

ABSTRACT
Aneuploidy is a common feature of human solid tumors and is often associated with poor prognosis. There is growing evidence that oncogenic signaling pathways, which are universally dysregulated in cancer, contribute to the promotion of aneuploidy. However, the mechanisms connecting signaling pathways to the execution of mitosis and cytokinesis are not well understood. Here, we show that hyperactivation of the ERK1/2 MAP kinase pathway in epithelial cells impairs cytokinesis, leading to polyploidization and aneuploidy. Mechanistically, deregulated ERK1/2 signaling specifically downregulates expression of the F-box protein Fbxw7β, a substrate-binding subunit of the SCFFbxw7 ubiquitin ligase, resulting in the accumulation of the mitotic kinase Aurora A. Reduction of Aurora A levels by RNA interference or pharmacological inhibition of MEK1/2 reverts the defect in cytokinesis and decreases the frequency of abnormal cell divisions induced by oncogenic H-RasV12. Reciprocally, overexpression of Aurora A or silencing of Fbxw7β phenocopies the effect of H-RasV12 on cell division. In vivo, conditional activation of MEK2 in the mouse intestine lowers Fbxw7β expression, resulting in the accumulation of cells with enlarged nuclei. We propose that the ERK1/2/ Fbxw7β/Aurora A axis identified in this study contributes to genomic instability and tumor progression.
KEYWORDS: Aneuploidy, Aurora A, cytokinesis, ERK1/2 MAP kinases, Fbxw7
Introduction
Aneuploidy is a very common feature of cancer, being present in 90% of human solid tumors and consistently associated with a worse prognosis.1,2 Despite its largely detrimental effect on cellular fitness, aneuploidy is strongly associated with cancer, possibly because of its ability to generate the phenotypic diversity necessary to adapt to the stringent selection conditions observed during in vivo tumor development.1,3,4 Aneuploidy also promotes additional genomic instability by itself leading to both numerical and structural chromosomal alterations.5-7
One proposed route to aneuploidy is through generation of an unstable tetraploid intermediate state.8,9 These tetraploid cells arise mainly from 3 mechanisms: cell fusion, endoreduplication, and cytokinesis failure or premature exit from mitosis. Consistent with an initiator role of tetraploidy in aneuploidy and tumorigenesis, cells with high chromosome numbers are frequently observed in early-stage cancers and many tumor cells exhibit a bimodal distribution of chromosome numbers with a near-tetraploid peak.8 Experimentally, tetraploid fibroblast or epithelial cells generate tumors in mice that grow much faster than their diploid counterparts.10-12 Tetraploidization may help tolerate the genetic imbalance resulting from chromosomal instability (CIN) and aneuploidy to promote transformation.3
The mechanisms that cause tetraploidy and aneuploidy are not clear but accumulating evidence points to a role of oncogenic signaling pathways.13 Specifically, hyperactive Ras signaling has been implicated in the induction of CIN but the precise molecular mechanisms involved remain unknown.13,14 We have recently reported that oncogenic Ras or sustained nuclear MEK/ERK1/2 signaling induces tetraploidization of epithelial cells.10 Here, we investigated the molecular basis of this oncogenic response. We now show that hyperactivation of ERK1/2 MAP kinases (MAPKs) specifically downregulates the F-box protein isoform Fbxw7β, resulting in the accumulation of Aurora A, cytokinesis failure and polyploidization. Transgenic expression of activated MEK2 in mouse intestinal epithelial cells similarly decreases Fbxw7β levels, concomitantly to the accumulation of cells with enlarged nuclei, indicative of polyploidy. Our results link the activation of a common oncogenic signaling pathway to the promotion of aneuploidy.
Results
Activated MEK1DD and H-RasV12 induce cytokinesis defects leading to polyploidization
To study the mechanisms underlying activated Ras or MEK-induced tetraploidization, we analyzed the cell cycle kinetics of asynchronously proliferating intestinal epithelial IEC-6 cells expressing H-RasV12 or MEK1DD (Fig. 1A). FACS analysis of phospho-histone H3 staining revealed an increased proportion of IEC-6-H-RasV12 and IEC-6-MEK1DD cells in late G2/M phase (Fig. 1B), consistent with impairment in mitotic progression or cytokinesis. To carefully analyze progression through mitosis, IEC-6 cell populations were transduced with GFP-histone H2B and imaged by time-lapse digital microscopy. Mitotic events were timed to score defects in chromosome movements, anaphase progression and cytokinesis. The duration of mitosis (nuclear envelope breakdown (NEBD) to complete ingression of the cytokinetic furrow) was unaffected by the expression of H-RasV12 or MEK1DD (Fig. 1C and 1D). Yet, a significant fraction of IEC-6-H-RasV12 and IEC-6-MEK1DD cells were binucleated (26.1% and 26.3% vs none detected in control cells), indicative of a failure in cytokinesis (Fig. S1). Cytokinetic furrow initiation and ingression occurred with normal kinetics, but we observed several regression/ingression cycles in H-RasV12 and MEK1DD-expressing cells (Fig. S1A and S1B), suggesting that binucleation arises from a defect in abscission.15 In support of this idea, these cells often exhibited long cytoplasmic bridges and cytokinesis lasted more than 8 times longer than in control cells (Fig. 1C, 1D, S1A and S1B).
Figure 1.

Activated H-RasV12 or MEK1DD induce cytokinesis defects. IEC-6 cells were infected with empty vector, MEK1DD or H-RasV12 and analyzed 2 weeks post-infection. (A) Immunoblot analysis of proliferating IEC-6 cell populations (n=4). (B) Flow cytometry analysis of phospho-histone H3 (p-H3) expression. Results are expressed as mean ± SEM (n = 3). (C) Timing of mitotic progression revealed by time-lapse video imaging. Mean ± SEM of 44 vector, 80 MEK1DD and 92 H-RasV12-expressing IEC-6 cells 2 weeks post-infection. (D) Time-lapse video imaging of representative mitotic progression of IEC-6 cell populations expressing GFP-tagged histone H2B. Scale bar, 5 μm. Arrow, intercellular bridge. ***, P < 0.005.
