

Abstract
The majority of patients with incontinentia pigmenti (IP) have a mutation in the nuclear factor-kappa-β essential modulator (NEMO) gene, and mice with a targeted deletion of NEMO exhibit skin pathology remarkably similar to the human disease. This study characterizes the retinal vascular abnormalities of NEMO-deficient mice, and compares this phenotype to known features of human IP. Nineteen heterozygous NEMO-deficient female mice, ages ranging from post-natal day 8 (P-8) through 6.5 months of life, were studied. Eyes were sectioned and stained either whole or as retinal flat mounts after incubation for enzyme histochemical demonstration of ADPase, which labels the vasculature. With maturation, retinal arteriolar abnormalities became evident at 3 months of age. Global assessment of the retinal vasculature with ADPase staining showed increased arteriolar tortuosity. Microscopic examination of sections of ADPase-incubated retinas revealed arteriolar luminal narrowing due to endothelial cell hypertrophy and increased basement membrane deposition. Venous morphology was normal. This study characterized the histological retinal phenotype of heterozygous NEMO-deficient female mice. Most striking were retinal arteriolar abnormalities, including luminal narrowing, endothelial cell hypertrophy, and basement membrane thickening. Retinal flat mounts revealed arteriolar tortuosity without evidence of vaso-occlusion or neovascularization.
Keywords: Incontinentia Pigmenti, NEMO, retinal vasculature
Incontinentia pigmenti (IP) is an X-linked genodermatosis that is lethal to males in utero. Affected females are recognized in infancy by characteristic skin lesions, but vascular abnormalities of the brain and retina often develop into the more disabling sequelae of the disease (reviewed in Goldberg, 2004). The retinal changes of IP have been described by ophthalmoscopy, angiography, and histopathology. These changes include a persistent fetal vasculature (PFV), a predictable pattern of vaso-occlusion, ischemia, and neovascular retinopathy ultimately complicated by vitreous hemorrhage and retinal detachment (Fard and Goldberg, 1998; Bell et al, 2007).
A genetics study in 2000 from the International Incontinentia Pigmenti Consortium showed that mutations in the X-linked gene for nuclear factor kappa-β (NF-kβ) essential modulator (NEMO) account for the majority of human IP (IP consortium, 2000). The NEMO protein is a regulatory subunit necessary to activate NF-kB, which is a transcription factor influencing the expression of a variety of genes controlling cell survival and proliferation as well as mediating inflammatory cascades (Hayden et al, 1998).
Soon thereafter, two reports of mice with targeted NEMO deletions were published (Schmidt-Supprian et al, 2000; Makris et al, 2000). As expected, NEMO deficient males died in utero, and heterozygous females were viable. These female mice displayed skin findings strikingly similar to human IP lesions on dermatological examination and on histologic study of skin biopsies. Although NEMO deficient mice are widely accepted as a model for the skin pathology of human IP, their retinal phenotype has remained unexplored (Nelson, 2006). Here we describe the histopathologic retinal phenotype of heterozygous NEMO deficient female mice, including arteriolar abnormalities that may underlie the vaso-occlusive retinopathy characteristic of human IP.
Transgenic NEMO+/− mice were generated by Makris and colleagues (Makris et al, 2000), and C57/B6 (background strain) wild-type control animals were age matched. All Johns Hopkins institutional guidelines regarding animal care and experimentation and the Association for Research in Vision and Ophthalmology’s statement for Use of Animals in Ophthalmic and Vision Science Research were followed. Transgenic eyes were fixed overnight in 2% paraformaldehyde in 0.1 M cacodylate buffer, pH 7.4 at 4°C and shipped to the Wilmer Institute. A total of 38 eyes from 19 transgenic animals were evaluated. For each mouse, the retina from one eye was processed for adenosine diphosphatase (ADPase) activity, and the second eye was fixed whole for sectioning.
