

Abstract
AIM: Advances in genetics and immunology have contributed to the current understanding of the pathogenesis of inflammatory bowel diseases (IBD).
METHODS: The current opinion on the pathogenesis of IBD suggests that genetically susceptible individuals develop intolerance to dysregulated gut microflora (dysbiosis) and chronic inflammation develops as a result of environmental insults. Environmental exposures are innumerable with varying effects during the life course of individuals with IBD. Studying the relationship between environmental factors and IBD may provide the missing link to increasing our understanding of the etiology and increased incidence of IBD in recent years with implications for prevention, diagnosis, and treatment. Environmental factors are heterogeneous and genetic predisposition, immune dysregulation, or dysbiosis do not lead to the development of IBD in isolation.
RESULTS: Current challenges in the study of environmental factors and IBD are how to effectively translate promising results from experimental studies to humans in order to develop models that incorporate the complex interactions between the environment, genetics, immunology, and gut microbiota, and limited high quality interventional studies assessing the effect of modifying environmental factors on the natural history and patient outcomes in IBD.
CONCLUSION: This article critically reviews the current evidence on environmental risk factors for IBD and proposes directions for future research.
Keywords: Environmental factors, Inflammatory bowel disease, Exposomes
Core tip: Environmental factors are heterogeneous with varying effects during the life course of individuals with inflammatory bowel diseases (IBD). Studying the relationship between environmental factors and IBD may provide the missing link to increasing our understanding of the etiology and increased incidence of IBD in recent years with implications for prevention, diagnosis, and treatment. However, the impact of modifying specific environmental factors on causation and established disease remain poorly studied with limited high quality data from interventional studies to guide clinical practice.
INTRODUCTION
Ulcerative colitis (UC) and Crohn’s disease (CD) are chronic idiopathic inflammatory bowel diseases (IBD). Although the exact pathogenesis of IBD remains unknown, part of the underlying mechanism is a deregulated host immune response to intestinal flora, in genetically susceptible individuals[1,2]. The greatest risk for developing IBD is having a family history of the disease[3]. The greatest risk is seen in monozygotic twins and if both parents suffer from IBD[1]. The estimated relative risk to a sibling of a patient with IBD is 13-36 and 7-17 for CD and UC respectively[2,4]. The first CD susceptibility gene, NOD2 gene within the IBD 1 locus, was a major discovery in 2001[2,3]. Hugot et al[3] found independent associations with CD for three different polymorphisms of the NOD2 gene. The risk of IBD varies depending on whether a subject has one copy (heterozygous) or both copies (homozygous) of the defective allele[2]. However, genetic susceptibility does not completely explain the variance in disease incidence, suggesting a strong role for environmental factors[3,4]. Environmental factors such as smoking, infection, drugs, stress, air pollution, water pollution, diet, and food additives have been investigated in IBD and other autoimmune diseases. These factors have been collectively referred to as exposomes[5]. The term exposome refers to all possible environmental exposures on a human being from conception to death[5]. Factors contributing to the exposome of humans are multifarious. Therefore, the study of the impact of exposomes on human health provides a complete view of human health and disease. Combining the study of exposomes with advances in genetics and immunology may unravel the etiology of diseases such as IBD, diabetes, and cancer, thus enabling the development of preventive interventions against specific exposomes[6]. This article critically reviews the most commonly studied environmental risk factors associated with IBD and proposes directions for future research on environmental factors in IBD.
MATERIALS AND METHODS
A systematic literature search was conducted in PubMed, EMBASE, and Cochrane Library from 1965 through May 2016. The search terms: “environmental factors” and “inflammatory bowel disease”; “exposomes” and “IBD”; “environment” and “crohn’s disease”; “environment” and “ulcerative colitis”; were used to identify relevant studies. In addition, bibliographies of the retrieved articles were searched to identify additional relevant articles on the most commonly studied environmental risk factors in IBD (Table 1). The 2009 Oxford Centre for Evidence-based Medicine (OCEBM) levels of Evidence (LOE) was used to assess the strength of evidence[7] (Table 2). The 2009 OCEBM LOE was chosen because it evaluates what type of evidence is likely to provide the strongest support from studies assessing etiology, prevention, therapy, harm and prognosis without explicitly making definitive recommendations unlike GRADE which is intended for appraising systematic reviews used in developing guidelines[7], Moreover, OCEBM LOE can be applied in situations where there are no systematic reviews available[7].
Table 1.
Commonly studied environmental factors in inflammatory bowel diseases
| Environmental factor |
| Lifestyle |
| Smoking |
| Sleep |
| Stress |
| Diet |
| Breastfeeding |
| Pharmacologic agents |
| Non-steroidal anti-inflammatory drugs |
| Antibiotics |
| Oral contraceptives |
| Vaccination |
| Gut Microbiome |
| Dysbiosis |
| Ecological factors |
| Air Pollution |
| Water Pollution |
| Low Vitamin D |
| Surgery |
| Appendectomy |
Table 2.
