM DTT, 0.02% sodium azide and 7% D2O, using a Bruker Avance 700 MHz spectrometer, equipped with 5 mm, triple resonance and z-axis gradient cryoprobes. For backbone chemical shift assignments of WT-p53(310–362), 2D 1H-15N TROSY-HSQC and 3D TROSY-HNCACB and TROSY-HN(CO)CACB experiments were performed on a U-[13C,15N,2H]-labeled protein. The 1H-15N assignments of the 2KQ-p53 and 2KR-p53 mutants were obtained by comparing the 2D 1H-15N TROSY-HSQC spectra of U-[15N]-labeled mutant proteins to that of U-[13C,15N,2H]-labeled wild-type protein.
Immunoblotting analysis
Cells were seeded and induced as for immunofluorescence but on a 60 mm culture dish. Twenty-four hours after induction, cells were harvested and split into pellets for immunoblotting or RT-PCR. Lysis for immunoblotting was performed as previously described.82 p53 protein was visualized by separating 40 µg of whole-cell lysate on a 12% polyacrylamide gel, transferring to nitrocellulose followed by immunoblotting with DO-1 antibody (hybridoma supernatant, 1:1 dilution with 5% milk) for 1 hour at room temperature. Other antibodies used were anti-p21 (C19, Oncogene Research Products), anti-pRb (XZ.131 supernatant), anti-HA (Covance), anti-Flag (Sigma-Aldrich), anti-pan-acetyl lysine (Cell Signaling Technology), anti-p53 Ac382 (a gift from Y. Taya), and anti-actin (Sigma-Aldrich).
Gluteraldehyde crosslinking
H1299 cells were transfected with pCDNA3 vectors expressing WT-p53, or mutants 2KR-p53, or 2KQ-p53 (500 ng). Twenty-four hours later, samples were harvested in TEGN buffer, and gluteraldehyde was added to a final concentration of .005% to .020%. Samples were incubated on ice for 20 minutes, and the crosslinking reaction was stopped by the addition of protein sample buffer. Multimeric complexes were separated by SDS-PAGE and analyzed by Western Blotting.
RT-PCR and qRT-PCR
RNA was extracted from cell pellets using RNeasy Mini Kit (Qiagen) and quantitated by ultraviolet spectrophotometry. cDNA was created from 1 µg of total RNA using the Superscript III First Strand Synthesis System for RT-PCR (Invitrogen), following the protocol for oligo-dT priming. RT-PCR was performed using a dNTP mix (Roche) and Taq 2000 DNA Polymerase (Stratagene). Conditions for linear amplification were established through template and cycle curves. The cycling conditions were as follows: a denaturation step at 95°C for 5 min followed by 20 cycles (for p21, GADPH, and PIG3) or 19 cycles (for Mdm2) at 95°C for 30 sec, 56°C for 30 sec and 70°C for 30 sec, with a final extension of 72°C for 7 min. PCR products were then separated on 2.5% agarose gels and bands were visualized with ethidium bromide and the Gel Logix 100 imaging system (Kodak). qRT-PCR was performed essentially as described.83 Sequences for the primers are available upon request.
Electrophoretic mobility shift assay (EMSA)
WT- p53 and 2KQ-p53 purified proteins from Sf9 cells (0 to 80 ug of total protein) were incubated for 25 minutes at room temperature in 1X EMSA buffer (12.5 mM Tris-HCl pH 6.8, 25 mM KCl, 10% glycerol, 0.05% Triton-X, 0.5 mg/ml BSA, 1 mM DTT, 250 ng mutant p21 oligonucleotide) with 10 ng of a p21 5′ binding site-containing 44 base pair oligonucleotide (5′-AGC TAG TAG AGC GAA TAT ATC CCA ATA TAT TGG CGT GCT GCA GC-3′) or a miR-34a p53 binding site-containing 44- mer (5′-CGG GCT CTG CCT GGG CTT GCC TGG GCT TGT TCC GAG CCG GGC TG-3′), labeled at the 5′ end with IRDye 800. Oligonucleotides were manufactured by Integrated DNA Technologies. The samples were run on a 4% native polyacrylamide gel at 200 Volts. The gel was visualized with a Licor Odyssey apparatus according to the manufacturer's protocols. Oligonucleotide sequences used for EMSA are available upon request.