Aurora A is a critical mediator of H-RasV12-induced cytokinesis failure
The mitotic kinase Aurora A is frequently overexpressed in human cancer, and its overexpression leads to cytokinesis failure, tetraploidization and genetic instability in cultured cells and mouse models.16,17 We thus sought to evaluate the role of Aurora A in H-RasV12-induced cytokinesis failure. Expression of H-RasV12 or MEK1DD in IEC-6 cells resulted in a marked up-regulation of Aurora A (Fig. 2A). Similarly to oncogenic Ras, overexpression of Aurora A was sufficient to cause cytokinesis failure and impair cell cycle progression of IEC-6 cells (Fig. 2B-F). Higher upregulation of Aurora A levels also resulted in multipolar spindles (Fig. 2F).
Figure 2.

Overexpression of Aurora A impairs cytokinesis and cell division. (A) Immunoblot analysis of Aurora A in proliferating IEC-6 cells infected with vector, MEK1DD or H-RasV12 (n = 3). (B to F) IEC-6 cells were infected with empty vector or Aurora A-encoding retrovirus and cell populations expressing moderate or high levels of Aurora A were selected for functional analysis. (B) Immunoblot analysis of Aurora A expression (n = 4). (C) Flow cytometry analysis of p-H3 expression. Results are expressed as mean ± SEM (n = 3). (D) Time-lapse video imaging of mitotic progression of IEC-6-H-RasV12 cells transduced with GFP-histone H2B and moderately overexpressing Aurora A. Scale bar, 5 μm. Arrow, intercellular bridge. (E) Timing of mitotic progression of control (Vector) IEC-6 cells (n = 44) or IEC-6 cells expressing moderate (n = 80) or high (n = 116) levels of Aurora A at 2 weeks post-infection. (F) Quantification of cell division defects. (G to I) IEC-6-H-RasV12 cells were infected or not with lentiviruses encoding non-target shRNA (shNT) or the indicated Aurora A shRNAs (shAurA). (G) Immunoblot analysis of Aurora A expression (n = 3). (H) Time-lapse video imaging of mitotic progression of IEC-6-H-RasV12 cells transduced with GFP-histone H2B and infected with Aurora A shRNA. (I) Quantification of cell division defects. ***, P < 0.005.
To directly test the contribution of Aurora A to H-RasV12-induced cytokinesis failure, we down-regulated the expression of Aurora A to close-to-normal levels using lentivirus-transduced Aurka shRNAs (Fig. 2G). Complete depletion of Aurora A impairs mitotic entry and leads to multiple mitosis defects 18 (unpublished observations). Reduction of Aurora A levels with 2 independent shRNAs reverted the defect in cytokinesis timing induced by H-RasV12, and markedly decreased the frequency of binucleated cells and cells exhibiting long cytoplasmic bridge, furrow regression and tripolar division (Fig. 2H and 2I). Overall, these results suggest that Aurora A is a key effector of oncogenic H-RasV12-mediated tetraploidization and aneuploidy.
Oncogenic H-RasV12 induces polyploidization through ERK1/2 MAPK signaling
Expression of activated MEK1DD up-regulates Aurora A levels (Fig. 2A) and induces cytokinesis failure (Fig. 1B-D). To assess the specific contribution of the ERK1/2 MAPK signaling branch to H-RasV12-induced polyploidy, IEC-6-H-RasV12 cells were treated with a low dose of the selective MEK1/2 inhibitor, PD184352. High doses of this compound induce G1 arrest. Treatment with 1 µM PD184352, which restrains ERK1/2 hyperactivation, decreased the levels of Aurora A to levels seen in non-transformed cells (Fig. 3A). Importantly, PD184352 markedly reduced the length of cytokinesis and the frequency of abnormal cell divisions (furrow regression, binucleation, multipolar spindles) of H-RasV12-expressing cells (Fig. 3B and 3C). Consistent with these observations, long term treatment of IEC-6-H-RasV12 cells with PD184352 significantly decreased the proportion of G2/M and polyploid/aneuploid cells (Fig. 3D-F). Thus, oncogenic H-RasV12 induces cytokinesis defects, polyploidization and aneuploidy largely through ERK1/2 signaling.
Figure 3.

H-RasV12-induced cytokinesis failure is mediated by the ERK1/2 pathway. (A) Immunoblot analysis of proliferating IEC-6-H-RasV12 cells treated with vehicle or 1 μM PD184352 (n = 4). (B) Timing of mitotic progression of IEC-6-H-RasV12 cells treated with vehicle (n = 84) or PD184352 (n = 82). Results are expressed as mean ± SEM. (C) Quantification of cell division defects. (D) Representative cell cycle profiles of IEC-6-H-RasV12 cells treated or not with PD184352 (n = 3). (E) Proportion of cells in each cell cycle phase (n = 3). (F) Proportion of cells with > 4N DNA content 7 d after infection of IEC-6 cells with H-RasV12 and treatment with vehicle or PD184352 (n = 3). ns, non significant; **, P < 0.01; ***, P < 0.005.
Hyperactivation of the ERK1/2 pathway downregulates Fbxw7β expression
We next investigated the molecular mechanism by which deregulated ERK1/2 signaling up-regulates Aurora A expression. The abundance of Aurora A is controlled at the level of transcription and protein turnover.17,19 Specifically, Aurora A is targeted for degradation at mitotic exit by the anaphase-promoting complex/cyclosome (APC/C) and co-activator Cdh1.20-22 Expression of H-RasV12 in Cdh1GT/GT MEFs further up-regulated the levels of Aurora A (Fig. S2A) and significantly increased the frequency of polyploid/aneuploid cells as compared to empty vector (Fig. S2B and S2C). These results argue that mechanisms other than Cdh1 inactivation contribute to Aurora A accumulation.
The SCFFbxw7 ubiquitin ligase also promotes Aurora A ubiquitination and degradation.23-25 We thus examined the effect of H-RasV12 or MEK1DD on the expression of Fbxw7 isoforms in IEC-6 cells. No change in expression of Fbxw7α was observed, but the levels of Fbxw7β mRNA and protein were markedly downregulated in cells with activated Ras or MEK1 (Fig. 4A and 4B). Actinomycin D-chase experiments revealed no effect on Fbxw7β mRNA stability, suggesting that regulation takes place predominantly at the transcriptional level (Fig. 4C). Treatment of IEC-6-H-RasV12 cells with PD184352 rescued the expression of Fbxw7β, concomitant to the reduction of Aurora A levels (Fig. 4D and 4E). These findings were generalized to human MCF10A mammary epithelial cells expressing H-RasV12 (Fig. S3).
Figure 4.