Oxford Centre for Evidence-Based Medicine evidence levels of evidence scale
| Level | Study questions on therapy/prevention, etiology/harm |
| 1a | Systematic review (with homogeneity of RCTs) |
| 1b | Individual RCT (with narrow confidence intervals) |
| 1c | All or none studies |
| 2a | Systematic review (with homogeneity) of cohort studies |
| 2b | Individual cohort study (including low quality RCT, e.g., < 80% follow-up) |
| 2c | Outcomes research: ecological studies |
| 3a | Systematic review (with homogeneity) of case control studies) |
| 3b | Individual case control study |
| 4 | Case-series (and poor quality cohort an case control studies ++) |
| 5 | Expert opinion without explicit critical appraisal or based on physiology, bench research or first principles |
Users can add a minus-sign “-” to denote the level of evidence that fails to provide a conclusive answer because: either a single result with a wide confidence interval, or a systematic review with troublesome heterogeneity. Provided by Ref. [7].
RESULTS
Hygiene
Strachan[8] proposed the hygiene hypothesis in 1989 to explain the dramatic rise in atopic diseases. The central principle of this hypothesis is that abnormal immune responses such as autoimmunity and allergy are the result of improvements in personal hygiene and smaller family sizes which have reduced exposure to microbial stimulation[8]. An expansion of this hypothesis is the “microflora” or altered microbiota hypothesis proposed by Noverr and Huffnagle and the IBD hygiene hypothesis[9,10]. The microflora hypothesis proposes that changes in the gut microbiota due to dietary changes and antibiotic use in western countries have altered microbial mediated mechanisms of immunological tolerance[9]. Taken together, these hypotheses suggest that environmental changes can impact the composition of gut microbiota and lead to disease[8-10]. However, recent evidence suggests that the hygiene hypothesis is not applicable to all populations worldwide, and it may be most relevant in countries experiencing increasing affluence or following migration from resource poor to more affluent countries[10].
Smoking
Cigarette smoking is the earliest environmental risk factor that has been consistently shown to be associated with IBD[11-13]. The mechanism by which smoking exerts its effect in IBD is poorly understood. However, putative mechanisms by which smoking modulates the immune system in UC may involve the reduction in tumor necrosis factor (TNF alpha) production via the action of nicotine on the nicotinic acetylcholine receptor a7 subunit, increased production IL-10 in response to carbon monoxide in cigarette smoke, increased mucin synthesis, decrease in IL-8 expression, hypoperfusion of the rectum and acutely damaged colonic tissue[12]. In CD, increased carbon monoxide from cigarette smoke may cause impairment in vasodilation capacity in chronically inflamed micro vessels, resulting in ischemia, and perpetuating ulceration and fibrosis[12]. Decreased total radical-trapping antioxidant potential and abnormalities of the microvasculature, and a defect in bacterial clearance or macrophage deficiency may also play a role[12].
Smoking and Crohn’s disease risk of disease: Smoking increases the risk of developing CD among current smokers [HR = 1.90 (95%CI: 1.42-2.53)][11]. The increased risk is associated with the number of pack years smoked (P trend < 0.0001), whereas smoking cessation is associated with a reduction in the risk [HR = 1.35 (95%CI: 1.05-1.73)][11]. Passive smoking exposure in childhood is no longer considered a risk factor for incident CD[14] (LOE 3A, 2B).
Risk of disease progression: Smoking increases the risk for advanced and difficult to treat to disease; It increases the risk of penetrating intestinal complications, strictures or fistulae, and need for surgical resections (first or second surgery)[13,15,16]. Smoking cessation results in decreased risk of CD, decreased risk of flares, decreased need for steroids and immunosuppressive therapy(IST)[13,17-19] (LOE 3b, 2b).
Risk of relapse: Current smoking is associated with higher relapse rates[13,15,17]. Smoking cessation is associated with a 32% reduction in the risk of a relapse as compared with continued smokers[13]. Smoking cessation can be achieved in IBD patients by utilizing appropriate counselling services, nicotine replacement therapy and pharmacologic agents such as bupropion and varenicline[12,18,19] (LOE 2b).
Smoking and ulcerative colitis risk of disease: In contrast to CD, current smoking is protective against UC, however the mechanism by which smoking exerts its protective effect in UC has not been clearly defined[12]. A case-control study of the effect of smoking on the risk of acquiring UC among 212 individuals showed that the relative risk of UC among former smokers increased in proportion to the cumulative number of cigarettes smoked before the onset of disease, suggesting a causal relationship between smoking and disease occurrence[20]. Analysis of 400 incident cases from the nurses’ health study showed that current smoking had a protective effect on the development of UC [multivariate HR = 0.86 (95%CI: 0.61-1.20)], whereas smoking cessation increased the risk of UC [HR = 1.56 (95%CI: 1.26-1.93)][11]. The risk of UC was significantly increased 2-5 years after smoking cessation [HR = 3.06 95%CI: (2.00-4.67) and remained elevated over 20 years[11] (LOE 2b, 3a).