Chromatin immunoprecipitation (ChIP)
Inducible cells were seeded to a 50% density in 10 cm plates and protein expression was induced for 24 hours as described above. Crosslinking, lysis, sonication, immunoprecipitation, purification, and semi-quantitative or qRT-PCR were performed as previously described.83,85 Primer sequences are available upon request.
Fluorescent-activated cell Sorting (FACS) analysis
For cell cycle analysis, 2 × 105 cells were seeded per 60-mm plate without tetracycline. Twenty-two hours after plating, the cells were washed and tetracycline added as needed to equilibrate protein levels. 0.5 mM 5-fluorouracil (5-FU) was added 24 hours after induction as indicated and 48 hours after that, cells were harvested by trypsinization and fixed overnight with methanol at −20°C as previously described.85 Fixed cells and fragmented DNA were spun down for 5 minutes at 1660 g, resuspended with 1 ml cold PBS, and rehydrated for 30 minutes on ice.86 After another spin, cells were resuspended in 0.5 ml of PBS solution containing RNase (50 µg/ml) and propidium iodide (PI) (60 µg/ml, Sigma), and incubated in the dark for 20 minutes at room temperature. Stained cells were analyzed in a fluorescence-activated cell sorter (FACSCalibur, Becton Dickinson), gating away the debris and aggregates. Cell cycle profiles and apoptotic sub-G1 content were quantitated using the ModFit LT program.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to thank Dr. Ella Freulich for her technical expertise and support, Dr. Masha Poyurovsky for her valuable input on the manuscript, Dr. Song Xiang (now at Shanghai's Institute for Nutritional Sciences) for his assistance with PyMOL, and other members of the Prives lab for helpful discussion and input.
Funding
This work was supported by NIH grant CA77742 and CA87497.
Supplemental Material
Supplemental Material may be downloaded here: publisher's website
References
- [1].Attardi LD. The role of p53-mediated apoptosis as a crucial anti-tumor response to genomic instability: lessons from mouse models. Mutat Res 2005; 569: 145-57; PMID:15603759; http://dx.doi.org/ 10.1016/j.mrfmmm.2004.04.019 [DOI] [PubMed] [Google Scholar]
- [2].Prives C, Hall PA. 1999. The p53 pathway. J Pathol 1999; 187:112-26; PMID:10341712; http://dx.doi.org/ 10.1002/(SICI)1096-9896(199901)187:1%3c112::AID-PATH250%3e3.0.CO;2-3 [DOI] [PubMed] [Google Scholar]
- [3].Ryan KM, Phillips AC, Vousden KH. Regulation and function of the p53 tumor suppressor protein. Curr Opin Cell Biol 2001; 13:332-7; PMID:11343904; http://dx.doi.org/ 10.1016/S0955-0674(00)00216-7 [DOI] [PubMed] [Google Scholar]
- [4].Sengupta S, Harris CC. p53: traffic cop at the crossroads of DNA repair and recombination. Nat Rev Mol Cell Biol 2005; 6:44-55; PMID:15688066; http://dx.doi.org/ 10.1038/nrm1546 [DOI] [PubMed] [Google Scholar]
- [5].Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell 2009; 137:413-31; PMID:19410540; http://dx.doi.org/ 10.1016/j.cell.2009.04.037 [DOI] [PubMed] [Google Scholar]
- [6].Janicke RU, Graupner V, Budach W, Essmann F. The do's and don'ts of p53 isoforms. Biol Chem 2009; 390:951-63; PMID:19453282; http://dx.doi.org/ 10.1515/BC.2009.093 [DOI] [PubMed] [Google Scholar]
- [7].Joerger AC, Fersht AR. Structural biology of the tumor suppressor p53. Annu Rev Biochem 2008; 77:557-82; PMID:18410249; http://dx.doi.org/ 10.1146/annurev.biochem.77.060806.091238 [DOI] [PubMed] [Google Scholar]
