

Abstract
Chromosomal microarrays (CMAs) are routinely used in both research and clinical laboratories; yet, little attention has been given to the estimation of genome-wide true and false negatives during the assessment of these assays and how such information could be used to calibrate various algorithmic metrics to improve performance. Low-throughput, locus-specific methods such as fluorescence in situ hybridization (FISH), quantitative PCR (qPCR), or multiplex ligation-dependent probe amplification (MLPA) preclude rigorous calibration of various metrics used by copy number variant (CNV) detection algorithms. To aid this task, we have established a comparative methodology, CNV-ROC, which is capable of performing a high throughput, low cost, analysis of CMAs that takes into consideration genome-wide true and false negatives. CNV-ROC uses a higher resolution microarray to confirm calls from a lower resolution microarray and provides for a true measure of genome-wide performance metrics at the resolution offered by microarray testing. CNV-ROC also provides for a very precise comparison of CNV calls between two microarray platforms without the need to establish an arbitrary degree of overlap. Comparison of CNVs across microarrays is done on a per-probe basis and receiver operator characteristic (ROC) analysis is used to calibrate algorithmic metrics, such as log2 ratio threshold, to enhance CNV calling performance. CNV-ROC addresses a critical and consistently overlooked aspect of analytical assessments of genome-wide techniques like CMAs which is the measurement and use of genome-wide true and false negative data for the calculation of performance metrics and comparison of CNV profiles between different microarray experiments.
Keywords: Microarray, Sensitivity, Specificity, Precision, ROC
1. Introduction
Chromosomal microarray (CMA) is a broad term used to describe testing for copy number variation utilizing either single nucleotide polymorphism (SNP) or comparative genomic hybridization (CGH) microarrays. CMA has been used in research for years and has also proven valuable in the clinical setting exhibiting a much higher diagnostic yield than conventional chromosome analysis and/or subtelomeric fluorescence in situ hybridization (FISH) for a range of developmental phenotypes [1–3]. The American College of Medical Genetics and Genomics (ACMG) recommends that CMA be used as a first tier diagnostic test in the evaluation of patients with multiple congenital anomalies not specific to a well-delineated genetic syndrome, non-syndromic developmental delay/intellectual disability, and autism spectrum disorders [2,4]. The ACMG has also published guidelines for the interpretation and reporting of constitutional copy number variants (CNVs) as well as for the design and performance expectations of microarrays used in clinical CMA testing [5]. The ACMG recommends that CMA platforms should be able to detect genome-wide CNVs of at least 400 Kb at 99% analytical sensitivity (with a lower limit at the 95% confidence interval >98%) and a false-positive rate of <1% [5]. In order to generate such performance metrics detected CNVs must be classified as either true or false positives but also regions called diploid by a CMA must also be classified at either true or false negatives.
Evaluation of a CMA platform typically involves the selection of several dozen or more well-characterized cases that contain a collection of appropriately sized (majority being <1 Mb), unique CNVs that are located throughout the genome. For smaller laboratories, or those just starting up the assay, the collection of enough appropriate cases can be difficult. We sought to develop a method that could aid the CMA evaluation process by allowing laboratories to calculate performance metrics in a high-throughput, low cost way that relied on fewer cases. This method can utilize both unique and common/polymorphic CNVs and is based on a per-probe approach to quantify outcome metrics such as true and false positive and negative results, as well as sensitivity/recall, specificity, and precision. Calculation of precision is critical given the disproportionate amount of true negative data in such genome-wide comparisons (most of the genome will not be affected by a CNV). Using two CMA designs that differ in the level of resolution we show that performance metrics can be adequately calculated with as few as 20 cases. This approach relies on comparing the CNV profiles of the same samples run on different CMA designs, however, by using a per-probe approach, an arbitrary decision concerning the minimal degree of overlap two CNVs must share in order to be called the “same” CNV is not necessary and thus provides for a much more granular analysis of copy number data. In addition, the high-throughput nature of this comparative methodology allows for the simultaneous calibration of algorithm metrics such as log2 ratio threshold, minimum number of probes to call a CNV, or others. We created several Perl scripts, CNV-ROC, that can be executed to accomplish this methodology.
In the clinical setting, CMA testing influences management of patients in many ways including generation of medical referrals, and providing guidance for diagnostic imaging and specific laboratory testing [6]. Given the complexity of CMA testing and the implications it holds for future patient management, the ability to assess quality control and performance metrics in a variety of ways can be very useful. Even if manufacturers of chromosomal microarrays have performed extensive validation, it is still necessary for individual laboratories to establish for themselves the performance characteristics in their own setting and with their unique set of samples. Thus, the establishment of a comprehensive and low cost method of measuring the performance of a given CMA platform is of great significance to the laboratory community.
