

Emerging evidence continues to support the functional importance of extracellular matrix (ECM) proteins in cellular signaling. In mineralizing tissues, including bone, cartilage and vasculature, ECM proteins not only provide the microenvironment for propagation of crystal growth but also support and transmit mechanical cues to the cells, and these cues govern many aspects of cell function, including proliferation and differentiation. When cells interact with the matrix and produce their own matrix proteins, it is a form of intercellular communication. Although this “matricrine” signaling receives less research attention than chemical forms of intercellular communication, such as autocrine and paracrine signaling, it is important in biomineralization in both health and disease.
In this issue of Circulation Research, Fu and colleagues1 highlight the important role of the non-collagenous ECM protein, cartilage oligomeric matrix protein (COMP) in calcific atherosclerosis. COMP, a member of the thrombospondin family of proteins (TSP-5), maintains cartilage structural integrity by binding collagen and other ECM proteins, such as aggrecan (chondroitin sulfate proteoglycan 1), aggregates of which give cartilage its springy resistance to compression.2–4 COMP overexpression enhances ECM organization and assembly by increasing total soluble glycosaminoglycan content and levels of aggrecan and collagen type II.5 Thus, COMP appears to control the assembly and maintenance of the tertiary architecture of extracellular matrix. Its homopentameric structure, like that of a spiny starfish, allows it to bind to multiple sites, bridging collagen fibrils to one another and bridging cells to matrix proteins and proteoglycans.6
ECM proteins interact with the intracellular cytoskeleton through mechanical links with integrins. As described elegantly by Ingber and colleagues, as a tensegrity model, the mechanical features of ECM are central determinants of cell shape7 and, thus, cell behavior. One robust example of the ability of ECM mechanical characteristics to control cell behavior consists of lineage determination. For instance, Simmons and colleagues showed that valvular cells undergo osteochondrogenic differentiation when grown on an ECM with a particular range of elastic modulus (25–30 kPa), whereas on a less compliant matrix (110 kPa), valvular cells undergo myofibroblastic differentiation.8 Similarly, the Anseth group showed that growth on a substrate with lower elastic modulus directs valvular myofibroblasts into a more dormant fibroblastic phenotype.9
One possible mechanism, by which matrix stiffness may control cell differentiation is through release of morphogens from sites on binding proteins that bridge cellular integrins with ECM proteins, as proposed by Hinz.10 In this paradigm (Figure 1), transforming growth factor-β (TGF-β) superfamily members, which include molecular morphogens, are held in, what we term, “spring-loaded” sites in latent binding proteins that are strategically located between integrins and anchored ECM proteins. Even a brief contraction of the cytoskeleton may allow a cell to “sense” local ECM properties by tugging on the chain of proteins (integrins/binding proteins/ECM proteins). If the cell tugs on a stiff ECM, the resistance produces tension that can unfold a binding protein “spring” to release the morphogens, which may then activate receptors on the cell surface. However, if the cell tugs on a compliant ECM, the matrix may simply “give,” offering no resistance, generating no tension to open the binding protein, and failing to release the morphogen.11 This concept of a “spring-loaded morphogen” is attractive since it may account for the many known effects of matrix elasticity on cellular differentiation.
Figure 1. Speculative model of “spring-loaded morphogen” release.
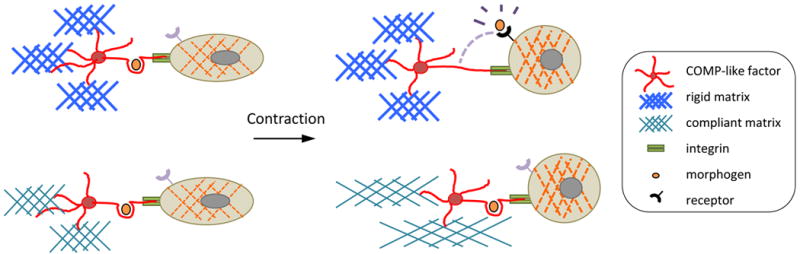
Based on the mechanism proposed by Hinz,10 this schematic shows that binding of cells via integrins to extracellular matrix proteins, such as collagen and homopentameric COMP, allows cells to detect substrate stiffness, by a brief cytoskeletal contraction that releases growth factors or morphogens. When the ECM is altered by inflammation, proteases, mechanical injury, and/or contraction of neighboring cells, cells may detect it by receptor activation by molecular factors upon contraction. Iterative feedback loops may arise if these factors induce synthesis of new matrix, which may change matrix stiffness and/or sensitivity to inflammatory factors.