- [8].Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer 2009; 9:749-58; PMID:19776744; http://dx.doi.org/ 10.1038/nrc2723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Chene P. The role of tetramerization in p53 function. Oncogene 2001; 20:2611-7; PMID:11420672; http://dx.doi.org/ 10.1038/sj.onc.1204373 [DOI] [PubMed] [Google Scholar]
- [10].Jimenez GS, Khan SH, Stommel JM, Wahl GM. p53 regulation by post-translational modification and nuclear retention in response to diverse stresses. Oncogene 1999; 18:7656-65; PMID:10618705; http://dx.doi.org/ 10.1038/sj.onc.1203013 [DOI] [PubMed] [Google Scholar]
- [11].Lomax ME, Barnes DM, Hupp TR, Picksley SM, Camplejohn RS. 1998. Characterization of p53 oligomerization domain mutations isolated from Li-Fraumeni and Li-Fraumeni like family members. Oncogene 1998; 17:643-9; PMID:9704930; http://dx.doi.org/ 10.1038/sj.onc.1201974 [DOI] [PubMed] [Google Scholar]
- [12].Ribeiro RC, Sandrini F, Figueiredo B, Zambetti GP, Michalkiewicz E, Lafferty AR, DeLacerda L, Rabin M, Cadwell C, Sampaio G, et al. 2001. An inherited p53 mutation that contributes in a tissue-specific manner to pediatric adrenal cortical carcinoma. Proc Natl Acad Sci U S A 2001; 98:9330-5; PMID:11481490; http://dx.doi.org/ 10.1073/pnas.161479898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Muscolini M, Montagni E, Caristi S, Nomura T, Kamada R, Di Agostino S, Corazzari M, Piacentini M, Blandino G, Costanzo A et al.. Characterization of a new cancer-associated mutant of p53 with a missense mutation (K351N) in the tetramerization domain. Cell Cycle 2009; 8(20):3396-405; PMID:19806023; http://dx.doi.org/ 10.4161/cc.8.20.9910 [DOI] [PubMed] [Google Scholar]
- [14].Muscolini M, Montagni E, Palermo V, Di Agostino S, Gu W, Abdelmoula-Souissi S, Mazzoni C, Blandino G, Tuosto L. The cancer-associated K351N mutation affects the ubiquitination and the translocation to mitochondria of p53 protein. J Biol Chem 2011; 286(46):39693-702; PMID:21953469; http://dx.doi.org/ 10.1074/jbc.M111.279539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Stommel JM, Marchenko ND, Jimenez GS, Moll UM, Hope TJ, Wahl GM. A leucine-rich nuclear export signal in the p53 tetramerization domain: regulation of subcellular localization and p53 activity by NES masking. Embo J 1999; 18:1660-72; PMID:10075936; http://dx.doi.org/ 10.1093/emboj/18.6.1660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Sakaguchi K, Sakamoto H, Lewis MS, Anderson CW, Erickson JW, Appella E, Xie D. Phosphorylation of serine 392 stabilizes the tetramer formation of tumor suppressor protein p53. Biochemistry 1997; 36:10117-24; PMID:9254608; http://dx.doi.org/ 10.1021/bi970759w [DOI] [PubMed] [Google Scholar]
- [17].Li AG, Piluso LG, Cai X, Wei G, Sellers WR, Liu X. Mechanistic insights into maintenance of high p53 acetylation by PTEN. Mol Cell 2006; 23:575-87; PMID:16916644; http://dx.doi.org/ 10.1016/j.molcel.2006.06.028 [DOI] [PubMed] [Google Scholar]
- [18].Kawaguchi Y, Ito A, Appella E, Yao TP. Charge modification at multiple C-terminal lysine residues regulates p53 oligomerization and its nucleus-cytoplasm trafficking. J Biol Chem 2006; 281:1394-400; PMID:16291740; http://dx.doi.org/ 10.1074/jbc.M505772200 [DOI] [PubMed] [Google Scholar]
- [19].Appella E, Anderson CW. Post-translational modifications and activation of p53 by genotoxic stresses. Eur J Biochem 2001; 268:2764-72; PMID:11358490; http://dx.doi.org/ 10.1046/j.1432-1327.2001.02225.x [DOI] [PubMed] [Google Scholar]