2. Methods
2.1. Microarray protocols
In order to test this quality control method, genomic DNA was isolated from residual clinical peripheral blood samples using the Puregene Blood Kit (Qiagen; Valencia, CA). DNA quantification and quality assessment was performed using a NanoDrop spectrophotometer (Thermo Scientific; Wilmington, DE) and agarose gel electrophoresis. For NimbleGen array-CGH experiments, labeling of the patient sample and the normal male or female reference (Promega; Madison, WI) was performed by primer extension of Cy3 and Cy5 labeled random nonamers (TriLink Biotechnologies; San Diego, CA), respectively. The labeled patient and reference DNA were quantitated and equal amounts were hybridized to a human CGH 385 K Whole-Genome Tiling version 2.0 array or a human CGH 720 K Whole-Genome Tiling version 3.0 array (Roche NimbleGen; Madison, WI) according to the manufacturer's instructions. Median probe spacing on the 385 K and 720 K arrays is approximately 7 Kb and 2.5 Kb, respectively. Post-hybridization procedures were performed according to the manufacturer's instructions. The NimbleScan software tool (version 2.5; Roche NimbleGen) was used for feature extraction, calculation of log2 ratio values, and calculation of several quality control metrics. CNV calling and data interpretation were performed using the Nexus Copy Number software tool (version 6.1, BioDiscovery; El Segundo, CA) and FASST2 algorithm supplied with the Nexus software suite (Supplemental Materials) [7]. Whereas in this study we chose to use the FASST2 algorithm for CNV detection any algorithm can be used with CNV-ROC. For the comparison with the Affymetrix CytoScan HD array the data from the 720 K NimbleGen array was converted from hg18 coordinates to hg19 coordinates.
For the Affymetrix CytoScan HD microarray experiments, processing of the patient sample was performed by end point PCR amplification using DNA taq polymerase (Clontech, Inc.; Mountain View, CA). The labeled patient DNA was hybridized to a human whole genome array containing 1.9 million non-polymorphic markers, as well as 750,000 SNP probes (Affymetrix; Santa Clara, CA), according to the manufacturer's instructions. Median probe spacing is approximately 1 Kb. Post-hybridization procedures were performed according to the manufacturer's instructions. The ChAS (Chromosome Analysis Software) tool (version 1.1.2; Affymetrix) was used for feature extraction, calculation of log2 ratio values, and calculation of several quality control metrics (Supplemental Materials). CNV calling and data interpretation were performed with the .CEL files using the Nexus Copy Number software tool and SNP-FASST2 algorithm supplied with the Nexus software suite (Supplemental Materials).
Samples undergoing testing had to meet several quality control thresholds for inclusion (Supplemental Materials). Several different log2 ratio thresholds were utilized for CNV detection, however 10 probes was the consistent minimum CNV size used for all array types. Based on this minimum number of probes, we conservatively estimated the approximate minimal size CNV detectable by each array type to be 100 Kb for the 385 K array, 50 Kb for the 720 K array, and 20 Kb for the CytoScan HD array. All experiments and analyses performed were replicated in a comparison of the 385 K array to the NimbleGen human CGH 2.1 M Whole-Genome Tiling version 2.0 array (Supplemental Materials).
2.2. Comparison samples and calculations
Forty samples were used for comparisons, 20 unique samples for the 385 K vs. 720 K microarray comparisons and 20 additional samples for the 720 K vs. CytoScan HD microarray comparisons. All cases were previously analyzed and interpreted by standard clinical criteria. The distribution included both normal (no known pathogenic CNVs) and abnormal cases in which a known pathogenic lesion was identified (i.e. 17q12 or 22q11.2 deletion). An appropriate mix of male and female patients, as well as small (100–400 Kb) and large (>400 Kb) duplications and deletions were present. The CNVs present in these cases covered a significant portion of the genome (Supplemental Tables 3 and 4). Additionally, each case contained between 10 and 40 common, polymorphic CNVs. Sensitivity, specificity, false positive rate, recall and precision were calculated using conventional methods. Sensitivity (which is identical to recall) was calculated as the ratio of true positives to the sum of the true positives and false negatives, specificity was calculated as the ratio of true negatives to the sum of the true negatives and false positives. The false positive rate was calculated as the ratio of false positives to the sum of the false positives and true negatives, and precision was calculated as the ratio of true positives to the sum of the true positives and false positives. The 95% confidence intervals were calculated according to the efficient-score method [8].
2.3. Common vs. unique CNVs
For this study, CNVs were divided into two groups: common, clinically benign CNVs and unique, clinically significant CNVs. Common CNVs were defined as annotated segmental duplications. A list of these features was obtained using the “Table Browser” feature of the UCSC Genome Bioinformatics site (http://genome.ucsc.edu/index.html). Many of these locations also overlapped with CNVs detected in HapMap samples as ascertained by Conrad and colleagues [9] using a 42 million feature NimbleGen array platform.