ADPase is an enzyme highly specific for the retinal vasculature that hydrolyzes adenosine 5′-diphosphate to adenosine monophosphate. It has been used histochemically to study human, mouse, and rat retinal vasculatures in flatmounts and for flat-embedded cross sectional analysis (Lutty and McLeod, 1992; Lutty et al, 1994, 1996). Prior to retinal dissection for ADPase reaction, the fixed eye cups were washed three times (10 minutes each) in 15% sucrose/0.1 M cacodylate buffer, then three times (10 minutes each) in 10% sucrose/0.1 M cacodylate buffer. The sensory retinas were then dissected out of the eyes, and washed overnight in 10% sucrose/0.1M cacodylate buffer at 4°C. ADPase enzyme histochemistry was performed by incubating the retinas at 37° C for one hour in 0.2 M Tris-maleate (pH 7.2), 3 mM lead nitrate, 6 mM magnesium chloride, 1% Triton X-100, and 1 mg of ADP per ml of incubating solution. Finally, post-incubation washes in 5% sucrose with 0.1 M cacodylate were performed, and the retinas were flat-embedded in glycol methacrylate for microscopy. When viewed in the flat perspective, ADPase positive vascular endothelial cells appear white under dark field illumination due to the lead ADPase reaction product. After imaging the vascular pattern, the embedded retinas were sectioned at 2 μm increments. Sections were counterstained with either toluene blue or hematoxylin and periodic acid/Shiff’s reagent (PAS) for light microscopic examination.
Fellow eyes from each mouse were fixed further in 25% Karnovsky’s fixative at 4°C. After washing and dehydrating with increasing ethanol concentrations, the eyes were embedded whole in glycol methacrylate as described previously (Lutty, 1994). Two micron thick sections were stained with hematoxylin and eosin or hematoxylin and PAS.
A total of 38 eyes from 19 female heterozygous NEMO deficient mice were examined. Mice ranged in age from 8 days post-natal to adult mice 6.5 months old. The results are summarized in Table 1. The younger mouse eyes (ages 8 days through 25 days) had normal appearing retinal vasculature in the flat perspective and upon sectioning (Table 1). However, by 3 months of age, abnormalities were observed in the flat-embedded, ADPase-incubated retinas. The retinal arterioles had changed from the straight and normal appearance at younger ages to tortuous and irregular configurations. These changes were universal by 3.5 months of age (6/6). In Figure 1, the vascular patterns in NEMO deficient mice at 17 days and 3.5 months old are compared to wild type age-matched controls.
Table 1.
Distribution of NEMO+/− mouse ages and retinal abnormalities
| Age | Number of mice | Arteriolar Tortuosity | Arteriolar Endothelial Cell Changes | Persistent Fetal Vasculature |
|---|---|---|---|---|
|
| ||||
| 8 days | 1 | – | – | |
| 10 days | 1 | – | – | |
| 17 days | 3 | – | – | Anterior Hyaloid Vessels (2/3) |
| 25 days | 1 | – | – | |
| 3 months | 4 | 1/4 eyes | 1/4 eyes | |
| 3.5 months | 6 | 6/6 eyes | 6/6 eyes | Persistent Posterior TVL (1/6) |
| 4 months | 2 | 2/2 eyes | 2/2 eyes | |
| 6.5 months | 1 | 1/1 eye | 1/1 eye | |
TVL = Tunica Vasculosa Lentis
Figure 1.
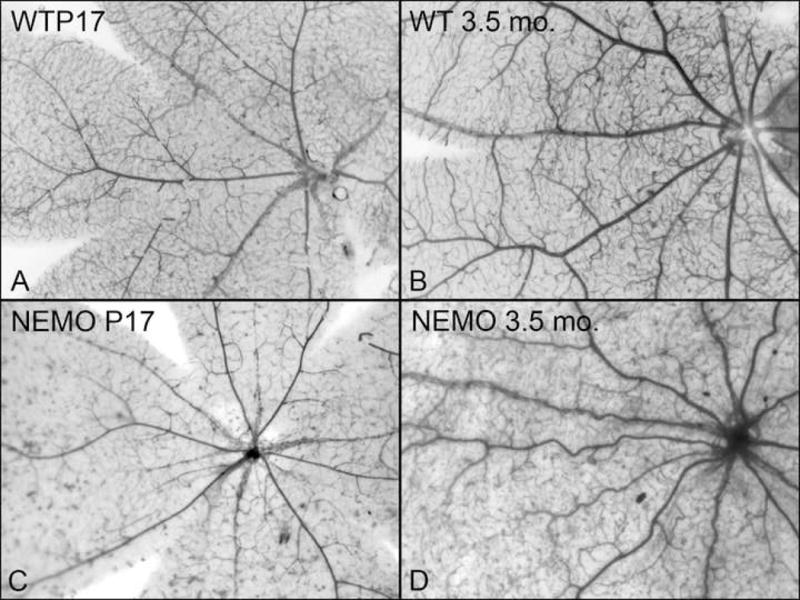
Microscopic sections showed that the arteriolar tortuousity was associated with cellular changes such as thickened and irregular walls with highly irregular lumens (Figure 2). This thickening appeared to be due to a redundancy of hypertrophic vascular endothelial cells and reduplication of their basement membrane, as seen on PAS staining. Evidence of arteriolar wall changes became universal (9/9 animals) at 3.5 months of age and thereafter. (Table 1). Electron microscopy was attempted to further define these findings, but was found not to be possible on glycol methacrylate embedded tissue.