Risk of disease progression: Current smoking is associated with benign disease course, low hospitalization rates, decreased need for steroids in UC suggesting a less severe clinical presentation and a better long term prognosis than in nonsmokers[21]. However, both hospitalization and colectomy occurred more frequently among smokers who quit before disease onset[22]. Furthermore, hospitalization and colectomy occurred most frequently in the heaviest smokers who quit before disease onset[22].
Risk of relapse: Current smoking is associated with lower relapse and colectomy rates than nonsmokers[22-24]. Overall, there is robust evidence from observational studies and meta-analyses on the association of smoking with IBD. Although smoking has been identified as a modifiable environmental risk factor for IBD, the specific mechanism by which it exacts its effect in UC and CD remains unclear (LOE 3b, 2b).
Environment, sanitation, industrialization and socio-economic status
Public health strategies such as vaccination and environmental sanitation as well as the increasing use of antibiotics has led to changes in the interaction between humans and microbes in the environment. Consequently, improvements in hygiene and health care can alter the composition of the gut microbiota and lead to a state of disequilibrium between protective and pathogenic bacteria (dysbiosis)[8,9].
Risk of disease: A population based cohort study of 2144660 Canadian immigrants showed that younger age at arrival to Canada was associated with increased risk of IBD in immigrants[25]. Canadian-born children of immigrants from Sub-Saharan Africa, Middle East/North Africa, South Asia, North America/Western Europe had a similar risk of IBD as children of nonimmigrants[25]. However, the incidence of IBD remained lower among children of immigrants from East Asia and the Pacific, indicating that the underlying risk is activated with earlier life exposure to the Canadian environment in certain groups[25]. A systematic review of case-control and cohort studies found a positive association between urban environment and both CD and UC [pooled IRRs for urban vs rural environment for UC and CD studies were 1.17 (1.03, 1.32) and 1.42 (1.26, 1.60), respectively], however differences in study design, study quality and lack of data from low prevalence regions (i.e., developing world) limit the generalizability of the results[26]. Data on the association of socio-economic status and risk of IBD is mixed; a population-based study from Canada reported that IBD patients are not of a higher socioeconomic status[27]. A study from France reported that the relative risk (RR) for CD and UC was higher in rural and peri-urban areas with no association with socio-economic status[28] while another study from France reported that RR of CD was higher in urban areas and areas with poor sanitary equipment[29]. However, no significant association was found between socioeconomic variables and incidence of UC[29]. Recent data from China, a rapidly industrializing nation with a historically low prevalence of IBD[30] showed that the incidence of CD was significantly higher in affluent areas than less affluent areas suggesting that high socio-economic status was associated with increased incidence of IBD[31]. Data are lacking on the effect of urbanization and socio-economic status on the risk of disease progression and relapse (LOE 2a, 2b, 2c).
Air pollution
Air pollution has dramatically increased in recent years, particularly in developing countries in Asia that are experiencing rapid industrialization and the highest increase in IBD incidence[31,32]. Exposure of the gut to air pollutants can occur via inhalation of gaseous pollutants, mucociliary clearance of particulate matter (PM) from the lungs and contamination of food and water sources (LOE 4, 2c).
Risk of disease: Kaplan et al[32] showed that residential exposures to Sulphur dioxide (SO2) and Nitric oxide (NO2) may increase the risk of early-onset UC and CD respectively. It has been hypothesized that the effect of air pollution and PM on the incidence of IBD is mediated by the intestinal microbiota; however, conclusive scientific evidence is lacking[33] (LOE 2c, 5).
Risk of disease progression: Ananthakrishnan et al[34] showed that an increase in the density of pollutant emission by 1-log was associated with a 40% increase in the rate of IBD hospitalizations (incidence RR = 1.40; 95%CI: 1.31-1.50) for both UC and CD hospitalization). Data are lacking on the effect of air pollution on relapse in patients with quiescent IBD (LOE 2c).
Water pollution
Risk of disease: Ingestion of pollutants and PM via water sources may induce systemic effects that may impact the incidence, frequency of flares, and success of therapy in IBD[35-38]. Antagonists of steroid receptors that may interfere with treatment have been found in bottled water[38]. Endocrine-disrupting chemicals(EDCs) such as pthalic acid( used as plasticizers for polyvinyl chloride, polystyrene, and many other polymers) and nonylphenols (used in manufacturing antioxidants, lubricating oil additives, laundry and dish detergents, emulsifiers, and solubilizers) may modify glucocorticoid action by altering steroid hormone metabolism through the pregnane X receptor(PXR)[35]. EDCs can influence several steroid receptor proteins such as the glucocorticoid receptor, androgen receptor, AHR, peroxisome proliferator-activated receptor gamma (PPAR-γ) as well as cytochrome P450 enzymes[35]. In-vitro studies suggest that the glucocorticoid receptor and PPAR-γ may play an important role in the pathogenesis of IBD[35-37]. However, there are no supporting in-vivo studies on the effect of EDCs on the risk of incident IBD, disease progression and relapse (LOE 5).