Hyperactivation of ERK1/2 signaling downregulates Fbxw7β expression. (A) Quantitative PCR analysis of Fbxw7 isoforms mRNA levels in IEC-6 cells infected with vector, MEK1DD or H-RasV12. Results are expressed as mean ± SEM (n = 3). (B) Immunoblot analysis of Fbwx7 isoforms protein expression (n = 3). (C) Actinomycin D-chase analysis of Fbxw7β mRNA stability in IEC-6 cells infected with vector, MEK1DD or H-RasV12. Each point corresponds to the mean ± SEM of 3 individual experiments. (D) Effect of PD184352 on mRNA levels of Fbxw7 isoforms in IEC-6-H-RasV12 cells. Mean ± SEM (n = 3). (E) Effect of PD184352 on protein levels of Fbxw7β and Aurora A in IEC-6-H-RasV12 cells (n = 3). ***, P < 0.005.
To address the physiopathological relevance of these cellular studies, we generated a bitransgenic mouse model conditionally expressing activated MEK2DD in the intestine under control of a doxycline-inducible system (Fig. S4). Induction of MEK2DD and ensuing hyperactivation of ERK1/2 signaling resulted in decreased expression of Fbxw7β in vivo (Fig. 5A), concomitant to an increase in the frequency of Ki-67-positive cells (Fig. 5B) and phospho-histone H3-positive cells (Fig. 5C) in colonic crypts. Notably, the average nucleus size was enlarged (with many nuclei exhibiting atypical morphology) in colon sections of Villin-rtTA;MEK2DD mice, a finding consistent with the presence of polyploid/aneuploid cells (Fig. 5D).
Figure 5.
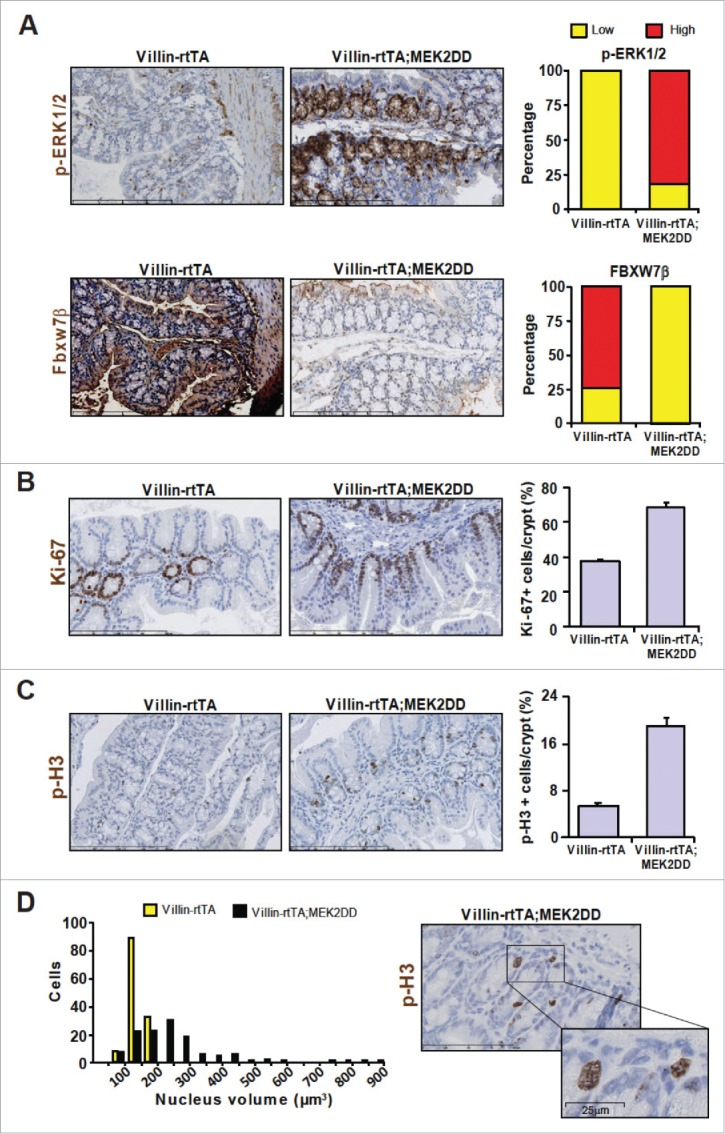
ERK1/2 activity inversely correlates with Fbxw7β expression in MEK2DD transgenic mice. (A) Immunohistochemistry staining for phospho-ERK1/2 and Fbxw7β in colon sections from control Villin-rtTA and Villin-rtTA;MEK2DD mice treated with doxycycline for 10 d. Right, visual quantification of phospho-ERK1/2 and Fbxw7β staining in control (n = 4) and MEK2DD (n = 11) mice. (B) Ki-67 staining of colon sections. Right, quantification of Ki-67-positive cells per crypt. Data represent the mean ± SEM of 150 crypts, 3 mice/group. (C) Phospho-H3 staining of colon sections. Right, quantification of phospho-H3-positive cells per crypt. Mean ± SEM of 150 crypts, 3 mice/group. (D) Distribution of nuclear size of colonic epithelial cells from Villin-rtTA and Villin-rtTA;MEK2DD mice. Nuclear volume was measured on phospho-histone H3 positive crypt cells. n = 150 cells per genotype.
Fbxw7β isoform regulates Aurora A levels and the accuracy of cell division
To specifically assess the impact of Fbxw7 on Aurora A expression and aneuploidy, the expression of Fbxw7 isoforms was silenced by RNAi in IEC-6 cells. Depletion of Fbxw7β using 2 distinct siRNAs caused the upregulation of Aurora A expression, associated with an increase in phospho-histone H3 levels (Fig. 6A). Silencing of Fbxw7α isoform had no detectable effect. Cycloheximide-chase experiments revealed that depletion of Fbxw7β increases the half-life of Aurora A from 1h43 ± 10 min to 6 h 45 ± 18 min in IEC-6 cells, consistent with a direct effect on protein turnover (Fig. 6B). Functionally, knock-down of Fbxw7β expression increased the fraction of IEC-6 cells in G2/M phase (Fig. 6C and S5) and led to the accumulation of cells with >4N DNA content (Fig. 6D). Fbxw7α depletion also resulted in the accumulation of cells with >4N DNA, without inducing the accumulation of phospho-histone H3-positive cells (Fig. 6C and 6D). Fbxw7α is known to be a major regulator of cyclin E stability.26,27 Ectopic expression of cyclin E impairs S phase progression, leading to polyploidy and CIN.28,29 In agreement with these observations, cell cycle analysis revealed that IEC-6 cells depleted of Fbxw7α accumulate in S phase, with no significant change in the proportion of G2/M phase cells (Fig. S5).