3. Results
3.1. Manual CNV-based comparison
To illustrate the advantages of computer-aided evaluation, we first performed a manual comparison between two NimbleGen array-CGH platforms of different resolution by visually examining each CNV called by Nexus Copy Number (version 6.1; FASST2 algorithm) on each array type for each case. For this assessment, only one log2 ratio threshold (0.3) was used for all cases on both array types (+0.3 for duplications and −0.3 for deletions). When we refer to a log2 ratio threshold it is implied that it is negative for deletions and positive for duplications. Special attention was given to identify whether or not any CNV detected by the higher resolution array type would have been expected to be detected on the lower resolution array type. Using the 720 K array as the surrogate “gold standard” for the 385 K array, CNVs of appropriate size (>100 Kb on each array type) were compared to one another to assess true and false positives and negatives on the 385 K array with respect to the 720 K array. True positives were defined as CNVs >100 Kb on the 385 K array that had a corresponding CNV present on the 720 K array; false positives were defined as CNVs >100 Kb on the 385 K array without any corresponding CNV of any size on the 720 K array; and false negatives were defined as CNVs >100 Kb on the 720 K array without any corresponding CNV on the 385 K array. True negatives were defined as those intervening regions between CNVs of >100 Kb on the 720 K array that had no corresponding CNV regions on the 385 K array. Analyses were performed using a CNV size threshold of 400 Kb and 100 Kb, as well as for both benign CNVs and clinically significant CNVs, and for clinically significant CNVs only (Supplemental Table 1). Based on the results of this per-CNV manual comparison of twenty cases and a size threshold of 400 Kb for all CNVs, a sensitivity of 80.9% (with a lower limit at the 95% confidence interval of 66.3%) and specificity of 99.2% (false positive rate of 0.8% with a higher limit at the 95% confidence interval of 2.2%) was achievable. However, when examining only those CNVs >400 Kb and clinically significant (known pathogenic lesions and novel CNVs of unclear clinical significance) a sensitivity of 100% and a specificity of 100% (false positive rate of 0%) were achieved. When testing the lower size threshold of 100 Kb, a sensitivity of 79.8% (with a lower limit at the 95% confidence interval of 74.4%) was achievable when using data from all CNVs (benign and clinically significant) greater than 100 Kb and a single log2 ratio threshold of 0.3. When examining CNVs >100 Kb and only those that were likely to be clinically significant we achieved the same 100% sensitivity and specificity as with the 400 Kb analysis.
Aside from the laborious nature of this type of manual analysis it is also clear that with a per-CNV based analysis and these few cases there are not enough CNVs to truly establish precise and meaningful sensitivity and specificity measures. When examining only clinically significant CNVs greater than 400 Kb a mere 18 CNVs were available for analysis. Even though the sensitivity and specificity were each 100%, the 95% confidence intervals were quite large (sensitivity with a lower limit at the 95% confidence interval of 78.1% and specificity with a lower limit at the 95% confidence interval of 99.0%). Additionally, when visually examining these CNVs it was easier to determine when two CNVs were the same despite changes in breakpoints or fragmented calls. Lastly, because of the extensive time and effort involved in performing this manual comparison only one log2 ratio threshold could be examined for each array type precluding any analysis that might identify a different log2 ratio threshold that would further increase sensitivity without a significant loss in specificity via receiver operator characteristic (ROC) analysis.
Our goal was to create a computer-aided approach to overcome the limitations described above so that laboratories could assess the performance characteristics of a whole-genome microarray to detect CNVs using two microarray platform designs that differed primarily in their level of resolution. To demonstrate the utility of this approach we used two different NimbleGen CGH microarray designs and designed several command line executable Perl scripts collectively called CNV-ROC. Both microarray designs were whole genome, unbiased CGH microarrays (not targeted arrays). To show that the method is robust we also repeated the analysis comparing one of the NimbleGen CGH microarrays to an array manufactured by Affymetrix (CytoScan HD) that contains both copy number and single nucleotide polymorphism probes. An additional assessment comparing yet another type of NimbleGen CGH microarray was also performed and the results were very similar (Supplemental Fig. 1).
3.2. Analysis of array metrics
Even though CNV-ROC can be used to calibrate any algorithmic metric, in lieu of an exhaustive effort we sought to determine which array metric would be most appropriate to calibrate during ROC analysis. We did this by creating a separate command line utility using Perl to computationally compare CNV calls produced by Nexus Copy Number using the FASST2 algorithm between two different arrays (Supplemental Materials). Whereas our method of calling CNVs was using the FASST2 algorithm any CNV calling algorithm would work with our methodology. The 720 K array was again treated as a “gold standard” to which the 385 K array data was then compared. A previously fluorescence in situ hybridization (FISH)-validated log2 ratio threshold value of 0.3 was used for the FASST2 algorithm when calling the CNVs for both array platforms. The analysis was performed for CNV calls of 400 Kb or greater and of 100 Kb or greater. In the 385 K CNV set, for 20 samples there were a total of 45 CNVs that were greater than 400 Kb and a total of 283 CNVs greater than 100 Kb (Table 1). For each experiment, every CNV in the 385 K array was checked for any overlapping CNV calls from 720 K array. From this, two sets of CNVs could be created for each size-cutoff experiment, one set that had one or more overlapping 720 K CNVs (true positives) and one set that had no overlapping 720 K CNVs (false positives). From each set, the median and mean values were calculated for the: CNV probes' log2 ratio, CNV size in base pairs, and number of probes in the CNV. In the 100 Kb CNV size cutoff analysis, CNVs with overlapping 720 K CNVs had a higher mean log2 ratio value (0.542 vs 0.506) and a higher median log2 value (0.477 vs 0.452) than the 385 K CNVs with no overlapping 720 K CNVs (ie true positive CNVs had a higher mean and median log2 ratio value than false positive CNVs). In the 400 Kb CNV size cutoff analysis, CNVs with overlapping 720 K CNVs also had a higher mean and median log2 ratio than those that had no overlapping 720 K CNVs. An unpaired t-test for groups of unequal size and variance was performed on the differing groups of log2 ratio means and found that the difference in log2 ratio means between the sets was statistically significantly different for the 400 Kb analysis (p = 6.43 × 10−11), but not significant for the 100 Kb analysis (p = 0.275). Subdividing these CNVs into deletions and duplications yielded statistically similar results for the 100 Kb (data not shown). Statistical tests could not be performed for duplications and deletions separately in the 400 Kb CNV cutoff group because of small numbers of CNVs. These analyses suggested that the single most appropriate threshold metric for future ROC analysis was the log2 ratio value and that our overall approach of using a higher resolution array to confirm a lower resolution array is appropriate. This latter conclusion is based on the observation that CNVs with overlap (true positives) were represented by significantly more probes than those CNVs without overlap (false positives) (p = 0.0454 for the 400 Kb analysis and p = 2.19 × 10−10 for the 100 Kb analysis). Thus, a CNV called with more probes had a higher probability of being confirmed with the other array. Based on this analysis we chose to only calibrate the log2 ratio threshold in this demonstration, however, it is possible to calibrate any algorithm metric using CNV-ROC.