- [20].Boehme KA, Blattner C. Regulation of p53 - insights into a complex process. Crit Rev Biochem Mol Biol 2009; 44(6):367-92; PMID:19929178; http://dx.doi.org/ 10.3109/10409230903401507 [DOI] [PubMed] [Google Scholar]
- [21].Kruse J-P, Gu W. Modes of p53 Regulation. Cell 2009; 137(4):609-22; PMID:19450511; http://dx.doi.org/ 10.1016/j.cell.2009.04.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Li M, Luo J, Brooks CL, Gu W. Acetylation of p53 inhibits its ubiquitination by Mdm2. J Biol Chem 2002; 277:50607-11; PMID:12421820; http://dx.doi.org/ 10.1074/jbc.C200578200 [DOI] [PubMed] [Google Scholar]
- [23].Jansson M, Durant ST, Cho EC, Sheahan S, Edelmann M, Kessler B, La Thangue NB. Arginine methylation regulates the p53 response. Nat Cell Biol 2008; 10:1431-9; PMID:19011621; http://dx.doi.org/ 10.1038/ncb1802 [DOI] [PubMed] [Google Scholar]
- [24].Joubel A, Chalkley RJ, Medzihradszky KF, Hondermarck H, Burlingame AL. Identification of new p53 acetylation sites in COS-1 cells. Mol Cell Proteomics 2009; 8:1167-73; PMID:19155208; http://dx.doi.org/ 10.1074/mcp.M800487-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kruse JP, Gu W. MSL2 promotes Mdm2-independent cytoplasmic localization of p53. J Biol Chem 2009; 284:3250-63; PMID:19033443; http://1dx.doi.org/ 10.1074/jbc.M805658200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kim W, Bennett EJ, Huttlin EL, Guo A, Li J, Possemato A, Sowa ME, Rad R, Rush J, Comb MJ, et al.. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol Cell 2011; 44(2):325-40; PMID:21906983; http://dx.doi.org/ 10.1016/j.molcel.2011.08.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].DeHart CJ, Chahal JS, Flint SJ, Perlman DH. Extensive post-translational modification of active and inactivated forms of endogenous p53. Mol Cell Proteomics 2014;13(1):1-17; PMID:24056736; http://dx.doi.org/ 10.1074/mcp.M113.030254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Shieh SY, Ahn J, Tamai K, Taya Y, Prives C. The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev 2000; 14:289-300; PMID:10673501 [PMC free article] [PubMed] [Google Scholar]
- [29].Itahana Y, Ke H, Zhang Y. p53 Oligomerization is essential for its C-terminal lysine acetylation. J Biol Chem 2009; 284:5158-64; PMID:19106109; http://dx.doi.org/ 10.1074/jbc.M805696200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Zupnick A, Prives C. Mutational analysis of the p53 core domain L1 loop. J Biol Chem 2006; 281:20464-73; PMID:16687402; http://dx.doi.org/ 10.1074/jbc.M603387200 [DOI] [PubMed] [Google Scholar]
- [31].Chen X, Ko LJ, Jayaraman L, Prives C. p53 levels, functional domains, and DNA damage determine the extent of the apoptotic response of tumor cells. Genes Dev 1996; 10:2438-51; PMID:8843196; http://dx.doi.org/ 10.1101/gad.10.19.2438 [DOI] [PubMed] [Google Scholar]
- [32].Menendez D, Inga A, Resnick MA. The Biological Impact of the Human Master Regulator p53 Can Be Altered by Mutations That Change the Spectrum and Expression of Its Target Genes. Mol Cell Biol 2006; 26:2297-308; PMID:16508005; http://dx.doi.org/ 10.1128/MCB.26.6.2297-2308.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci U S A 1992; 89:5547-51; PMID:1319065; http://dx.doi.org/ 10.1073/pnas.89.12.5547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Gossen M, Freundlieb S, Bender G, Muller G, Hillen W, Bujard H. Transcriptional activation by tetracyclines in mammalian cells. Science 1995; 268(5218):1766-9; PMID:7792603; http://dx.doi.org/ 10.1126/science.7792603 [DOI] [PubMed] [Google Scholar]