Figure 6.

Fbxw7β isoform regulates Aurora A levels and the accuracy of cell division. (A) Immunoblot analysis of proliferating IEC-6 cells transfected with siRNAs targeted to Fbxw7α or Fbxw7β isoforms (n = 4). (B) Cycloheximide-chase analysis of Aurora A stability. IEC-6 cells were either left untreated or transfected with Fbxw7β siRNA (B2) or infected with H-RasV12. After 72 h, cycloheximide (50 μg/ml) (CHX) was added and the cells were harvested at the indicated times. Aurora A expression was monitored by immunoblotting. Right, quantification of 3 experiments (mean ± SEM). (C and D) Proportion of cells expressing p-H3 (C) or having >4N DNA content (D) 72 h after transfection of IEC-6 cells with Fbxw7α or Fbxw7β siRNAs. Results are expressed as mean ± SEM (n = 4). (E) Immunoblot analysis of Aurora A in proliferating IEC-6 cells infected with retrovirus-encoding Fbxw7α or Fbxw7β (n = 3).
Similarly, depletion of Fbxw7β in human MCF10A cells using validated siRNAs 27 targeting either the specific 5′ exon of β isoform or the common exon 10 of FBXW7 gene upregulated Aurora A expression, while depletion of α or γ isoform had no significant effect (Fig. S6A and S6B). Silencing of Fbxw7β in MCF10A cells also increased the proportion of cells in G2/M and the frequency of polyploid/aneuploid cells (Fig. S6C-F). Reciprocally, overexpression of Fbxw7β isoform, but not Fbxw7α, in IEC-6-H-RasV12 cells was sufficient to down-regulate Aurora A expression (Fig. 6E). These results identify Fbxw7β isoform as a specific regulator of Aurora A.
Discussion
Aneuploidy is a hallmark of human solid tumors. Paradoxically, large-scale genomics studies have revealed that mutations in genes encoding mitotic regulators are very rare, arguing that changes in the expression or activity of mitotic proteins likely account for the high prevalence of chromosome segregation defects observed in tumors. There is growing evidence that oncogenic signaling pathways, which are universally dysregulated in cancer, not only drive unrestrained cell proliferation and resistance to apoptosis, but also contribute to CIN and aneuploidy.13 However, the precise mechanisms by which oncogenic signaling events alter the function of mitosis or cytokinesis regulators remain poorly understood. We have shown previously that hyperactivation of the ERK1/2 MAPK pathway induces tetraploidization of intestinal epithelial cells, enhancing their tumorigenic potential.10 Our study now provides mechanistic understanding into how this oncogenic signaling pathway impacts on the regulatory machinery of cell division. We found that aberrant ERK1/2 signaling specifically downregulates the F-box protein Fbxw7β, resulting in the accumulation of the mitotic kinase Aurora A and cytokinesis failure.
Activating mutations in RAS genes occur in ∼30% of cancers and are often acquired early in the tumorigenic process.30,31 Several observations suggest that oncogenic Ras signaling may contribute to cancer progression by promoting genomic instability.13,14 Overexpression of oncogenic Ras mutants in various model cell lines was shown to promote chromosome mis-segregation, centrosome amplification, ploidy changes, and induction of CIN. Oncogenic Ras also increases aneuploidization in mice.14,32 How Ras signaling links to CIN and aneuploidy has remained largely unclear. Early work showed that treatment with MEK1/2 inhibitor PD98059 decreases the frequency of Ras-induced micronuclei in thyroid PCCL3 cells, pointing to the involvement of the ERK1/2 signaling branch.33 We show here that hyperactivation of ERK1/2 by H-RasV12 is both necessary and sufficient to impair cytokinesis execution, leading to polyploidization. Importantly, we have identified the mitotic kinase Aurora A as a key effector of this response, providing a mechanistic link between Ras signaling and the cell division machinery. The Aurora A kinase controls multiple steps of the cell division cycle, including centrosome maturation, mitosis entry, bipolar spindle formation, and cytokinesis.16,17 Its overexpression induces cytokinesis failure and tetraploidization 34-36; this study). Aurora A is frequently overexpressed in human cancers as a result of gene amplification, but also by other mechanisms such as transcriptional activation and protein stabilization by upregulation of interacting proteins.16,17 Our identification of the ERK1/2 pathway as an upstream regulator of Aurora A turnover adds an additional layer of complexity to Aurora A regulation with potentially important implications for cancer biology.
The destruction of Aurora A protein is controlled by multiple ubiquitin ligases, including SCFbxw7.17,23-25 The SCFbxw7 ligase is a tumor suppressor protein that targets multiple oncoproteins for ubiquitylation and degradation through its substrate-binding subunit Fbxw7.37,38 The FBXW7 gene produces 3 mRNA transcripts, Fbxw7α, β and γ, which consist of a specific first exon linked to 10 shared exons, allowing for isoform-specific transcriptional control.39 Much remain to be learned about the isoform-specific regulation and substrates of the 3 Fbxw7 isoforms. Here, we uncovered a unique mechanism of regulation of SCFbxw7 by hyperactive ERK1/2 signaling. We found that activated Ras or MEK markedly downregulates Fbxw7β mRNA and protein expression in epithelial cells without affecting Fbxw7α isoform. This effect is reversed by treatment with the MEK1/2 inhibitor PD184352. Acute expression of activated MEK2 in transgenic mouse intestinal cells also decreases the levels of Fbxw7β protein. FBXW7 is mutated in 11% of non-hypermutated colorectal carcinomas.40 However, genetic alterations in the Ras-ERK1/2 pathway are found in 55% of these tumors, suggesting that impairment of Fbxw7 function may be more prevalent than predicted by genomic analysis.
Specific downregulation of Fbxw7β isoform by H-RasV12 signaling or RNAi silencing caused the accumulation of Aurora A by enhancing its stability. Reciprocally, pharmacological inhibition of the ERK1/2 pathway or ectopic expression of Fbxw7β lowered Aurora A levels. A similar inverse relationship between Fbxw7β and Aurora A was observed in human glioma cell lines.41 Concomitant to the upregulation of Aurora A, silencing of Fbxw7β in intestinal and breast epithelial cells increased the proportion of polyploid cells. In support of our results, co-deletion of Fbxw7 and Trp53 in the mouse intestine was reported to cause aggressive adenocarcinomas that exhibit a CIN phenotype.42 In conclusion, we propose that the ERK1/2/Fbxw7β/Aurora A axis identified in this study may contribute to the genomic instability and malignant progression of epithelial cancers.