Figure 2.
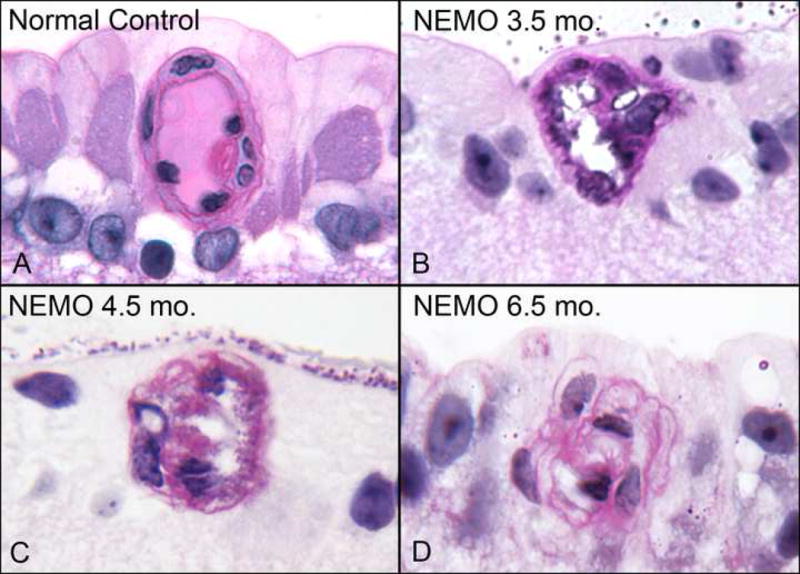
Additionally, the abnormal number and size of endothelial cells along the arteriolar walls resulted in impingement on the vascular lumen, as shown in examples from 3.5 to 6.5 months of age (Figure 2). There was loss of the normal configuration of flattened end-to-end endothelial cells, which ordinarily maximizes intravascular space. In extreme cases at 6.5 months of age, cross sections of arterioles revealed only a slit-like lumen remaining (Figure 2). Evidence of frank vaso-occlusion or neo-vascularization was not seen in the studied eyes, and inflammatory or eosinophilic infiltrates in the retinas were not present.
The fetal vasculature in the vitreous and anterior chamber of NEMO-deficient mice was examined. Persistent vessels of the anterior hyaloid system were present in some 17-day old (P17) NEMO-deficient mouse eyes, and non-regressed posterior tunica vasculosa lentis (TVL) was present in a 3.5 month old eye. While examples of anterior hyaloid vessels were seen in P17 control mice as well, persistence of the TVL in the vitreous chamber at 3.5 months is markedly abnormal.
We describe the histopathologic retinal phenotype of heterozygous NEMO deficient female mice, which includes retinal arteriolar changes and vascular tortuosity with sparing of the retinal venules. The mouse findings appear to be an abridged version of human IP pathology, as arteriolar luminal narrowing is present without evidence of full vaso-occlusion. While complete vaso-occlusions are often seen in human IP (Goldberg, 1974), we saw no evidence of retinal non-perfusion or neovascularization (NV), which can readily be recognized after ADPase incubation, in the NEMO deficient mice. Both nonperfusion and NV have been reported in other mouse models of proliferative retinal disease (Lutty et al, 1994) and in human IP (Chao et al, 2000).