Table 1.
Computer-aided single log2 ratio threshold comparison of two different array resolution designs.
| Type of CNV | Total number of all CNVs | Number of common CNVs | Number of unique CNVs |
|---|---|---|---|
| >100 Kb 385 K CNVs with 720 K Overlap | 211 | 107 | 104 |
| >100 Kb 385 K CNVs without 720 K Overlap | 72 | 32 | 40 |
| >400 Kb 385 K CNVs with 720 K Overlap | 43 | 24 | 19 |
| >400 Kb 385 K CNVs without 720 K Overlap | 2 | 2 | 0 |
3.3. Computer-aided probe-based simultaneous CNV comparison and metric calibration
Our next goal was to use CNV-ROC to find the optimal log2 ratio that could be used to call CNVs and maximize both genome-wide sensitivity and specificity. This optimal log2 ratio could be calculated from a ROC analysis, which plots the sensitivity vs. false positive rate as the threshold of an experimental metric is varied (in this case the log2 ratio threshold at which to call a CNV in the 385 K array). CNV-ROC uses a per-probe approach to both compare one microarray against the other as well as calibrate one specific metric (log2 ratio value) to optimize sensitivity and specificity. Amongst the advantages of a per-probe based approach is the drastic increase in data points for ROC analysis compared to a per-CNV approach, however, the vast majority of these data points are true negatives. This creates an unbalanced classification problem that can lead to false positive rates and specificities that are of less value. Thus, in addition to plotting traditional ROC curves of sensitivity vs. false positive rate CNV-ROC also creates and analyzes precision vs. recall curves.
Whereas this analysis could be performed on a per-CNV basis, as with the manual analysis, the number of CNVs would be the same and still lacking in ability to calculate robust performance metrics. In addition, per-CNV based comparison approaches suffer from the problem of having to arbitrarily choose a percentage of overlap to classify a CNV as “matching” one from another platform. These considerations led us to investigate a novel per-probe approach that compared probes in CNV calls from the 385 K array to calls from the 720 K array and assigned the 385 K probes “truth-values” based on CNVs at the corresponding locations in the 720 K array. This approach drastically increases the number of data points available for comparative analysis and does not rely on assigning an arbitrary percent overlap to establish identity. The Nexus Copy Number FASST2 algorithm was run with log2 ratio thresholds varying between 0.20 and 0.70 in increments of 0.05 to create 11 sets of CNV calls for both the 385 K and 720 K arrays (Fig. 1). We chose to report our results at a CNV size of 400 Kb, however, CNV-ROC is capable of performing this analysis at any user defined size threshold. Because of the higher probe density, CNVs called in the 720 K array were allowed to confirm CNVs from the 385 K array as true positives. Smaller 720 K array CNVs (<400 Kb) were not used to penalize the 385 K array (false negatives), because we were only assessing the performance of the 385 K array at a resolution of >400 Kb. Only 720 K CNVs greater than 400 Kb could be used to call a false negative. We also allowed CNVs >400 Kb on the 720 K array to confirm smaller CNVs on the 385 K array while also allowing CNVs <400 Kb on the 720 K array to confirm larger CNVs on the 385 K array. These latter two situations were only used to calculate true positives and not false positives. We made this decision to account for two situations: (1) when a CNV on the 385 K array was called just below the 400 Kb size threshold but the same CNV was called on the 720 K array at just over 400 Kb or vice versa, and (2) when a large CNV (>400 Kb) gets fragmented into several smaller CNVs (<400 Kb) by the CNV detection algorithm and as such would not be considered in the comparison because of the size threshold. In CNV-ROC we provide an option to analyze CMA data both with and without this option. Probes from the 385 K array with poor coverage (no 720 K probes located within a buffer of 15 Kb) were discarded from the analysis. In the ROC analysis, a single 720 K log2 threshold was used as a gold standard while 385 K arrays for each log2 ratio threshold value were analyzed against it. CNV-ROC re-runs the analysis for every 720 K log2 ratio threshold value (Fig. 1). All 385 K probes and 720 K probes were labeled by CNV-ROC as either a CNV (deletion or duplication) or normal (not a CNV) using the log2 ratio threshold and the CNV calls made by the FASST2 algorithm (although any CNV calling algorithm could be used with CNV-ROC). For each log2 ratio threshold tested against the gold standard, all 385 K probes for each sample were assigned a truth-value by CNV-ROC. The truth-value for each 385 K probe was assigned as a true-positive or true-negative if one of the 720 K array probes in its buffer range (15 Kb) had the same outcome (gain, loss, normal/no CNV), otherwise the 385 K probe was labeled as a false-positive or false-negative. CNV-ROC is capable of accurately computing this information, given a paired set of microarray data, in a few hours on a typical desktop workstation (Supplemental Protocol).