Vitamin D
Vitamin D has immuno-regulatory properties in several autoimmune diseases via its genomic actions on the vitamin D receptor (VDR)[39-41]. There is accumulating evidence that vitamin D may play an integral role in the incidence and disease activity in IBD[42-62] (LOE 5).
Risk of disease: Vitamin D3 (1, 25(OH) D3) decreases production of regulatory Th17 cells and influences the function of natural killer T cells[41,45]. Experimental studies in VDR and IL-10 knockout mice have shown increased expression of inflammatory cytokines in the colon and increased susceptibility to experimental models of colitis[43,46,47]. Subsequent administration of 1,25(OH)2D3 ameliorated colitis and suppressed tumor necrosis factor alpha-related gene expression in the colon of the mice[43,47,61]. However, genome-wide association studies (GWAS) of VDR polymorphisms and IBD have yielded conflicting results; some studies support a positive association while others do not[50,53,58]. Epidemiologic data suggests that the incidence of IBD is higher in residents of northern latitudes compared with southern latitudes, possibly explained by differences in ultraviolet light exposure[42,44,49,55]. However, other studies report no geographic/latitudinal difference[42]. Evidence from the Nurses’ Health Study showed that women in the highest quartile of predicted vitamin D level had a 40% reduction in the risk of CD over 22 years of follow-up compared with those in the lowest quartile (HR = 0.54, 95%CI: 0.30-0.99)[42]. However, there was no effect on the risk of UC[42] (LOE 2a, 3b).
Risk of disease progression: Evidence in support of the association of vitamin D deficiency with greater disease activity is more compelling[48,51,52,57,59,60]. Two small open label studies showed that supplementation with Vitamin D was associated with lower CDAI scores[51,60]. Recently, a large prospective cohort study showed that low vitamin D was associated with higher morbidity and disease severity in patients with UC and CD[59] (LOE 2b).
Risk of relapse: One RCT showed that Vitamin D supplementation was associated with a reduced risk of relapse over the subsequent 12 mo of follow-up compared with those receiving placebo[51]. A meta-analysis of 14 case-control studies showed that IBD was significantly associated with having higher odds of vitamin D deficiency[44].
On balance, the effect of vitamin D on the incidence of IBD is unclear. Vitamin D deficiency has been associated with several chronic diseases and the association with IBD is not causal[54,56]. What we do know is that Vitamin D deficiency is common in IBD patients and long-standing deficiency has been associated with reduced bone mineral density[56]. There is robust evidence that deficiency is associated with disease activity[55-59] and vitamin D supplementation appears to improve CDAI and QOL[51,52,57,60]. Large, prospective, double-blinded RCTs are necessary to define the role of vitamin D therapies in prevention and treatment of IBD. However, in the absence of such studies it is prudent to screen for and treat vitamin D deficiency or insufficiency in patients with IBD (LOE1a, 2a).
Gut microbiome
The gut microbiome consists of a vast number of microorganisms belonging to over 1000 species[63]. The human gut is sterile at birth; bacterial colonization occurs within the first hours of life and increases in number and diversity, depending on the mode of delivery, type of infant feeding, and environmental exposure[64]. Microbial diversity quickly increases in early childhood and leads to the development of the adult gut microbiome[64]. Composition of the gut microbiota is influenced by numerous factors such as antibiotic use, host genetics, diet, phylogeny of the host and intestinal inflammation[64-69].The majority of the bacteria in the adult gut belong to three phyla, Firmicutes, Proteobacteria, and Bacteriodetes[65]. Over time, humans have co-evolved with gut microbes to exist in a symbiotic relationship. The human gut provides the optimal environment for the microbiota to thrive while the microbiota provide physiological benefits such as fermentation of indigestible carbohydrates; synthesis of short chain fatty acids (SCFA) and certain vitamins; biotransformation of conjugated bile acids; degradation of dietary oxalates and resistance to colonization by pathogenic micro-organisms[66,67]. Studies have demonstrated that the gut microbiota have a significant effect on the development of the immune system[68,69] (LOE 1a, 2b, 5).