Materials and methods
Reagents, plasmids and antibodies
The MEK1/2 inhibitor PD184352 was a gift from Parke-Davis. Plasmids pBabe-HA-H-RasV12 and pBabe-HA-MEK1DD have been described.10 pBabe-puro-Aurora A (hAur-A, plasmid 8510) 43 and pCLNR-Neo-H2B-GFP (pCLNR-H2BG, plasmid 17735) 44 retroviral constructs were purchased from Addgene. Fbxw7α and Fbxw7β constructs were kindly provided by KI Nakayama (Kyushu University) and subcloned into pMSCV-hygro vector for retroviral infections.
Commercial antibodies were from the following sources: anti-ERK1/2 from Upstate Biotechnology; anti-phospho-ERK1/2 and anti-phospho-histone H3 from Cell Signaling Technology; anti-HA, anti-α-tubulin, anti-Fbxw7 and anti-GAPDH from Santa-Cruz Biotechnology; anti-Aurora A from Invitrogen. Antibodies for immunohistochemistry: anti-phospho-ERK1/2 from Cell Signaling, anti-Ki-67 from Novus Biologicals and anti-Fbxw7β from LifeSpan Biosciences.
Cell culture, transfections and infections
The rat intestinal epithelial cell line IEC-6 was cultured as previously described.10 Human mammary epithelial MCF10A cells were cultured in F12 medium supplemented with 5 ng/ml EGF, 10% fetal bovine serum, 0.5 μg/ml hydrocortisone, 10 μg/ml insulin, 2 mM glutamine and antibiotics. Cdh1GT/GT MEFs were obtained from S. Kuninaka (Keio University School of Medicine) and cultured as described.45
IEC-6 and MCF10A cells were transfected with siRNAs using Lipofectamine 2000 reagent (Invitrogen). Cells were infected with retroviral vectors as described 46 and polyclonal populations of infected cells were selected either for 3 d with 4 µg/ml puromycine, 5 d with 300 µg/ml hygromycin or 14 d with 400 µg/ml G418. IEC-6 cell populations co-expressing MEK1DD, H-RasV12 or Aurora A constructs with H2B-GFP were obtained by simultaneous infection with pBabe-puro-MEK1DD or pBabe-puro-H-RasV12 or pBabe-puro-Aurora A and pCLNR-Neo-H2B-GFP, followed by selection with puromycin and G418. Infection with shRNA-expressing lentiviruses was carried out as described 47 and cells were selected with puromycin.
RNA interference reagents
The following TRC lentiviral shRNA constructs for mouse/rat Aurka gene were purchased from Sigma: TRCN0000025139 (shAurA-39) and TRCN0000025141 (shAurA-41).
The target sequences of the rat Fbxw7 siRNAs were:
rFbxw7α si2, 3′-AUCUACUGAAACAAAGCGGdTdT-5′
rFbxw7α si3, 5′-CGAACUGGAGGCUCUCUGAdTdT-3′
rFbxw7β si4, 5′-GUUAGAAUCUGUGACAUACdTdT-3′ 24
rFbxw7β si6, 5′-CAUAGUCUCCUCCAAUAAUdTdT-3′.
The target sequences of the human FBXW7 siRNAs were:
hFbxw7 Exon 10, 5′-ACAGGACAGUGUUUACAAAdTdT-3′
hFbxw7α, 5′-GUGAAGUUGUUGGAGUAGAdTdT-3′
hFbxw7β, 5′-UAUGGGUUUCUACGGCACAdTdT-3′,
hFbxw7γ, 5′-CUACUCUAAACCAUGGCUUdTdT-3′.
Immunoblotting analysis
Cell lysis and immunoblot analysis were performed as described previously.48
Live cell imaging
IEC-6 cell populations co-expressing MEK1DD, H-RasV12 or Aurora A with H2B-GFP were plated on 3-cm clear plates (Greiner). After 24 h, the cells were washed twice with PBS and cultured in 2 ml of imaging medium (Gibco) supplemented with 5 mM Hepes (pH 7.4). All live images were acquired using an epifluorescence microscope (personalDV, Applied Precision). The cells were imaged using a 60X/1.42 NA plan Apo objective. For every field of cells, a stack 9 z-slice spaced 1.5 µm was collected every 3 min using a 0.15 sec exposure at 10% neutral density filtering in the fluorescence channel for 24 h, in addition to a DIC reference image. Image sequences were viewed and annotated as overlays between maximum intensity projections of the z-stacks with the DIC images using SoftWoRx 3.6.2 and custom written MATLAB programs.
Cell cycle analysis
For cell cycle analysis, exponentially proliferating cells were pulsed with 10 μM BrdU for 2 h. The cells were trypsinized, washed in PBS, fixed in cold 70% ethanol, and kept at −20°C until flow cytometry analysis. For staining, the cells were washed with blocking buffer (0.5% bovine serum albumin in PBS), and DNA was denatured with 2 N HCl in PBS for 20 min. The cells were washed again and incubated for 2 min in 0.1 M sodium borate (pH 8.5) to neutralize remaining HCl. After washing with dilution buffer (blocking buffer with 0.5% Tween 20), the cells were incubated with anti-BrdU antibody (2.5 mg/ml) and/or anti-phospho-histone H3 (pH3) antibody (2.5 mg/ml) for 60 min, followed by washing with dilution buffer and incubation with Alexa Fluor 350–conjugated anti-mouse IgG and/or FITC-conjugated anti-rabbit IgG for 60 min. The cells were then washed with PBS and incubated on ice for 30 min in propidium iodide (PI) buffer (0.1% sodium citrate, 50 mg/ml PI, and 0.2 mg/ml RNase) in the dark. Fluorescence was recorded on a BD LSR II cytometer and the cell cycle distribution or the percentage of phospho-histone H3 positive cells was analyzed using the FACSDiva software (BD Biosciences).
Real-time quantitative PCR
Total RNA was extracted from cells using the RNeasy Mini Kit (Qiagen) and reverse transcribed using the High Capacity cDNA Reverse Transcription Kit with random primers (Applied Biosystems). For the rat Fbxw7 α and β isoforms, gene expression levels were measured using custom designed assays. For all other genes, gene expression was measured using Universal ProbeLibrary assays (Roche). The sequences of primers and probes are available upon request. Analysis of PCR product amplification was performed on the ABI PRISM 7900HT Sequence Detection System (Applied Biosystems). The hypoxanthine guanine phosphoribosyl transferase (HPRT) and GAPDH genes were used as endogenous controls for rat and human cells. The relative level of target gene expression was quantified by using the ΔΔCT method.
mRNA stability assay
IEC-6 cells were treated with actinomycin D (10 µg/ml) for the indicated times. Total RNA was extracted and the expression of Fbxw7β mRNA was measured by quantitative PCR.