The timing of observed retinal changes in NEMO-deficient mice is different from human disease. In human IP, retinal disease often presents in neonates or early childhood (Berlin et al, 2002; Chao et al, 2000), but the mouse changes were not seen until 3 months of age, and were not consistently present until age 3.5 months or older. The cause for this delay is unclear, and may simply reflect the milder retinal phenotype that appears to exist in mice.
The clinical presentation of human IP varies widely in severity, and this is likely related to accompanying genetic differences among IP patients. A recent publication reviews the almost 70 NEMO gene mutations connected to human IP, and stresses the variability in clinical disease among them. Additionally, the report highlights a lack of correlation between mutation severity and disease course, pointing out that small NEMO mutations can generate more severe clinical consequences than larger deletions (Fusco et al, 2008). The mouse mutation studied here is a gene deletion that generates no NEMO protein (Makris et al, 2000), so the study of only a single mouse genotype may explain some of the differences between our mouse retinal findings and the features of human IP.
Skin biopsies from human IP patients reveal an eosinophilic infiltrate, and there is evidence that perivascular inflammation contributes to the central nervous system pathology of IP (Berlin et al, 2002). Similarly, skin biopsies from NEMO heterozygous mice reveal a dermal and epidermal infiltration of neutrophils and eosinophils (Makris et al, 2000); however, inflammatory changes and peri-vascular infiltration of eosinophils were absent in the retinas we studied. While this does not discount a possible inflammatory vasculitis underlying some human retinal changes, it suggests that other sequelae of NF-kB inactivation may contribute to retinal vasculopathy.
One plausible molecular explanation for NF-kB’s ability to affect blood vessels and endothelial cells is highlighted by recent work in zebrafish (Santoro et al, 2007). Genetic studies of these organisms identified the gene birc1 as a regulator of vessel integrity and endothelial cell survival, and additional biochemical experiments then revealed that the birc1 mechanism is mediated through an NF-kB pathway. NEMO gene expression regulates NF-kB activation, and may similarly influence the survival of endothelial cells in the retinal vasculature of NEMO-deficient mice. Such an influence would be consistent with the apoptosis-driven mechanism of skin pathology in NEMO-deficient mice (Makris et al, 2000). An apoptosis assay (Promega DeadEnd Fluorometric TUNEL System) was attempted on NEMO-deficient retinal tissue, but it proved not reliable in our limited available glycol methacrylate-embedded tissue. Apoptotic endothelial cell death, and subsequent turnover, remains one possible explanation for the abnormalities seen in NEMO-deficient retinal arterioles since the endothelial cells in arterioles were highly abnormal (Figure 2). However, this seems unlikely since all vessels have ADPase activity, which suggests the endothelial cells are viable (Lutty and McLeod, 1992).
In summary, the retinal features we describe in NEMO deficient mice are similar, but not identical, to findings in human IP patients. Future studies are necessary to understand the phenotypic differences from human retinal disease, and to elucidate the molecular mechanisms underlying the vascular changes in both mice and humans.
Acknowledgments
This work was supported by National Eye Institute grant R01EY09357 (GL), EY 01765 (Wilmer), an unrestricted grant from Research to Prevent Blindness Inc., and the Guerrieri Fund for Retinal Research at the Wilmer Eye Institute. Dr. Oster was the recipient of a Mitchell Prize at the Wilmer Institute. The authors greatly appreciate Dr. Constantin Makris’ contribution of the mice investigated in this study.