Risk of disease: Animal studies have shown that bacterial colonization of the gut is critical for the development of intestinal inflammation in IBD[68,69]. Additionally, GWAS have identified 163 genetic risk loci associated with IBD, including 28 shared between CD and UC[2,70]. Although host genetics play a critical role in disease pathogenesis, non-genetic factors also play a substantial role in the development of IBD[70]. Many of the genetic risk alleles associated with IBD are involved in regulation of the innate or adaptive immune system[2,70]. Taken together, the accruing evidence supports the notion that IBD is due to an abnormal immune response to microbial stimulation in genetically susceptible individuals[68-71]. Clinical observations also support the role of gut microbiota in IBD[72-74]. First, IBD typically affects intestinal regions with the highest concentration of bacteria and the use of antibiotics can be effective in the management of IBD[73]. Second, studies of fecal diversion revealed recurrence of colitis with reintroduction of the fecal stream[73,74]. Furthermore, significant alterations of the gut microbiota have been associated with IBD, leading to the notion that dysbiosis leads to disease pathogenesis[72,75-77]. A case control study using metagenomic analysis of microbial community structure showed a statistically significant difference in temporal stability and microbial diversity between the microbial compositions of IBD patients and non-IBD controls[77]. Furthermore, the authors reported that microbiota of IBD patients displayed a reduction in the levels of two phyla of bacteria, Firmicutes and Bacteriodetes, compared with non-IBD controls[77]. New sequencing technologies may provide opportunities to increase our understanding of how dysbiosis may affect IBD[77-79]. However, factors that trigger this change in the delicate balance of the gut microbiota are poorly understood. Prospective studies are required to determine if the loss of certain classes of bacteria can predict individuals with a greater risk of developing IBD (LOE 1a, 3b, 4, 3b, 5).
Risk of disease progression: Dysbiosis has been associated with active disease and relapse in a limited number of studies. Sokol et al[80] demonstrated that Fecalibacterium prausnitzii (Firmicutes) and Bifidobacteria are underrepresented in patients with active IBD and infectious colitis. Reduction in the numbers of F. prausnitzii was associated with a higher risk of postoperative recurrence of ileal CD[81] Lower numbers of F. prausnitzii on resected ileal mucosa of CD patients was associated with endoscopic recurrence at 6 mo[81]. A decrease in F prausnitzii was also associated with the time to relapse after infliximab withdrawal in another study[82]. In patients with UC, quantities of different species of lactobacillus were significantly lower in patients with active inflammation compared with patients in remission[83]. These studies imply that a reduction in intestinal F. prausnitzii, Bifidobacteria, and Lactobacillus species may be important in the initiation of CD or UC respectively (LOE 2b).
Risk of relapse: A cohort study of CD patients demonstrated lower rates of Firmicutes in relapsers compared with non-relapsers[82]. A low rate of F. prausnitzii and a low rate of Bacteroides independently predicted relapse in CD patients[82]. Furthermore, a decrease in Firmicutes was shown to correlate with the time-to-relapse after infliximab withdrawal[82]. A cohort study of UC patients showed that low levels of F. prausnitzii was associated with a four-fold increase in the risk of relapse[84]. The recovery of the F. prausnitzii population after relapse was associated with maintenance of clinical remission in a cross-sectional study of UC patients[84] (LOE 2b).
Infection
The gut bacterial composition of patients with IBD significantly differs from that of non-IBD controls[78,80,81]. However, studies to identify microbial pathogens that cause IBD has met with limited success[70]. Although various pathogenic organisms have been suggested, no pathogen has been consistently implicated in IBD[70]. Pathobionts are symbiotic organisms within the gut that typically do not elicit an inflammatory response, however, under specific environmental conditions, pathobionts have the potential to cause inflammation leading to disease[70,76]. This has led to the notion that the etiologic agents for dysbiosis in IBD patients are not necessarily pathogens, but rather disproportionate populations of pathobionts[70,75,76]. Animal and human studies have shown that deficiencies in T-regulatory-cell (T-Reg-cell) populations or function are central to the pathogenesis of rheumatoid arthritis, asthma, type 1 diabetes and IBD[85,86]. Pro-inflammatory TH17 cells have been shown to antagonize FOXP3+ Treg-cells and the numbers and function of certain TReg-cell populations are reduced in germ-free animals[87,88]. Dysbiosis involves an abnormal change in the composition of the gut microbiota whereby either the numbers of symbionts are reduced and/or pathobionts are increased. The various causes for this change in microbiota are not entirely clear, however, the result is non-specific gut inflammation, which can trigger IBD in genetically susceptible individuals. This gut inflammation can be exacerbated by opportunistic organisms such as viral, bacterial and fungal infections. Thus, prior infections may lead to chronic IBD in patients with a genetic predisposition while coexisting IBD may predispose patients to enteric infections (LOE 3b, 5).