Fig. 1.
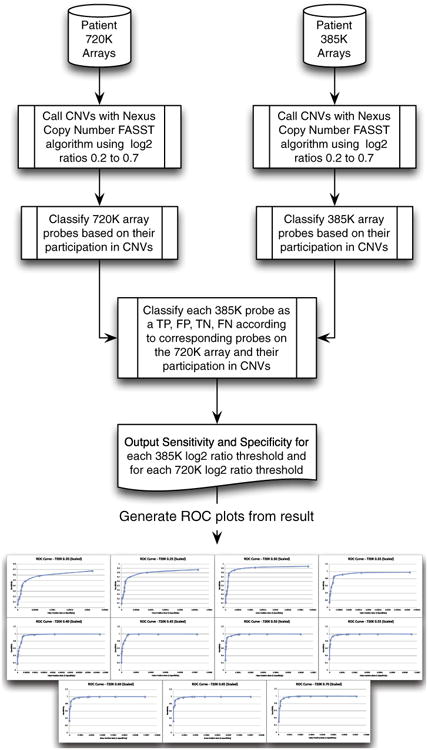
Demonstration of a probe based ROC analysis performed using arrays of different resolution.
CNV-ROC produced a separate ROC curve for every 720 K gold standard using a different log2 ratio threshold (Fig. 1). From the ROC curves an optimal value for the log2 ratio threshold, or “sweet spot”, is determined by CNV-ROC by finding the closest point on the curve to the ROC coordinate (0, 1) (Fig. 2 and Supplemental Table 2). The optimal log2 ratio thresholds for the 385 K array fell between 0.30 and 0.50 across all 720 K array log2 ratio threshold conditions. Optimal log2 ratio thresholds for deletions and duplications were similar but not exactly the same (Table 2). Whereas this analysis was capable of elucidating the optimal log2 ratio value to use to maximize both sensitivity and specificity, the values obtained for specificity were clearly biased toward the very large number of true negative probes (non-CNV probes) present in the array comparisons. This resulted in specificities that routinely exceeded 99.999% across most 385 K log2 ratios thresholds tested. To account for this unbalanced classification issue we also calculated the performance metrics of precision and recall and plotted those values against one another similar to the more traditional ROC analysis (Supplemental Fig. 2). Precision is calculated using only true and false positives, is equivalent to the positive predictive value, and is a more meaningful performance metric than specificity or false positive rate using a per-probe approach. An alternative transformation attempted was to normalize the number of TN probes by dividing the total number of true negatives by the average number of probes present in CNVs >400 Kb. This seemed appropriate given that a requirement for any true or false positive probe was inclusion within a CNV that was >400 Kb. This transformation effectively reduced the number of true negative probes into “chunks” of true negative values equivalent to actual CNV-sized segments. This option is also available in CNV-ROC if desired by the user.
Fig. 2.
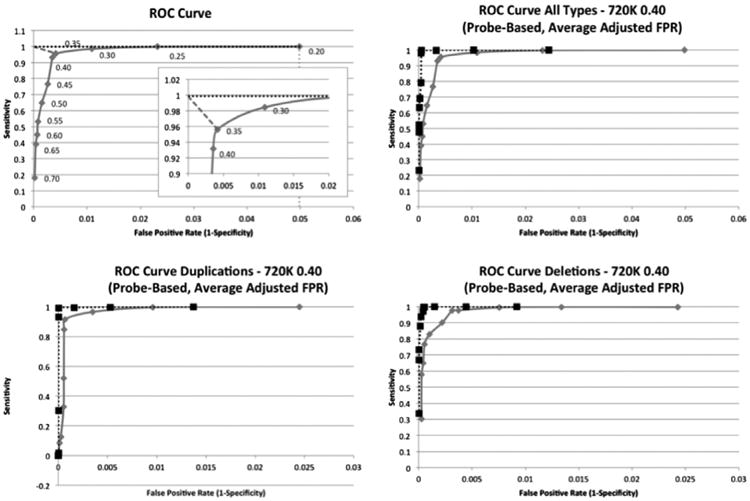
The top left image is a demonstration of how the optimal point is determined based on a normalized graph. Each point is labeled with the log2 ratio that was used to call CNVs compared from the 385 K array to the CNVs called in the 720 K array with a 0.40 log2 ratio. The dotted vertical line represents the normalized maximum FPR and the dotted horizontal line represents the normalized maximum sensitivity. The dashed diagonal line in the top left corner represents the shortest distance to the normalized optimal point. A larger graph of the “knee” is also shown. The top right graph and bottom two graphs are ROC curves for 385 K CNVs greater than 400 Kb that use the 720 K 0.40 log2 threshold as a gold standard. The top right graph represents an analysis including all CNV types, the bottom left graph represents an analysis for just CNV duplications, and the bottom right graph represents an analysis for just CNV deletions. All analyses are performed on a per-probe basis and have had their FPR normalized using the average number of probes found in 385 K CNVs >400 Kb. The solid lines represent the analyses performed using all probes and the dotted lines represent the analyses that exclude probes from common CNV regions.
Table 2.