MEK2DD transgenic mice
The MEK2DD transgenic construct was generated by cloning the human MEK2S222D/S226D (MEK2DD) mutant cDNA downstream of a heptameric TetO element fused to a minimal CMV promoter (Fig. S4a). The construct was injected into fertilized eggs and 6 transgenic founders were identified by Southern blotting and further analyzed for inducibility and repressibility of the transgene. MEK2DD founder lines were bred to Villin-rtTA transgenic mice, which express the reverse tetracycline-regulated transactivator (rtTA) specifically in the gastrointestinal epithelium.49 Bitransgenic Villin-rtTA;MEK2DD mice were found to express MEK2DD exclusively upon doxycycline induction in a dose-dependent manner (Fig. S4b). As expected, MEK2DD expression led to a marked increased in the activating phosphorylation of ERK1/2 (Fig. 5a). To evaluate the impact of MEK2 hyperactivation in the intestine, bitransgenic Villin-rtTA;MEK2DD and littermate monotransgenic Villin-rtTA control mice were fed with 500 μg/ml doxycycline at 6 weeks of age for the indicated times. All mice were housed under specific-pathogen free conditions, and experiments were performed in accordance with the Canadian Council on Animal Care guidelines and with Université de Montréal Institutional Animal Care and Use Committee approval.
Immunohistochemistry
The colons from transgenic mice were dissected, fixed in 10% formalin, embedded in paraffin, and processed for histopathological analysis. For immunohistochemical staining, colon specimens were deparaffinized in an automated immunostainer (Discovery XT, Ventana Medical Systems). Heat-induced epitope retrieval (HIER) and blocking steps were performed using proprietary reagents (Cell Conditionner 1 and 2, Inhibitor D and Blocker D included in DABmap detection kit (Ventana Medical Systems)). Sections were then incubated with anti-phospho-ERK1/2 (Cell Signaling Technology; 1:100), anti-Fbxw7β (LifeSpan Biosciences; 1:100), or anti-phospho-histone H3 (Cell Signaling Technology; 1:50) antibody for 1 h at room temperature. Bound antibody was detected with biotin-conjugated anti-rabbit IgG secondary antibody at 1:100 dilution (Jackson ImmunoResearch Laboratories). Nuclei were counterstained with haematoxylin. For measurement of nucleus volume, colon sections were stained with anti-phospho-histone H3 antibody to label cells in G2/M phase. Microscopic fields were randomly selected and the diameter of the nucleus was measured along the major and minor axes. The mean value of the 2 diameters was used to calculate the nucleus volume using the equation v = (4/3) π r3. A total of 150 phospho-histone H3 positive cells were scored for each genotype. To assess the correlation of phospho-ERK1/2 and Fbxw7β staining, colon sections were scored blinded by 2 readers.
Statistical analysis
Results are expressed as the mean ± SEM. Unpaired t test was performed unless otherwise indicated (Excel).
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank K.I. Nakayama, S. Kuninaka, X. Yang and P. Roux for reagents, L. Gaboury, J. Hinsinger and C. Danila for immunohistochemistry analysis, and A.S. Maddox, V. Archambault, M. Therrien, A. Verreault and G. Sauvageau for helpful discussions.
Funding
This work was supported by grants from the Cancer Research Society (S.M.), the Canadian Institutes of Health Research (S.M.) and the National Cancer Institute of Canada (P.S.M.). SD is recipient of studentships from the Cole Foundation and Canadian Institutes of Health Research, and J.F.D. is a fellow of the Swiss National Science Foundation. P.S.M. and S.M. hold the Canada Research Chair in Cell Division and Chromosomal Organization and Cellular Signaling, respectively.
References
- [1].Holland AJ, Cleveland DW. Losing balance: the origin and impact of aneuploidy in cancer. EMBO Rep 2012; 13:501-14; PMID:22565320; http://dx.doi.org/ 10.1038/embor.2012.55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].McGranahan N, Burrell RA, Endesfelder D, Novelli MR, Swanton C. Cancer chromosomal instability: therapeutic and diagnostic challenges. EMBO Rep 2012; 13:528-38; PMID:22595889; http://dx.doi.org/ 10.1038/embor.2012.61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Gordon DJ, Resio B, Pellman D. Causes and consequences of aneuploidy in cancer. Nat Rev Genet 2012; 13:189-203; PMID:22269907 [DOI] [PubMed] [Google Scholar]
- [4].Pfau SJ, Amon A. Chromosomal instability and aneuploidy in cancer: from yeast to man. EMBO Rep 2012; 13:515-27; PMID:22614003; http://dx.doi.org/ 10.1038/embor.2012.65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Crasta K, Ganem NJ, Dagher R, Lantermann AB, Ivanova EV, Pan Y, Nezi L, Protopopov A, Chowdhury D, Pellman D. DNA breaks and chromosome pulverization from errors in mitosis. Nature 2012; 482:53-8; PMID:22258507; http://dx.doi.org/ 10.1038/nature10802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Janssen A, van der Burg M, Szuhai K, Kops GJ, Medema RH. Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science 2011; 333:1895-8; PMID:21960636; http://dx.doi.org/ 10.1126/science.1210214 [DOI] [PubMed] [Google Scholar]
- [7].Sheltzer JM, Blank HM, Pfau SJ, Tange Y, George BM, Humpton TJ, Brito IL, Hiraoka Y, Niwa O, Amon A. Aneuploidy drives genomic instability in yeast. Science 2011; 333:1026-30; PMID:21852501; http://dx.doi.org/ 10.1126/science.1206412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Davoli T, de Lange T. The causes and consequences of polyploidy in normal development and cancer. Annu Rev Cell Dev Biol 2011; 27:585-610; PMID:21801013; http://dx.doi.org/ 10.1146/annurev-cellbio-092910-154234 [DOI] [PubMed] [Google Scholar]