Role of Bacterial infections: Bacterial infections have been implicated as triggers of relapse in IBD[89-95]. Clostridium difficile infection (CDI) in patients with IBD is associated with significant morbidity[90,93] Hospitalized patients with CDI-IBD have a 4-fold greater mortality risk than patients who do not have IBD-CDI[89,93]. However, there is no evidence to suggest that prior CDI predisposes patients to the development of IBD. Enteric infections such as adherent-invasive E-coli, Salmonella and Campylobacter, Mycobacterium avium species have been hypothesized to increase the risk of development of CDI and IBD[70,91,92,94]. However, the apparent increase in IBD risk after enteric infections, particularly in the year after the diagnosis of infection, may be due to a detection bias (LOE 2c, 2c, 5).
Role of viral infections: Cytomegalovirus, ebstein barr virus and human herpes virus have been implicated in exacerbations of IBD or superimposed infection in IBD[96-107]. Human papillomavirus has been linked with squamous cell carcinoma in patients with IBD[98]. However there is no conclusive evidence of a causal link between viruses and IBD. In a recent study using metagenomic analysis, patients with herpes viridae sequences in their colon demonstrated increased expression of human endogenous viral sequences and differences in the diversity of their microbiome[106]. Although the study provided a promising approach to better understand virus-host and phage-bacteria interactions in IBD, further studies are needed to define the contribution of viral infections in the etiopathogenesis of IBD (LOE 3b, 3c, 4, 5).
Role of Parasitic infections: Parasites such as helminths are thought to play an immunomodulatory role in IBD and loss of helminth infections has been proposed as a possible explanation for the reduced incidence and prevalence of IBD in developing countries; the “IBD hygiene hypothesis[108]. Animal, clinical and epidemiological studies support this hypothesis and several distinct immuno-regulatory mechanisms have been described[108-112]. Helminths promote IL10 secretion, a regulatory cytokine that down regulates Th1 responses and colitis in murine models of IBD[110]. Helminths increase the number of Treg-cells in MLNs and the intestinal lining, and promote lamina propria T cells to make more IL10 and TGFβ[112]. Helminths promote the growth of IL4-producing, Th2 cells which inhibit Th1 responses supporting the importance of worm-induced Th2 cytokines for disease control[109]. Helminths have also been shown to increase the number of Treg-cells that help maintain the gut lining in a state of immune tranquility and limit the potential for IBD[111,112]. Recently, an elegant experimental study showed that helminth infection protected mice deficient in the CD susceptibility geneNod2 from intestinal inflammation by inhibiting colonization with Bacteroides species via a TH-2 dependent pathway, which promoted the establishment of protective microbiota enriched in Clostridiales[109]. Additionally, that study showed that individuals from helminth-endemic regions harbored a similar protective microbiota, and that deworming treatment reduced Clostridiales and increased Bacteroidales, further supporting the role of parasitic infection in the IBD -hygiene hypothesis whereby certain individuals are genetically susceptible to changes in the gut microbiome[109].
One RCT assessed the safety and efficacy of modified Trichuris Suis in patients with CD and reported no significant treatment-related side effects[113]. A subsequent systematic review concluded that there was insufficient evidence to allow any firm conclusions regarding the efficacy and safety of helminths for treating IBD[114]. Further RCTs are required to assess the efficacy and safety of helminth therapy in IBD.
Diet
The human diet is influenced by environmental and cultural practices. Diet can influence intestinal inflammation via several pathways, such as, altering the gut microbiome, affecting gastrointestinal permeability, and direct effect of dietary constituents acting as food antigens[115] (LOE 5).
Risk of disease: Evidence from animal[116,117] and epidemiologic studies[115,118] suggests that dietary factors play an important role in gut inflammation and the risk of developing IBD (LOE 2b,3a,5).
Fats
Risk of disease: A systematic review of 19 studies (18 case-control and one cohort; n = 2609; 1340 UC and 1269 CD patients) reported increased risk of developing UC with high intake of total fat, polyunsaturated fatty acid (PUFAs), omega-6 fatty acids, and increased risk of CD with high intake of PUFAs, omega-6 fatty acids, saturated fats[118]. Consumption of high-fat diet worsened dextran sodium sulfate-induced colitis in mice, possibly by increasing colonic epithelial non classical natural killer T cells, and reducing circulating Treg-cells[117]. A high saturated fat diet altered bile acid composition, and increased expansion of sulfate-reducing bacteria (Bilophilia wadsworthia), which in-turn can produce greater amounts of mucosally toxic hydrogen sulfide and induce colitis in IL-10 deficient mice[116]. The results of these animal studies have not been translated to humans (LOE 2b, 5).
Fiber
Risk of disease: A study using IL-10 deficient mice showed that consumption of soluble fibers reduced intestinal inflammation[119]. In humans, high intake of dietary fiber, particularly fruits and cruciferous vegetables was associated with decreased risk of CD, but not UC (HR = 0.59; 95%CI: 0.39-0.90)[120]. The protective effect of fiber was observed to be statistically significant in those consuming more than 22.1 g/d in another study[118]. Additionally, high intake of fruits was associated with a 73%-80% decreased risk of CD in the same study[118] (LOE 2b, 2b, 5).