Optimal log2 ratio threshold determined for the 385 K array at every 720 K array log2 ratio threshold value using CNV-ROC.
| 720 K gold standard log2 ratio | All CNV types | Deletions | Duplications |
|---|---|---|---|
| 0.20 | 0.30 | 0.30 | 0.25 |
| 0.25 | 0.30 | 0.30 | 0.25 |
| 0.30 | 0.30 | 0.35 | 0.30 |
| 0.35 | 0.35 | 0.40 | 0.30 |
| 0.40 | 0.35 | 0.40 | 0.35 |
| 0.45 | 0.40 | 0.45 | 0.35 |
| 0.50 | 0.50 | 0.50 | 0.40 |
| 0.55 | 0.50 | 0.50 | 0.45 |
| 0.60 | 0.50 | 0.50 | 0.50 |
| 0.65 | 0.50 | 0.50 | 0.65 |
| 0.70 | 0.50 | 0.50 | 0.65 |
The optimal log2 ratio was determined for deletions, duplications, and a combination of both deletions and duplications. Overall, larger log2 ratio threshold are more appropriate for duplications than for deletions. The larger range for duplications is due to smaller numbers of CNV duplications.
Once performance metrics were calculated, we determined the most appropriate log2 ratio threshold for the 385 K array platform by maximizing the trade-off between genome-wide sensitivity and FPR as well as precision and recall. In performing this analysis we looked at all CNVs taken together as well as only those CNVs that would be presumed to have clinical significance in constitutional/congenital genetic disorders. If we include all CNVs in our analysis of the 385 K array we are unable to reach sensitivity and FPR thresholds of the magnitude mentioned in the ACMG recommendation manuscript. However, when we include only those probes from CNVs that are considered clinically significant, we determined that the best log2 ratio threshold to call duplications is 0.35, which produces a sensitivity of 99.4% with a lower limit of the 95% confidence interval of 98.2%, a specificity of 99.9% with a lower limit of the 95% confidence interval of 99.9%, and a precision of 99.6% with a lower limit of the 95% confidence interval of 98.5%. Furthermore, we determined that the best log2 ratio threshold to call deletions is 0.40, which produces a sensitivity of 99.7% with a lower limit of the 95% confidence interval of 99.1%, a specificity of 99.9% with a lower limit of the 95% confidence interval of 99.9%, and a precision of 99.5% with a lower limit of the 95% confidence interval of 98.9%. If we used just one log2 ratio threshold to call both duplications and deletions, the optimal log2 ratio threshold was found to be 0.35 with a sensitivity, specificity, and precision of 99.3% (lower 95% of 98.7%), 99.9% (lower 95% of 99.9%), and 99.5% (lower 95% of 99.0%). These log2 ratio threshold values were the same whether we used the sensitivity vs. FPR plots or the precision vs. recall plots. Furthermore, these values are very similar to those obtained by manual analysis, however, have much smaller 95% confidence intervals.
Given that NimbleGen microarrays are no longer in production, and in an effort to show that this methodology is robust, we also tested whether a higher resolution Affymetrix microarray could be used in a comparison with a lower resolution NimbleGen array. With 20 new samples we used CNV-ROC with the Affymetrix CytoScan HD microarray which has ∼1.9 million copy number and ∼750,000 single nucleotide polymorphism (SNP) probes and the NimbleGen 720 K array. We used the FASST2 algorithm to call CNVs on the NimbleGen array and the SNP-FASST2 algorithm on the Affymetrix CEL files to call CNVs on the CytoScan HD array (Supplemental Materials). As both arrays were much higher resolution we decreased our CNV size threshold to 100 Kb. The buffer region size was changed from 15 Kb to 6 Kb based on the resolution of the 720 K array. Lastly, based on the results from the NimbleGen only comparison we choose to only test log2 ratio values between 0.20 and 0.50. The results were similar, although noticeably lower for duplications, and showed that at a size resolution of 100 Kb, when only clinically significant CNVs were considered, a sensitivity, specificity, and precision of 85.8% (lower 95% of 84.9%), 99.9% (lower 95% of 99.9%), and 97.7% (lower 95% of 97.2%) could be obtained for duplications with a log2 ratio threshold of 0.40, and 99.0% (lower 95% of 98.3%), 99.9% (lower 95% of 99.9%), and 93.7% (lower 95% of 92.4%) for deletions with a log2 ratio of 0.40. (Supplemental Table 6 and Supplemental Fig. 3).
4. Discussion
We have shown that clinical laboratories with access to two different resolution chromosomal microarray designs can perform a cost-effective comparative analysis to calibrate specific algorithmic metrics and establish the performance characteristics of their particular platform in their laboratory setting. This analysis is run with a command line utility created in Perl, CNV-ROC, designed to run on a desktop PC, typically taking only a few hours to run, but possibly upwards of one day depending on the sizes of arrays being used and the log2 threshold ranges used in analyses. Whereas we chose to perform this comparison on NimbleGen whole-genome tiled CGH arrays and Affymetrix CytoScan HD arrays, the methodology used is robust and should be applicable to other oligonu-cleotide-based designs. In fact, there is no design issue that would preclude the use of CNV-ROC to compare CNV calls from sequencing data as well as from chromosomal microarrays.