Canfield and colleagues first described the presence of COMP in calcific atherosclerosis in 1998.12 They demonstrated that COMP is present in the fibrous tissue and in areas of microscopic calcium deposits in atherosclerotic lesions.12, 13 More recently, Du and colleagues showed that COMP deficiency markedly exacerbates – and its ectopic expression greatly reduces – vascular calcification. These findings suggest that it has a compensatory, negative feedback role.14 In vascular cells, COMP is also interacts with integrins, a key participant in matricrine signaling.15 One may speculate, based on the function of COMP in cartilage tissue, that it acts as a tertiary mechanical bridge in ECM, providing greater strength and greater resistance to stretch. If so, then a substrate deficient in COMP may be more compliant (a lower elastic modulus), which is associated with osteochondrogenic differentiation. Conversely, it is conceivable that, if COMP were overexpressed in valvular cells, valve stiffness would increase and direct the cells to a myofibroblastic lineage, which may lead to scar-like contracture and retraction, as seen in human aortic stenosis.
Recent evidence raises the possibility that COMP may be involved in a spring-loaded-morphogen mechanism, given its binding to integrins, its binding to the morphogen, bone morphogenetic protein-2, and its involvement in presenting such factors to cell surface receptors. Similar to the findings by Fu and colleagues in this issue of Circulation Research,1 Di Cesare and colleagues showed that COMP interacts with cells via the integrin receptor, alpha 5 beta 1.6 Interestingly, given that Du et al. showed that COMP binds to BMP-2,14 an alternative mechanical phenomenon may take place (Figure 2). In this alternative model, mechanical tension may alter the ability of COMP to present BMP-2 to its cell surface receptor. When a cell contracts on a more flexible (compliant) substrate, COMP may be free to present BMP-2 to the cell. In contrast, on a rigid substrate, COMP may be held back or stretched into an elongated configuration that does not allow ligand presentation. This conjecture is supported by the finding that COMP overexpression alters sensitivity of C3H10T1/2 mesenchymal cells to BMP-2.16
Figure 2. Speculative model of “tethered morphogen” release.
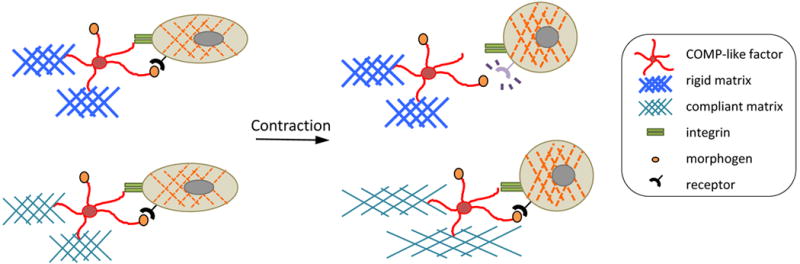
In this model, based on the findings by Fu et al,1 Di Cesare et al,6 and Du et al,14 mechanical tension may alter the ability of COMP to present a morphogen, such as BMP-2, to its cell surface receptor. On a more flexible (compliant) substrate, COMP may be free to present BMP-2 to the cell. In contrast, if the cell contracts on a rigid substrate, ligand presentation by COMP may be lost. While this phenomenon would have the opposite effect on morphogen binding as the spring-loaded paradigm, both phenomena would allow cells to convert mechanical to chemical signals in order to “sense” matrix stiffness.
Fu and colleagues1 also provide strong support for earlier findings, highlighting the role of inflammation in atherosclerotic calcification. The authors demonstrate that bone marrow-derived inflammatory cells are increased in the circulation of COMP-deficient hyperlipidemic mice. Inflammation, which has long been associated epidemiologically with increased risk of cardiovascular mortality,17, 18 precedes vascular matrix calcification, and inflammatory cells, specifically monocyte-macrophages, are found in close proximity to calcific atherosclerotic lesions.19 Inflammation also has substantial effects on matrix remodeling, promoting degradation by metalloproteinase induction and promoting new matrix by increasing expression and synthesis of certain ECM proteins. Li and colleagues showed that production of metalloproteinases by activated macrophages may increase plaque vulnerability by destabilizing calcified nodules.20
Inflammation is also associated with cellular mechanisms of cardiovascular disease. Fu and colleagues highlight the important contributions and additive effects of mesenchymal and hematopoetic lineage cells in calcific vasculopathy.1 Their findings are in agreement with previous lineage tracing studies showing that both VSMC and marrow-derived cells contribute to biomineralization in atherosclerotic lesions.21 Direct effects of activated monocyte/macrophages on vascular cell calcification were first described in 2000 through the potent pro-osteochondrogenic cytokine, TNF-a.22, 23 In hyperlipidemic mice, TNF-a is expressed locally in valvular leaflets, where calcified nodules occur.24 In definitive studies tying these concepts together, the Towler laboratory showed that TNF-a neutralizing antibodies (infliximab) reduced aortic calcium accumulation and osteochondrogenic differentiation of aortic myofibroblastic cells in a mouse model of calcific atherosclerosis.25
In the past, autocrine and paracrine molecular signaling mechanisms have been considered the primary means by which cells share information in inflammation. The findings by Fu and colleagues1 support the importance of matricrine signaling, a less appreciated mechanism of intercellular communication, but an important additional layer of control for cellular behavior and differentiation.