Risk of disease progression: Plantago ovata seeds (soluble fiber) were shown to have anti-inflammatory activity in HLA-B27 transgenic mice[121]. This animal model was further tested in a RCT that showed that Plantago ovata may be as effective as mesalamine for maintenance of remission in patients with UC[122]. However, the result of that study has not been validated by other RCTs (LOE 1b, 2b).
Carbohydrates Risk of disease: A systematic review showed no consistent association between total carbohydrate intake and IBD risk, even in studies reporting greater than double the recommended daily intake (130 mg total carbohydrates per day)[118] (LOE 3a).
Animal protein (red meat, processed meat, poultry, dairy products)
Risk of disease: The association between high meat intake and IBD is unclear, majority of studies show a positive association of total protein intake and IBD (87% to 148% increased risk)[118]. However, the association between meat intake and risk of IBD was statistically significant in only two studies (LOE 2b).
Risk of disease progression and relapse: The amino acid tryptophan supplies a crucial intermediate metabolite for the action of aryl hydrocarbon receptor (AhR) which suppresses immune responses in dendritic cells. A deficiency in AhR increases production of proinflammatory Th17 cells[123,124]. High tryptophan availability causes lactobacilli to switch their metabolism and produce an AhR ligand-indole-3-aldehyde that contributes IL-22 induction and subsequent IL-22 mediated attenuation of colitis[123] Similarly, indole-3-carbinol present in fruits and vegetables (such as cauliflower, broccoli, cabbage), activates the AhR and ameliorates colitis in mice[120]. Thus animal proteins may modulate inflammation in IBD via the actions of specific amino-acids or their metabolites on immune function. However, diet-based interventions to maintain remission in CD and UC have shown limited benefit in maintaining remission or preventing relapse[125-128] (LOE 1b, 3b, 5).
Food antigens
Risk of disease: Food antigens may act as important stimuli of the mucosal immune system leading to the pathogenesis of IBD. Patients with IBD report intolerance to several food items[125]. However, the abundance of different potential food antigens and lack of high quality evidence on the effect of specific food antigens on the risk of disease in IBD make the association difficult to characterize (LOE 4).
Risk of disease progression and relapse: The role of food antigens on disease progression was studied in 40 patients with CD[129]. Food specific IgG4 levels were used to select which foods to exclude in the intervention diet. The daily stool frequency significantly decreased by 11% in patients randomized to the intervention diet compared with the sham diet[129]. Additionally, patients on the intervention diet had reduced abdominal pain and improved general well-being[129]. The results of this study should to be interpreted with caution because of the presence of several confounders and a high dropout rate.
Food additives
Risk of disease: Food additives such as Aluminum, titanium dioxide (TiO2), and Microparticles/nanoparticles have been implicated in murine models of colitis[130,131]. Aluminum is a component of several processed foods, toothpaste, deodorants, and cosmetics. Oral administration of aluminum at toxic levels worsened intestinal inflammation in mice with 2,4,6-trinitrobenzene sulfonic (TNBS) acid- and dextran sodium sulfate (DSS)-induced colitis and chronic colitis in interleukin 10 deficient mice[131]. Aluminum impaired intestinal barrier function and enhanced intestinal bacterial translocation, favoring development of granulomas in mice[131] (LOE 5, 5).
Risk of disease progression and relapse: Microparticles are used as food additives, anticaking agents, or food colorants and accumulation of microparticles have been demonstrated in Peyer’s patches[130]. NLRP3 is a multiprotein complex containing caspase-1, which activates the proinflammatory cytokines IL-1b and IL-18. TiO2 particles were shown to activate the NLRP3 inflammasome[130]. Thus, TiO2 can be absorbed by intestinal epithelial cells and may aggravate inflammation in susceptible individuals[130]. The aforementioned evidence suggests that food additives and microparticles may worsen intestinal inflammation and contribute to disease progression or relapse in individuals with IBD (LOE 5).
Diet and the gut microbiome: There is increasing evidence demonstrating an association between diet and the gut microbiome[132-136]. Analysis of fecal 16S rRNA sequences from 60 mammalian species revealed clustering according to diet (herbivore, carnivore, and omnivore) and host phylogeny[135]. The functional evolution of the gut microbiome in relation to the human diet was demonstrated in another study by using shotgun metagenomics sequencing[134]. Differences in microbial genes that encode for enzymes involved in carbohydrate and amino acid metabolism have been demonstrated between herbivores and carnivores[136]. This suggests that long-term co-evolution of humans and gut microbiota has modified the composition of the human gut microbiome[132] (LOE 4).