Several technical problems are present when trying to calculate performance metrics for a CMA platform. The results from a CMA platform need to be confirmed with a separate test and ideally at a resolution at which CMA provides its greatest utility. Readily available technologies that could be used to confirm the results from CMA include FISH, quantitative PCR (qPCR), and multiplex ligation-dependent probe amplification (MLPA). The shortcoming of these approaches is that none of them is a genome wide screen. Comparison of microarray CNV findings with these technologies provides for identification of true and false positives but is generally lacking in true and false negatives. Without the later values, sensitivity and specificity cannot strictly be calculated and any comparison would be intrinsically biased toward overestimates of these performance metrics [10]. These values could be calculated, for a limited, specific set of genetic loci but this is not an economical way to assess the genome-wide coverage of CMA. Conventional chromosome analysis (karyotyping) interrogates the entire genome, however, the resolution of chromosome analysis is such that only lesions of >5–10 Mb can be consistently and reliably confirmed. Our method described here allows for calculation of true and false positives (as is done in most FISH, qPCR, or MLPA comparisons), as well as true and false negatives across the entire genome. Previous publications have used these locus-specific technologies to confirm CMA findings [11–13]. Whereas each of these publications illustrates the importance of CMA results confirmation none provide a demonstrable method for the calculation of true and false negative values from CMA data.
Despite the high-throughput nature of our method, and the ability to calculate true and false negatives, it is not without limitations. The gold standard we used for our comparison is another CMA design. Our analysis of probe numbers in true and false positive CNVs suggests that this comparison is valid, yet does not address what is the most appropriate log2 ratio value to use with the higher resolution array. To compensate for this, we tested the lower resolution arrays against the higher resolution arrays at various log2 ratio thresholds and calculated the most appropriate log2 ratio threshold to use for the lower resolution array at every higher resolution array log2 ratio threshold. This provided us with a range for the optimum log2 ratio threshold for the lower resolution array design. This range for the 385 K array, which was different for duplications and deletions (duplication range: 0.25–0.65, deletion range: 0.30–0.50), allowed us to determine what the optimal log2 ratio threshold should be when using the 385 K array design to achieve a certain sensitivity and specificity for simultaneous testing for both deletions and duplications. Our data also show that comparing two arrays of markedly different designs and chemistry results in lower performance metric values. This was especially true for sensitivity and duplications. Whereas the use of CNV-ROC for calibration of algorithmic metrics in such settings works as anticipated, finer tuning of CNV-ROC metrics, such as buffer region size and common CNV regions, as well as CNV calling parameters, such as minimum number of probes to call a CNV, is likely needed to optimize such comparisons.
We initially thought that the false positive rate might be better represented if the ROC analysis were performed for regions instead of probes, however conducting region-based analyses proved challenging due to the incomplete classification of non-CNV regions with a truth-value. The arrays could be quantified into CNV regions and non-CNV regions, but precise classification of the regions as different algorithmic metrics were altered proved difficult. We found that a probe-based analysis provided roughly uniform spacing and coverage between differing array types making it easier to assign truth-values to the probes. Comparison of two CNVs to one another to determine if they are the same is a difficult task. Breakpoint uniformity is often very poor and differences in probe coverage between different arrays leads to loss of CNVs and reducing the power of the analysis [14]. The use of a per-probe approach overcame these limitations. Fragmentation of CNV calls, however, still proved a problem when performing the comparative analysis at specific CNV size thresholds. To compensate for this phenomenon, as well as address subtle differences in breakpoints, we added an option to CNV-ROC that allows CNVs smaller than the specified size threshold to confirm comparison CNVs in the other array that are above the size threshold. Furthermore, given the nature of the per-probe analysis, larger CNVs will contribute more to the performance metrics calculations than smaller ones given the larger number of probes contained within larger CNVs. This behavior is not unintended as it reduces performance metrics more severely for large, likely clinically significant, CNVs that are discordant (false positives or false negatives).
Calculation of performance metrics with CNV-ROC is capable of using common, polymorphic CNVs. By including both unique and common CNVs we greatly increase the total number of CNV-related probes available for evaluation. An additional advantage of this approach is that following this type of comparison the microarray platform under consideration has been tested against a wide range of polymorphic CNVs in addition to those that are currently sought after in constitutional genetic conditions. Whereas polymorphic CNVs are generally not considered significant in terms of developmental disorders, they have been shown in several publications to convey quantifiable risk of common diseases [15,16]. For the comparison between the two NimbleGen arrays, both of which are comparative genomic hybridization arrays, we observed that the probe-based sensitivity of our array platform is lower when considering all CNVs than when considering just those that are unique and likely clinically significant. We believe the explanation for this is that most common CNVs are part of segmental duplications and are very polymorphic in the general population. This makes the choice of reference DNA very important. We chose to use reference DNA that is a combination of genomic DNA from several individuals of the same sex. It is likely that within this mix of individuals there are different genotypes at several segmental duplication loci. This results in “blunted” log2 ratio values of patient to reference signal at probes representing these loci. These lower ratios are often right at the log2 ratio threshold and are occasionally not detected on one or the other platform. Given that we are dealing with higher and lower resolution platforms, it is more common for these instances to be missed on the lower resolution platform and be classified as false negatives, thus lowering the sensitivity. All the false negatives we identified were within highly polymorphic regions of the genome and considered to be clinically benign.
Here we describe an approach and a command line utility, CNV-ROC, which allows clinical laboratories to calibrate various algorithmic metrics as well as calculate probe-based performance metrics.
Supplementary Material
Acknowledgments
The authors would like to thank the members of the University of Iowa Hospitals and Clinics Shivanand R. Patil Cytogenetics and Molecular Laboratory.