- [9].Ganem NJ, Storchova Z, Pellman D. Tetraploidy, aneuploidy and cancer. Curr Opin Genet Dev 2007; 17:157-62; PMID:17324569; http://dx.doi.org/ 10.1016/j.gde.2007.02.011 [DOI] [PubMed] [Google Scholar]
- [10].Duhamel S, Hebert J, Gaboury L, Bouchard A, Simon R, Sauter G, Basik M, Meloche S. Sef downregulation by Ras causes MEK1/2 to become aberrantly nuclear localized leading to polyploidy and neoplastic transformation. Cancer Res 2012; 72:626-35; PMID:22298595; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-2126 [DOI] [PubMed] [Google Scholar]
- [11].Fujiwara T, Bandi M, Nitta M, Ivanova EV, Bronson RT, Pellman D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature 2005; 437:1043-7; PMID:16222300; http://dx.doi.org/ 10.1038/nature04217 [DOI] [PubMed] [Google Scholar]
- [12].Davoli T, de Lange T. Telomere-driven tetraploidization occurs in human cells undergoing crisis and promotes transformation of mouse cells. Cancer cell 2012; 21:765-76; PMID:22698402; http://dx.doi.org/ 10.1016/j.ccr.2012.03.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Orr B, Compton DA. A double-edged sword: how oncogenes and tumor suppressor genes can contribute to chromosomal instability. Front Oncol 2013; 3:164; PMID:23825799; http://dx.doi.org/ 10.3389/fonc.2013.00164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kamata T, Pritchard C. Mechanisms of aneuploidy induction by RAS and RAF oncogenes. Am J Cancer Res 2011; 1:955-71; PMID:22016838 [PMC free article] [PubMed] [Google Scholar]
- [15].Barr FA, Gruneberg U. Cytokinesis: placing and making the final cut. Cell 2007; 131:847-60; PMID:18045532; http://dx.doi.org/ 10.1016/j.cell.2007.11.011 [DOI] [PubMed] [Google Scholar]
- [16].Marumoto T, Zhang D, Saya H. Aurora-A - a guardian of poles. Nat Rev Cancer 2005; 5:42-50; PMID:15630414; http://dx.doi.org/ 10.1038/nrc1526 [DOI] [PubMed] [Google Scholar]
- [17].Nikonova AS, Astsaturov I, Serebriiskii IG, Dunbrack RL Jr., Golemis EA. Aurora A kinase (AURKA) in normal and pathological cell division. Cell Mol Life Sci 2013; 70:661-87; PMID:22864622; http://dx.doi.org/ 10.1007/s00018-012-1073-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Marumoto T, Honda S, Hara T, Nitta M, Hirota T, Kohmura E, Saya H. Aurora-A kinase maintains the fidelity of early and late mitotic events in HeLa cells. J Biol Chem 2003; 278:51786-95; PMID:14523000; http://dx.doi.org/ 10.1074/jbc.M306275200 [DOI] [PubMed] [Google Scholar]
- [19].Vader G, Lens SM. The Aurora kinase family in cell division and cancer. Biochim Biophys Acta 2008; 1786:60-72; PMID:18662747 [DOI] [PubMed] [Google Scholar]
- [20].Castro A, Arlot-Bonnemains Y, Vigneron S, Labbe JC, Prigent C, Lorca T. APC/Fizzy-Related targets Aurora-A kinase for proteolysis. EMBO Rep 2002; 3:457-62; PMID:11964384; http://dx.doi.org/ 10.1093/embo-reports/kvf095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Littlepage LE, Ruderman JV. Identification of a new APC/C recognition domain, the A box, which is required for the Cdh1-dependent destruction of the kinase Aurora-A during mitotic exit. Genes Dev 2002; 16:2274-85; PMID:12208850; http://dx.doi.org/ 10.1101/gad.1007302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Taguchi S, Honda K, Sugiura K, Yamaguchi A, Furukawa K, Urano T. Degradation of human Aurora-A protein kinase is mediated by hCdh1. FEBS Lett 2002; 519:59-65; PMID:12023018; http://dx.doi.org/ 10.1016/S0014-5793(02)02711-4 [DOI] [PubMed] [Google Scholar]
- [23].Fujii Y, Yada M, Nishiyama M, Kamura T, Takahashi H, Tsunematsu R, Susaki E, Nakagawa T, Matsumoto A, Nakayama KI. Fbxw7 contributes to tumor suppression by targeting multiple proteins for ubiquitin-dependent degradation. Cancer Sci 2006; 97:729-36; PMID:16863506; http://dx.doi.org/ 10.1111/j.1349-7006.2006.00239.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kwon YW, Kim IJ, Wu D, Lu J, Stock WA Jr., Liu Y, Huang Y, Kang HC, DelRosario R, Jen KY, et al.. Pten regulates Aurora-A and cooperates with Fbxw7 in modulating radiation-induced tumor development. Mol Cancer Res 2012; 10:834-44; PMID:22513362; http://dx.doi.org/ 10.1158/1541-7786.MCR-12-0025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Mao JH, Perez-Losada J, Wu D, Delrosario R, Tsunematsu R, Nakayama KI, Brown K, Bryson S, Balmain A. Fbxw7/Cdc4 is a p53-dependent, haploinsufficient tumour suppressor gene. Nature 2004; 432:775-9; PMID:15592418; http://dx.doi.org/ 10.1038/nature03155 [DOI] [PubMed] [Google Scholar]
- [26].Grim JE, Gustafson MP, Hirata RK, Hagar AC, Swanger J, Welcker M, Hwang HC, Ericsson J, Russell DW, Clurman BE. Isoform- and cell cycle-dependent substrate degradation by the Fbw7 ubiquitin ligase. J Cell Biol 2008; 181:913-20; PMID:18559665; http://dx.doi.org/ 10.1083/jcb.200802076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].van Drogen F, Sangfelt O, Malyukova A, Matskova L, Yeh E, Means AR, Reed SI. Ubiquitylation of cyclin E requires the sequential function of SCF complexes containing distinct hCdc4 isoforms. Mol Cell 2006; 23:37-48; PMID:16818231; http://dx.doi.org/ 10.1016/j.molcel.2006.05.020 [DOI] [PubMed] [Google Scholar]
- [28].Minella AC, Swanger J, Bryant E, Welcker M, Hwang H, Clurman BE. p53 and p21 form an inducible barrier that protects cells against cyclin E-cdk2 deregulation. Curr Biol 2002; 12:1817-27; PMID:12419181; http://dx.doi.org/ 10.1016/S0960-9822(02)01225-3 [DOI] [PubMed] [Google Scholar]
- [29].Spruck CH, Won KA, Reed SI. Deregulated cyclin E induces chromosome instability. Nature 1999; 401:297-300; PMID:10499591; http://dx.doi.org/ 10.1038/45836 [DOI] [PubMed] [Google Scholar]