- [35].Clore GM, Ernst J, Clubb R, Omichinski JG, Kennedy WM, Sakaguchi K, Appella E, Gronenborn AM. Refined solution structure of the oligomerization domain of the tumour suppressor p53. Nat Struct Biol 1995; 2:321-33; PMID:7796267; http://dx.doi.org/ 10.1038/nsb0495-321. [DOI] [PubMed] [Google Scholar]
- [36].Karni-Schmidt O, Friedler A, Zupnick A, McKinney K, Mattia M, Beckerman R, Bouvet P, Sheetz M, Fersht A, Prives C. Energy-dependent nucleolar localization of p53 in vitro requires two discrete regions within the p53 carboxyl terminus. Oncogene 2007; 26:3878-91; PMID:17237827; http://dx.doi.org/ 10.1038/sj.onc.1210162 [DOI] [PubMed] [Google Scholar]
- [37].Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature 2003; 426 (6968):895-9; PMID:14685250; http://dx.doi.org/ 10.1038/nature02263 [DOI] [PubMed] [Google Scholar]
- [38].Blattner C, Tobiasch E, Litfen M, Rahmsdorf HJ, Herrlich P. DNA damage induced p53 stabilization: no indication for an involvement of p53 phosphorylation. Oncogene 1999; 18:1723-32; PMID:10208433; http://dx.doi.org/ 10.1038/sj.onc.1202480 [DOI] [PubMed] [Google Scholar]
- [39].Haupt Y, Oren M. p53-mediated apoptosis: mechanisms and regulation. Behring Inst Mitt 1996; 97:32-59; PMID:8950466 [PubMed] [Google Scholar]
- [40].Brugarolas J, Chandrasekaran C, Gordon JI, Beach D, Jacks T, Hannon GJ. Radiation-induced cell cycle arrest compromised by p21 deficiency. Nature 1995; 377:552-7; PMID:7566157; http://dx.doi.org/ 10.1038/377552a0 [DOI] [PubMed] [Google Scholar]
- [41].Deng C, Zhang P, Harper JW, Elledge SJ, Leder P. Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell 1995; 82:675-84; PMID:7664346; http://dx.doi.org/ 10.1016/0092-8674(95)90039-X [DOI] [PubMed] [Google Scholar]
- [42].Waldman T, Kinzler KW, Vogelstein B. p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res 1995; 55:5187-90; PMID:7585571 [PubMed] [Google Scholar]
- [43].He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y, Xue W, Zender L, Magnus J, Ridzon D, et al.. A microRNA component of the p53 tumour suppressor network. Nature 2007; 447:1130-4; PMID:17554337; http://dx.doi.org/ 10.1038/nature05939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Tarasov V, Jung P, Verdoodt B, Lodygin D, Epanchintsev A, Menssen A, Meister G, Hermeking H. Differential regulation of microRNAs by p53 revealed by massively parallel sequencing: miR-34a is a p53 target that induces apoptosis and G1-arrest. Cell Cycle 2007; 6:1586-93; PMID:17554199; http://dx.doi.org/ 10.4161/cc.6.13.4436 [DOI] [PubMed] [Google Scholar]
- [45].Zhao L, Samuels T, Winckler S, Korgaonkar C, Tompkins V, Horne MC, Quelle DE. Cyclin G1 has growth inhibitory activity linked to the ARF-Mdm2-p53 and pRb tumor suppressor pathways. Mol Cancer Res 2003; 1:195-206; PMID:12556559 [PubMed] [Google Scholar]
- [46].Laptenko O, Prives C. Transcriptional regulation by p53: one protein, many possibilities. Cell Death Differ 2006; 13:951-61; PMID:16575405; http://dx.doi.org/ 10.1038/sj.cdd.4401916 [DOI] [PubMed] [Google Scholar]