Acknowledgments
Sources of Funding:
This work was supported by grants HL114709 and HL121019 from the Heart, Lung and Blood Institute (NHLBI) of the National Institutes of Health.
Footnotes
Disclosures:
None
References
- 1.Fu Y, Gao C, Liang Y, Wang M, Huang Y, Ma W, Li T, Jia Y, Yu F, Zhu W, Cui Q, Li Y, Xu Q, Wang X, Kong W. Shift of macrophage phenotype due to cartilage oligomeric matrix protein deficiency drives atherosclerotic calcification. Circ Res. 2016 doi: 10.1161/CIRCRESAHA.115.308021. [DOI] [PubMed] [Google Scholar]
- 2.Oldberg A, Antonsson P, Lindblom K, Heinegard D. Comp (cartilage oligomeric matrix protein) is structurally related to the thrombospondins. J Biol Chem. 1992;267:22346–22350. [PubMed] [Google Scholar]
- 3.Newton G, Weremowicz S, Morton CC, Copeland NG, Gilbert DJ, Jenkins NA, Lawler J. Characterization of human and mouse cartilage oligomeric matrix protein. Genomics. 1994;24:435–439. doi: 10.1006/geno.1994.1649. [DOI] [PubMed] [Google Scholar]
- 4.Chen FH, Herndon ME, Patel N, Hecht JT, Tuan RS, Lawler J. Interaction of cartilage oligomeric matrix protein/thrombospondin 5 with aggrecan. J Biol Chem. 2007;282:24591–24598. doi: 10.1074/jbc.M611390200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haleem-Smith H, Calderon R, Song Y, Tuan RS, Chen FH. Cartilage oligomeric matrix protein enhances matrix assembly during chondrogenesis of human mesenchymal stem cells. J Cell Biochem. 2012;113:1245–1252. doi: 10.1002/jcb.23455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DiCesare PE, Morgelin M, Mann K, Paulsson M. Cartilage oligomeric matrix protein and thrombospondin 1. Purification from articular cartilage, electron microscopic structure, and chondrocyte binding. Eur J Biochem. 1994;223:927–937. doi: 10.1111/j.1432-1033.1994.tb19070.x. [DOI] [PubMed] [Google Scholar]
- 7.Ingber DE. Cellular mechanotransduction: Putting all the pieces together again. FASEB J. 2006;20:811–827. doi: 10.1096/fj.05-5424rev. [DOI] [PubMed] [Google Scholar]
- 8.Yip CY, Chen JH, Zhao R, Simmons CA. Calcification by valve interstitial cells is regulated by the stiffness of the extracellular matrix. Arterioscler Thromb Vasc Biol. 2009;29:936–942. doi: 10.1161/ATVBAHA.108.182394. [DOI] [PubMed] [Google Scholar]
- 9.Wang H, Haeger SM, Kloxin AM, Leinwand LA, Anseth KS. Redirecting valvular myofibroblasts into dormant fibroblasts through light-mediated reduction in substrate modulus. PLoS One. 2012;7:e39969. doi: 10.1371/journal.pone.0039969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hinz B. It has to be the alphav: Myofibroblast integrins activate latent tgf-beta1. Nat Med. 2013;19:1567–1568. doi: 10.1038/nm.3421. [DOI] [PubMed] [Google Scholar]
- 11.Engler AJ, Sen S, Sweeney HL, Discher DE. Cell. 2006;126:677–89. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 12.Canfield AE, Farrington C, Dziobon MD, Boot-Handford RP, Heagerty AM, Kumar SN, Roberts IS. The involvement of matrix glycoproteins in vascular calcification and fibrosis: An immunohistochemical study. J Pathol. 2002;196:228–234. doi: 10.1002/path.1020. [DOI] [PubMed] [Google Scholar]
- 13.Farrington C, Roberts IS, Heagerty AM, Canfield AE. The expression of cartilage oligomeric matrix protein, thrombospondin-1, bone sialoprotein and osteopontin in calcified and non-calcified arterial lesions. Biochem Soc Trans. 1998;26:S3. doi: 10.1042/bst026s003. [DOI] [PubMed] [Google Scholar]