- [30].Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer 2007; 7:295-308; PMID:17384584; http://dx.doi.org/ 10.1038/nrc2109 [DOI] [PubMed] [Google Scholar]
- [31].Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A, et al.. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res 2011; 39:D945-50; PMID:20952405; http://dx.doi.org/ 10.1093/nar/gkq929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Baker DJ, Dawlaty MM, Wijshake T, Jeganathan KB, Malureanu L, van Ree JH, Crespo-Diaz R, Reyes S, Seaburg L, Shapiro V, et al.. Increased expression of BubR1 protects against aneuploidy and cancer and extends healthy lifespan. Nat Cell Biol 2013; 15:96-102; PMID:23242215; http://dx.doi.org/ 10.1038/ncb2643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Saavedra HI, Knauf JA, Shirokawa JM, Wang J, Ouyang B, Elisei R, Stambrook PJ, Fagin JA. The RAS oncogene induces genomic instability in thyroid PCCL3 cells via the MAPK pathway. Oncogene 2000; 19:3948-54; PMID:10951588; http://dx.doi.org/ 10.1038/sj.onc.1203723 [DOI] [PubMed] [Google Scholar]
- [34].Anand S, Penrhyn-Lowe S, Venkitaraman AR. AURORA-A amplification overrides the mitotic spindle assembly checkpoint, inducing resistance to Taxol. Cancer Cell 2003; 3:51-62; PMID:12559175; http://dx.doi.org/ 10.1016/S1535-6108(02)00235-0 [DOI] [PubMed] [Google Scholar]
- [35].Meraldi P, Honda R, Nigg EA. Aurora-A overexpression reveals tetraploidization as a major route to centrosome amplification in p53−/− cells. EMBO J 2002; 21:483-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wang X, Zhou YX, Qiao W, Tominaga Y, Ouchi M, Ouchi T, Deng CX. Overexpression of aurora kinase A in mouse mammary epithelium induces genetic instability preceding mammary tumor formation. Oncogene 2006; 25:7148-58; PMID:16715125; http://dx.doi.org/ 10.1038/sj.onc.1209707 [DOI] [PubMed] [Google Scholar]
- [37].Welcker M, Clurman BE. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer 2008; 8:83-93; PMID:18094723; http://dx.doi.org/ 10.1038/nrc2290 [DOI] [PubMed] [Google Scholar]
- [38].Davis RJ, Welcker M, Clurman BE. Tumor suppression by the Fbw7 ubiquitin ligase: mechanisms and opportunities. Cancer Cell 2014; 26:455-64; PMID:25314076; http://dx.doi.org/ 10.1016/j.ccell.2014.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Spruck CH, Strohmaier H, Sangfelt O, Muller HM, Hubalek M, Muller-Holzner E, Marth C, Widschwendter M, Reed SI. hCDC4 gene mutations in endometrial cancer. Cancer Res 2002; 62:4535-9; PMID:12183400 [PubMed] [Google Scholar]
- [40].Cancer Genome Atlas N . Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012; 487:330-7; PMID:22810696; http://dx.doi.org/ 10.1038/nature11252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Gu Z, Inomata K, Ishizawa K, Horii A. The FBXW7 beta-form is suppressed in human glioma cells. Biochem Biophys Res Commun 2007; 354:992-8; PMID:17274947; http://dx.doi.org/ 10.1016/j.bbrc.2007.01.080 [DOI] [PubMed] [Google Scholar]
- [42].Grim JE, Knoblaugh SE, Guthrie KA, Hagar A, Swanger J, Hespelt J, Delrow JJ, Small T, Grady WM, Nakayama KI, et al.. Fbw7 and p53 cooperatively suppress advanced and chromosomally unstable intestinal cancer. Mol Cell Biol 2012; 32:2160-7; PMID:22473991; http://dx.doi.org/ 10.1128/MCB.00305-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Crane R, Kloepfer A, Ruderman JV. Requirements for the destruction of human Aurora-A. J Cell Sci 2004; 117:5975-83; PMID:15536123; http://dx.doi.org/ 10.1242/jcs.01418 [DOI] [PubMed] [Google Scholar]
- [44].Kanda T, Sullivan KF, Wahl GM. Histone-GFP fusion protein enables sensitive analysis of chromosome dynamics in living mammalian cells. Curr Biol 1998; 8:377-85; PMID:9545195; http://dx.doi.org/ 10.1016/S0960-9822(98)70156-3 [DOI] [PubMed] [Google Scholar]
- [45].Naoe H, Araki K, Nagano O, Kobayashi Y, Ishizawa J, Chiyoda T, Shimizu T, Yamamura K, Sasaki Y, Saya H, et al.. The anaphase-promoting complex/cyclosome activator Cdh1 modulates Rho GTPase by targeting p190 RhoGAP for degradation. Mol Cell Biol 2010; 30:3994-4005; PMID:20530197; http://dx.doi.org/ 10.1128/MCB.01358-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Rodier G, Makris C, Coulombe P, Scime A, Nakayama K, Nakayama KI, Meloche S. p107 inhibits G1 to S phase progression by down-regulating expression of the F-box protein Skp2. J Cell Biol 2005; 168:55-66; http://dx.doi.org/ 10.1083/jcb.200404146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Voisin L, Julien C, Duhamel S, Gopalbhai K, Claveau I, Saba-El-Leil MK, Rodrigue-Gervais IG, Gaboury L, Lamarre D, Basik M, et al.. Activation of MEK1 or MEK2 isoform is sufficient to fully transform intestinal epithelial cells and induce the formation of metastatic tumors. BMC Cancer 2008; 8:337; PMID:19014680; http://dx.doi.org/ 10.1186/1471-2407-8-337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Servant MJ, Coulombe P, Turgeon B, Meloche S. Differential regulation of p27(Kip1) expression by mitogenic and hypertrophic factors: Involvement of transcriptional and posttranscriptional mechanisms. J Cell Biol 2000; 148:543-56; PMID:10662779; http://dx.doi.org/ 10.1083/jcb.148.3.543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Roth S, Franken P, van Veelen W, Blonden L, Raghoebir L, Beverloo B, van Drunen E, Kuipers EJ, Rottier R, Fodde R, et al.. Generation of a tightly regulated doxycycline-inducible model for studying mouse intestinal biology. Genesis 2009; 47:7-13; PMID:18942097; http://dx.doi.org/ 10.1002/dvg.20446 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.