- [47].Anderson ME, Woelker B, Reed M, Wang P, Tegtmeyer P. Reciprocal interference between the sequence-specific core and nonspecific C-terminal DNA binding domains of p53: implications for regulation. Mol Cell Biol 1997; 17:6255-64; PMID:9343386; http://dx.doi.org/ 10.1128/MCB.17.11.6255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].McKinney K, Mattia M, Gottifredi V, Prives C. p53 linear diffusion along DNA requires its C terminus. Mol Cell 2004; 16:413-24; PMID:15525514; http://dx.doi.org/ 10.1016/j.molcel.2004.09.032 [DOI] [PubMed] [Google Scholar]
- [49].Kaeser MD, Iggo RD. Chromatin immunoprecipitation analysis fails to support the latency model for regulation of p53 DNA binding activity in vivo. Proc Natl Acad Sci U S A 2002; 99:95-100; PMID:11756653; http://dx.doi.org/ 10.1073/pnas.012283399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Tang Y, Luo J, Zhang W, Gu W. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol Cell 2006; 24:827-39; PMID:17189186; http://dx.doi.org/ 10.1016/j.molcel.2006.11.021 [DOI] [PubMed] [Google Scholar]
- [51].Lavin MF, Gueven N. The complexity of p53 stabilization and activation. Cell Death Differ 2006; 13:941-50; PMID:16601750; http://dx.doi.org/ 10.1038/sj.cdd.4401925 [DOI] [PubMed] [Google Scholar]
- [52].Atz J, Wagner P, Roemer K. Function, oligomerization, and conformation of tumor-associated p53 proteins with mutated C-terminus. J Cell Biochem 2000; 76:572-84; PMID:10653977; http://dx.doi.org/ 10.1002/(SICI)1097-4644(20000315)76:4%3c572::AID-JCB6%3e3.0.CO;2-6 [DOI] [PubMed] [Google Scholar]
- [53].Kulikov R, Winter M, Blattner C. Binding of p53 to the Central Domain of Mdm2 Is Regulated by Phosphorylation. J Biol Chem 2006; 281:28575-83; PMID:16870621; http://dx.doi.org/ 10.1074/jbc.M513311200 [DOI] [PubMed] [Google Scholar]
- [54].Christophorou MA, Ringshausen I, Finch AJ, Swigart LB, Evan GI. The pathological response to DNA damage does not contribute to p53-mediated tumour suppression. Nature 2006; 443:214-7; PMID:16957739; http://dx.doi.org/ 10.1038/nature05077 [DOI] [PubMed] [Google Scholar]
- [55].Liu G, Parant JM, Lang G, Chau P, Chavez-Reyes A, El-Naggar AK, Multani A, Chang S, Lozano G. Chromosome stability, in the absence of apoptosis, is critical for suppression of tumorigenesis in Trp53 mutant mice. Nat Genet 2004; 36:63-8; PMID:14702042; http://dx.doi.org/ 10.1038/ng1282 [DOI] [PubMed] [Google Scholar]
- [56].Saller E, Tom E, Brunori M, Otter M, Estreicher A, Mack DH, Iggo R. Increased apoptosis induction by 121F mutant p53. Embo J 1999; 18:4424-37; PMID:10449408; http://dx.doi.org/ 10.1093/emboj/18.16.4424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Nakamura Y, Futamura M, Kamino H, Yoshida K, Nakamura Y, Arakawa H. Identification of p53-46F as a super p53 with an enhanced ability to induce p53-dependent apoptosis. Cancer Sci 2006; 97:633-41; PMID:16827804; http://dx.doi.org/ 10.1111/j.1349-7006.2006.00214.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Pan Y, Haines DS. Identification of a tumor-derived p53 mutant with novel transactivating selectivity. Oncogene 2000; 19:3095-100; PMID:10871862; http://dx.doi.org/ 10.1038/sj.onc.1203663 [DOI] [PubMed] [Google Scholar]
- [59].Ludwig RL, Bates S, Vousden KH. Differential activation of target cellular promoters by p53 mutants with impaired apoptotic function. Mol Cell Biol 1996; 16:4952-60; PMID:8756654; http://dx.doi.org/ 10.1128/MCB.16.9.4952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Friedlander P, Haupt Y, Prives C, Oren M. A mutant p53 that discriminates between p53-responsive genes cannot induce apoptosis. Mol Cell Biol 1996; 16:4961-71; PMID:8756655; http://dx.doi.org/ 10.1128/MCB.16.9.4961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Kong XT, Gao H, Stanbridge EJ. Mechanisms of differential activation of target gene promoters by p53 hinge domain mutants with impaired apoptotic function. J Biol Chem 2001; 276:32990-3000; PMID:11395510; http://dx.doi.org/ 10.1074/jbc.M103681200 [DOI] [PubMed] [Google Scholar]