- 14.Du Y, Wang Y, Wang L, Liu B, Tian Q, Liu CJ, Zhang T, Xu Q, Zhu Y, Ake O, Qi Y, Tang C, Kong W, Wang X. Cartilage oligomeric matrix protein inhibits vascular smooth muscle calcification by interacting with bone morphogenetic protein-2. Circ Res. 2011;108:917–928. doi: 10.1161/CIRCRESAHA.110.234328. [DOI] [PubMed] [Google Scholar]
- 15.Wang L, Zheng J, Du Y, Huang Y, Li J, Liu B, Liu CJ, Zhu Y, Gao Y, Xu Q, Kong W, Wang X. Cartilage oligomeric matrix protein maintains the contractile phenotype of vascular smooth muscle cells by interacting with alpha(7)beta(1) integrin. Circ Res. 2010;106:514–525. doi: 10.1161/CIRCRESAHA.109.202762. [DOI] [PubMed] [Google Scholar]
- 16.Kipnes J, Carlberg AL, Loredo GA, Lawler J, Tuan RS, Hall DJ. Effect of cartilage oligomeric matrix protein on mesenchymal chondrogenesis in vitro. Osteoarthritis Cartilage. 2003;11:442–454. doi: 10.1016/s1063-4584(03)00055-4. [DOI] [PubMed] [Google Scholar]
- 17.Koenig W, Sund M, Frohlich M, Fischer HG, Lowel H, Doring A, Hutchinson WL, Pepys MB. C-reactive protein, a sensitive marker of inflammation, predicts future risk of coronary heart disease in initially healthy middle-aged men: Results from the monica (monitoring trends and determinants in cardiovascular disease) augsburg cohort study, 1984 to 1992. Circulation. 1999;99:237–242. doi: 10.1161/01.cir.99.2.237. [DOI] [PubMed] [Google Scholar]
- 18.Dweck MR, Jones C, Joshi NV, Fletcher AM, Richardson H, White A, Marsden M, Pessotto R, Clark JC, Wallace WA, Salter DM, McKillop G, van Beek EJ, Boon NA, Rudd JH, Newby DE. Assessment of valvular calcification and inflammation by positron emission tomography in patients with aortic stenosis. Circulation. 2012;125:76–86. doi: 10.1161/CIRCULATIONAHA.111.051052. [DOI] [PubMed] [Google Scholar]
- 19.Aikawa E, Nahrendorf M, Figueiredo JL, Swirski FK, Shtatland T, Kohler RH, Jaffer FA, Aikawa M, Weissleder R. Osteogenesis associates with inflammation in early-stage atherosclerosis evaluated by molecular imaging in vivo. Circulation. 2007;116:2841–2850. doi: 10.1161/CIRCULATIONAHA.107.732867. [DOI] [PubMed] [Google Scholar]
- 20.Li R, Mittelstein D, Lee J, Fang K, Majumdar R, Tintut Y, Demer LL, Hsiai TK. A dynamic model of calcific nodule destabilization in response to monocyte- and oxidized lipid-induced matrix metalloproteinases. Am J Physiol Cell Physiol. 2012;302:C658–665. doi: 10.1152/ajpcell.00313.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Naik V, Leaf EM, Hu JH, Yang HY, Nguyen NB, Giachelli CM, Speer MY. Sources of cells that contribute to atherosclerotic intimal calcification: An in vivo genetic fate mapping study. Cardiovasc Res. 2012;94:545–554. doi: 10.1093/cvr/cvs126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tintut Y, Patel J, Territo M, Saini T, Parhami F, Demer LL. Monocyte/macrophage regulation of vascular calcification in vitro. Circulation. 2002;105:650–655. doi: 10.1161/hc0502.102969. [DOI] [PubMed] [Google Scholar]
- 23.Tintut Y, Patel J, Parhami F, Demer LL. Tumor necrosis factor-alpha promotes in vitro calcification of vascular cells via the camp pathway. Circulation. 2000;102:2636–2642. doi: 10.1161/01.cir.102.21.2636. [DOI] [PubMed] [Google Scholar]
- 24.Li X, Lim J, Lu J, Pedego TM, Demer L, Tintut Y. Protective role of smad6 in inflammation-induced valvular cell calcification. J Cell Biochem. 2015;116:2354–2364. doi: 10.1002/jcb.25186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Al-Aly Z, Shao JS, Lai CF, Huang E, Cai J, Behrmann A, Cheng SL, Towler DA. Aortic msx2-wnt calcification cascade is regulated by tnf-alpha-dependent signals in diabetic ldlr−/− mice. Arterioscler Thromb Vasc Biol. 2007;27:2589–2596. doi: 10.1161/ATVBAHA.107.153668. [DOI] [PubMed] [Google Scholar]