References
- Bell WR, Green WR, Goldberg MF. Histopathologic and trypsin digestion studies of the retina in incontinentia pigmenti. Ophthalmology. 2008;15:893–897. doi: 10.1016/j.ophtha.2007.08.027. [DOI] [PubMed] [Google Scholar]
- Berlin AL, Paller AS, Chan LS. Incontinentia pigmenti: A review and update on the molecular basis of pathophysiology. J Am Acad Dermatol. 2002;47:169–187. doi: 10.1067/mjd.2002.125949. [DOI] [PubMed] [Google Scholar]
- Chao AN, Lai CC, Kao LY, Hsu JF, Yang KJ, Chen TL. Incontinentia pigmenti: a florid case with a fulminant clinical course in a newborn. Retina. 2000;20:558–560. doi: 10.1097/00006982-200005000-00026. [DOI] [PubMed] [Google Scholar]
- Fard AK, Goldberg MF. Persistence of fetal vasculature in the eyes of patients with incontinentia pigment. Arch Ophthal. 1998;116:682–684. [PubMed] [Google Scholar]
- Fusco F, Pescatore A, Bal E, Ghoul A, Paciolla M, Lioi MB, D’Urso M, Rabia SH, Bodemer C, Bonnefront JP, Munnich A, Miano MG, Smahi A, Ursini MV. Alterations of the IKBKG locus and diseases: An update and a report of 13 novel mutations. Human Mutation. 2008;29:595–604. doi: 10.1002/humu.20739. [DOI] [PubMed] [Google Scholar]
- Goldberg MF. The blinding mechanisms of incontinentia pigmenti. Trans Am Ophth Soc. 1974;92:167–176. [PMC free article] [PubMed] [Google Scholar]
- Goldberg MF. The skin is not the predominant problem in incontinentia pigmenti. Arch Dermatol. 2004;140:748–750. doi: 10.1001/archderm.140.6.748. [DOI] [PubMed] [Google Scholar]
- Hayden MS, West AP, Ghosh S. NF-kappaB and the immune response. Oncogene. 2006;25:6758–6780. doi: 10.1038/sj.onc.1209943. [DOI] [PubMed] [Google Scholar]
- International Incontinentia Pigmenti (IP) Consortium. Nature. 2000;405:466–472. doi: 10.1038/35013114. [DOI] [PubMed] [Google Scholar]
- Ito M, Yoshioka M. Regression of the hyaloid vessels and papillary membrane of the mouse. Anat Embryol. 1999;200:403–411. doi: 10.1007/s004290050289. [DOI] [PubMed] [Google Scholar]
- Lutty GA, McLeod DS, Pachnis A, Costantini F, Fabry ME, Nagel RL. Retinal and choroidal neovascularization in a transgenic mouse model of sickle cell disease. Am J Pathol. 1994;145:490–497. [PMC free article] [PubMed] [Google Scholar]
- Lutty GA, McLeod DS. A new technique for visualization of the human retinal vasculature. Arch Ophthalmol. 1992;110:267–276. doi: 10.1001/archopht.1992.01080140123039. [DOI] [PubMed] [Google Scholar]
- Lutty GA, Phelan A, McLeod DS, Fabry ME, Nagel RL. A rat model for sickle cell-mediated vaso-occlusion in retina. Microvasc Res. 1996;52:270–280. doi: 10.1006/mvre.1996.0064. [DOI] [PubMed] [Google Scholar]
- Makris C, Godfrey VL, Krähn-Senftleben G, Takahashi T, Roberts JL, Schwarz T, Feng L, Johnson RS, Karin M. Female mice heterozygous for IKK gamma/NEMO deficiencies develop a dermatopathy similar to the human X-linked disorder incontinentia pigmenti. Mol Cell. 2000;5:969–979. doi: 10.1016/s1097-2765(00)80262-2. [DOI] [PubMed] [Google Scholar]
- Nelson DL. NEMO, NFkappaB signaling and incontinentia pigmenti. Curr Opin Genet Dev. 2006;16:282–288. doi: 10.1016/j.gde.2006.04.013. [DOI] [PubMed] [Google Scholar]
- Santoro MM, Samuel T, Mitchell T, Reed JC, Stainier DY. Birc2 (cIap1) regulates endothelial cell integrity and blood vessel homeostasis. Nat Genet. 2007;39:1397–1402. doi: 10.1038/ng.2007.8. [DOI] [PubMed] [Google Scholar]
- Schmidt-Supprian M, Bloch W, Courtois G, Addicks K, Israel A, Rajewsky K, Pasparakis M. NEMO/IKKy-deficient mice model incontinentia pigmenti. Mol Cell. 2000;5:980–992. doi: 10.1016/s1097-2765(00)80263-4. [DOI] [PubMed] [Google Scholar]