- [62].Resnick MA, Inga A. Functional mutants of the sequence-specific transcription factor p53 and implications for master genes of diversity. Proc Natl Acad Sci USA 2003; 100:9934-9; PMID:12909720; http://dx.doi.org/ 10.1073/pnas.1633803100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Li X, Dumont P, Della Pietra A, Shetler C, Murphy ME. The codon 47 polymorphism in p53 is functionally significant. J Biol Chem 2005; 280:24245-51; PMID:15851479; http://dx.doi.org/ 10.1074/jbc.M414637200 [DOI] [PubMed] [Google Scholar]
- [64].Baptiste N, Prives C. p53 in the cytoplasm: a question of overkill? Cell 2004; 116:487-9; PMID:14980216; http://dx.doi.org/ 10.1016/S0092-8674(04)00164-3 [DOI] [PubMed] [Google Scholar]
- [65].Bergamaschi D, Samuels Y, Sullivan A, Zvelebil M, Breyssens H, Bisso A, Del Sal G, Syed N, Smith P, Gasco M, Crook T, Lu X. iASPP preferentially binds p53 proline-rich region and modulates apoptotic function of codon 72-polymorphic p53. Nat Genet 2006; 38:1133-1141; PMID:16964264; http://dx.doi.org/ 10.1038/ng1879 [DOI] [PubMed] [Google Scholar]
- [66].Samuels-Lev Y, O'Connor DJ, Bergamaschi D, Trigiante G, Hsieh JK, Zhong S, Campargue I, Naumovski L, Crook T, Lu X. ASPP proteins specifically stimulate the apoptotic function of p53. Mol Cell 2001; 8:781-94; PMID:11684014; http://dx.doi.org/ 10.1016/S1097-2765(01)00367-7 [DOI] [PubMed] [Google Scholar]
- [67].D'Orazi G, Cecchinelli B, Bruno T, Manni I, Higashimoto Y, Saito S, Gostissa M, Coen S, Marchetti A, Del Sal G, et al.. Homeodomain-interacting protein kinase-2 phosphorylates p53 at Ser 46 and mediates apoptosis. Nat Cell Biol 2002; 4:11-9; PMID:11780126; http://dx.doi.org/ 10.1038/ncb714 [DOI] [PubMed] [Google Scholar]
- [68].Okamura S, Arakawa H, Tanaka T, Nakanishi H, Ng CC, Taya Y, Monden M, Nakamura Y. p53DINP1, a p53-inducible gene, regulates p53-dependent apoptosis. Mol Cell 2001; 8:85-94; PMID:11511362; http://dx.doi.org/ 10.1016/S1097-2765(01)00284-2 [DOI] [PubMed] [Google Scholar]
- [69].Yoshida K, Liu H, Miki Y. Protein kinase C delta regulates Ser46 phosphorylation of p53 tumor suppressor in the apoptotic response to DNA damage. J Biol Chem 2006; 281:5734-40; PMID:16377624; http://dx.doi.org/ 10.1074/jbc.M512074200 [DOI] [PubMed] [Google Scholar]
- [70].Weinberg RL, Veprintsev DB, Bycroft M, Fersht AR. Comparative binding of p53 to its promoter and DNA recognition elements. J Mol Biol 2005; 348:589-96; PMID:15826656; http://dx.doi.org/ 10.1016/j.jmb.2005.03.014 [DOI] [PubMed] [Google Scholar]
- [71].Timofeev O, Schlereth K, Wanzel M, Braun A, Nieswandt B, Pagenstecher A, Rosenwald A, Elsässer HP, Stiewe T. p53 DNA binding cooperativity is essential for apoptosis and tumor suppression in vivo. Cell Rep 2013; 3(5):1512-25; PMID:23665223; http://dx.doi.org/ 10.1016/j.celrep.2013.04.008 [DOI] [PubMed] [Google Scholar]
- [72].Rowan S, Ludwig RL, Haupt Y, Bates S, Lu X, et al.. Specific loss of apoptotic but not cell-cycle arrest function in a human tumor derived p53 mutant. EMBO J. 1996; 15(4):827-38; PMID:8631304 [PMC free article] [PubMed] [Google Scholar]