Footnotes
Author contributions: CG, BD designed the study and wrote the manuscript; HM performed all the array-CGH experiments and performed the manual validation analysis; CG wrote the code for the command line utility in Perl; WW contributed to the development of the command line utility; BD, VS, TC contributed to the clinical and bioinformatics development of the methodology; BD, CG, HM, WW, VS, TC proof-read and edited the manuscript; VS, TC, BD coordinated the project. All authors read and approved the final manuscript.
Appendix A. Supplementary material: Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.jbi.2015.01.001.
Contributor Information
Corey W. Goodman, Email: corey-goodman@uiowa.edu.
Heather J. Major, Email: heather-major@uiowa.edu.
William D. Walls, Email: daniel-walls@uiowa.edu.
Val C. Sheffield, Email: val-sheffield@uiowa.edu.
Thomas L. Casavant, Email: tomc@eng.uiowa.edu.
Benjamin W. Darbro, Email: benjamin-darbro@uiowa.edu.
References
- 1.Hochstenbach R, van Binsbergen E, Engelen J, Nieuwint A, Polstra A, Poddighe P, et al. Array analysis and karyotyping: workflow consequences based on a retrospective study of 36,325 patients with idiopathic developmental delay in the Netherlands. Eur J Med Genet. 2009;52:161–9. doi: 10.1016/j.ejmg.2009.03.015. [DOI] [PubMed] [Google Scholar]
- 2.Shen Y, Dies KA, Holm IA, Bridgemohan C, Sobeih MM, Caronna EB, et al. Clinical genetic testing for patients with autism spectrum disorders. Pediatrics. 2010;125:e727–35. doi: 10.1542/peds.2009-1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vissers LE, de Vries BB, Veltman JA. Genomic microarrays in mental retardation: from copy number variation to gene, from research to diagnosis. J Med Genet. 2010;47:289–97. doi: 10.1136/jmg.2009.072942. [DOI] [PubMed] [Google Scholar]
- 4.Manning M, Hudgins L. Array-based technology and recommendations for utilization in medical genetics practice for detection of chromosomal abnormalities. Genet Med. 2010;12:742–5. doi: 10.1097/GIM.0b013e3181f8baad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kearney HM, South ST, Wolff DJ, Lamb A, Hamosh A, Rao KW. American College of Medical Genetics recommendations for the design and performance expectations for clinical genomic copy number microarrays intended for use in the postnatal setting for detection of constitutional abnormalities. Genet Med. 2011;13:676–9. doi: 10.1097/GIM.0b013e31822272ac. [DOI] [PubMed] [Google Scholar]
- 6.Coulter ME, Miller DT, Harris DJ, Hawley P, Picker J, Roberts AE, et al. Chromosomal microarray testing influences medical management. Genet Med. 2011;13:770–6. doi: 10.1097/GIM.0b013e31821dd54a. [DOI] [PubMed] [Google Scholar]
- 7.BioDiscovery I. Nexus Copy Number Version 7.5 User Manual. 2014 [Google Scholar]
- 8.Newcombe RG. Two-sided confidence intervals for the single proportion: comparison of seven methods. Stat Med. 1998;17:857–72. doi: 10.1002/(sici)1097-0258(19980430)17:8<857::aid-sim777>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 9.Conrad DF, Pinto D, Redon R, Feuk L, Gokcumen O, Zhang Y, et al. Origins and functional impact of copy number variation in the human genome. Nature. 2010;464:704–12. doi: 10.1038/nature08516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cottrell CE, Al-Kateb H, Bredemeyer AJ, Duncavage EJ, Spencer DH, Abel HJ, et al. Validation of a next-generation sequencing assay for clinical molecular oncology. J Mol Diagn: JMD. 2014;16:89–105. doi: 10.1016/j.jmoldx.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shaffer LG, Beaudet AL, Brothman AR, Hirsch B, Levy B, Martin CL, et al. Microarray analysis for constitutional cytogenetic abnormalities. Genet Med. 2007;9:654–62. doi: 10.1097/gim.0b013e31814ce3d9. [DOI] [PubMed] [Google Scholar]
- 12.Shen Y, Wu BL. Microarray-based genomic DNA profiling technologies in clinical molecular diagnostics. Clin Chem. 2009;55:659–69. doi: 10.1373/clinchem.2008.112821. [DOI] [PubMed] [Google Scholar]
- 13.Yu S, Bittel DC, Kibiryeva N, Zwick DL, Cooley LD. Validation of the Agilent 244K oligonucleotide array-based comparative genomic hybridization platform for clinical cytogenetic diagnosis. Am J Clin Pathol. 2009;132:349–60. doi: 10.1309/AJCP1BOUTWF6ERYS. [DOI] [PubMed] [Google Scholar]
- 14.Pinto D, Darvishi K, Shi X, Rajan D, Rigler D, Fitzgerald T, et al. Comprehensive assessment of array-based platforms and calling algorithms for detection of copy number variants. Nat Biotechnol. 2011;29:512–20. doi: 10.1038/nbt.1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Craddock N, Hurles ME, Cardin N, Pearson RD, Plagnol V, Robson S, et al. Genome-wide association study of CNVs in 16,000 cases of eight common diseases and 3,000 shared controls. Nature. 2010;464:713–20. doi: 10.1038/nature08979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gamazon ER, Nicolae DL, Cox NJ. A study of CNVs as trait-associated polymorphisms and as expression quantitative trait loci. PLoS Genet. 2011;7:e1001292. doi: 10.1371/journal.pgen.1001292. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.