- [73].Sykes SM, Mellert HS, Holbert MA, Li K, Marmorstein R, et al.. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol Cell 2006; 24(6):841-51; PMID:17189187; http://dx.doi.org/ 10.1016/j.molcel.2006.11.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Korotchkina LG, Leontieva OV, Bukreeva EI, Demidenko ZN, Gudkov AV, et al.. The choice between p53-induced senescence and quiescence is determined in part by the mTOR pathway. Aging 2010; 2(6):344-52; PMID:20606252; http://dx.doi.org/ 10.18632/aging.100160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Blagosklonny MV. Geroconversion: irreversible step to cellular senescence. Cell Cycle 2014; 13(23):3628-35; PMID:25483060; http://dx.doi.org/ 10.4161/15384101.2014.985507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Okamoto K, Beach D. Cyclin G is a transcriptional target of the p53 tumor suppressor protein. EMBO J 1994; 13:4816-22; PMID:7957050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Ho JS, Ma W, Mao DY, Benchimol S. p53-Dependent transcriptional repression of c-myc is required for G1 cell cycle arrest. Mol Cell Biol 2005; 25:7423-31; PMID:16107691; http://dx.doi.org/ 10.1128/MCB.25.17.7423-7431.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Pelengaris S, Khan M, Evan G. c-MYC: more than just a matter of life and death. Nat Rev Cancer 2002; 2:764-76; PMID:12360279; http://dx.doi.org/ 10.1038/nrc904 [DOI] [PubMed] [Google Scholar]
- [79].Mitchell KO, El-Deiry WS. Overexpression of c-Myc inhibits p21WAF1/CIP1 expression and induces S-phase entry in 12-O-tetradecanoylphorbol-13-acetate (TPA)-sensitive human cancer cells. Cell Growth Differ 1999; 10:223-30; PMID:10319992 [PubMed] [Google Scholar]
- [80].Marhin WW, Chen S, Facchini LM, Fornace AJ Jr, Penn LZ. Myc represses the growth arrest gene gadd45. Oncogene 1997; 14:2825-34; PMID:9190899; http://dx.doi.org/ 10.1038/sj.onc.1201138 [DOI] [PubMed] [Google Scholar]
- [81].Piluso LG, Wei G, Li AG, Liu X. Purification of acetyl-p53 using p300 co-infection and the baculovirus expression system. Protein Expr Purif 2005; 40:370-8; PMID:15766879; http://dx.doi.org/ 10.1016/j.pep.2004.12.015 [DOI] [PubMed] [Google Scholar]
- [82].Urist M, Tanaka T, Poyurovsky MV, Prives C. p73 induction after DNA damage is regulated by checkpoint kinases Chk1 and Chk2. Genes Dev 2004; 18:3041-54; PMID:15601819; http://dx.doi.org/ 10.1101/gad.1221004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Beckerman R, Donner AJ, Mattia M, Peart MJ, Manley JL, Espinosa JM, Prives C. A role for Chk1 in blocking transcriptional elongation of p21 RNA during the S-phase checkpoint. Genes Dev 2009; 23:1364-77; PMID:19487575; http://dx.doi.org/ 10.1101/gad.1795709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Mattia M, Gottifredi V, McKinney K, and Prives C. p53-Dependent p21 mRNA elongation is impaired when DNA replication is stalled. Mol Cell Biol 2007; 27:1309-20; PMID:17158927; http://dx.doi.org/ 10.1128/MCB.01520-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Baptiste N, Friedlander P, Chen X, Prives C. The proline-rich domain of p53 is required for cooperation with anti-neoplastic agents to promote apoptosis of tumor cells. Oncogene 2002; 21:9-21; PMID:11791172; http://dx.doi.org/ 10.1038/sj.onc.1205015 [DOI] [PubMed] [Google Scholar]
- [86].Berger M, Haupt Y. Flow cytometric analysis of p53-induced apoptosis. Methods Mol Biol 2003; 234:245-56; PMID:12824537 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.