Risk of disease: De Filippo et al[132] examined the fecal microbiome of Italian children (age 1-6 years) compared to that of children (age 1-6 years) from rural sub-Saharan Africa (Burkina Faso) and showed that there were similarities in the genera of gut bacteria present in the gut among children aged 1-2 years from both groups, possibly explained by breast feeding. However, there were considerable differences in the gut microbiome between the African children, fed a traditional high-fiber diet, and the European children, fed a modern Western diet among children older than 2 years[137]. African children showed a significant enrichment in Bacteroidetes and depletion in Firmicutes (P < 0.001), with abundance of bacteria from the genus Prevotella and Xylanibacter (known to contain a set of bacterial genes for cellulose and xylan hydrolysis), completely lacking in the European children. In addition, significantly more SCFAs (P < 0.001) were found in African children than European children. Also, Enterobacteriaceae (Shigella and Escherichia) were significantly underrepresented in African children compared with European children (P < 0.05)[132]. Wu et al[136] demonstrated that long-term agrarian dietary patterns are associated with an enterotype dominated by Prevotella, a genus frequently observed in people from rural Africa, underscoring the impact of diet on the microbiome in healthy human patients. Wu et al[136] also demonstrated that a long-term diet high in animal protein and fats and low in carbohydrates, similar to a “Westernized” diet, is associated with high quantities of Bacteroides and low quantities of Prevotella, further supporting the impact of diet on the gut microbiome. Further studies on the link between diet, gut microbiota, and the development of IBD are needed to provide important insights into the association of a “westernized diet” with the increasing incidence of IBD (LOE 4).
Non-steroidal anti-inflammatory drugs
Cyclooxygenase (COX) is an important enzyme that is found in two isoforms in the body COX-1 and COX-2. COX-1 is present at constant levels in some tissues, whereas COX-2 is an inducible enzyme that can be up regulated by inflammation. COX-1 produces prostaglandins (PG) in the intestine to maintain the gut epithelial barrier and COX-2 mediates inflammation. Prostaglandins (PGs) promote inflammation, mucus production, and vascular flow in the gut. non-steroidal anti-inflammatory drugs (NSAIDS)’s inhibit both COX-1 and COX-2, whereas COX-2 inhibitors selectively inhibit COX-2.
Risk of disease: Inflammation of the colon leads to an increase in PG synthesis and upregulation of COX-2. Thus it would seem logical that COX-2 inhibition could have a protective role in IBD. However, the role of other mediators has not yet been defined and the true effect of COX-2 inhibition has not been fully elucidated. It can also be argued that since COX-1 protects the gut epithelium, NSAIDS could cause or worsen IBD by breaking down the barrier between the immune system and intestinal luminal antigens. However, no causal relationship between NSAID use and incident IBD has been established.
Risk of disease progression: High-dose NSAIDs have been associated with an increase in disease activity in patients with CD or UC, while low-doses of NSAIDs were not associated with a higher disease activity index (DAI) score among CD patients[137]. In contrast, another study reported no association between NSAID use and increased disease activity in IBD, suggesting that NSAID use in IBD deserves further study before recommending that patients refrain from their use under all circumstances[138].
Risk of relapse: Nonselective NSAIDs were associated with a 17%-28% relapse rate within 9 days of ingestion in patients with quiescent IBD[139]. In another study, the adjusted odds ratio between NSAID use and relapse was 6.31 (95%CI, 1.16-34.38, P = 0.03)[140]. Similarly, Cox-2 inhibitors have been associated with relapse in patients with CD and UC suggesting that the all classes of NSAIDs are associated with relapse in IBD patients[141,142]. However, other studies have reported no increase in flares and a beneficial safety profile during short-term treatment of IBD-associated arthritis and arthralgia[143,144] (LOE 2b, 4).
Antibiotics
Risk of disease: Antibiotic use can induce selection pressure and alter the gut microbiome[145-147]. A case-control study showed that treatment for pneumonia before the first 5 years of life increased the risk for childhood and adult-onset CD[145]. Another study found that individuals diagnosed with IBD were more likely to have been prescribed antibiotics 2-5 years before their diagnosis[146]. A recent meta-analysis showed that exposure to antibiotics was significantly associated with newly diagnosed CD (OR = 1.74, 95%CI: 1.35-2.23) but not UC (OR = 1.08, 95%CI: 0.91-1.27)[147] (LOE 2a, 3b, 3b).
Risk of disease progression: In a systematic review of RCTs, antibiotics were superior to placebo at inducing remission in patients with active CD (RR of CD not in remission = 0.85; 95%CI: 0.73-0.99, P = 0.03)[71]. Antibiotics were also superior to placebo for reducing fistula drainage in CD patients with perianal fistulae (RR = 0.8, 95%CI: 0.66-0.98)[71]. In active UC, antibiotics were superior to placebo for inducing remission (RR of UC not in remission = 0.64, 95%CI: 0.43-0.96)[71].
Risk of relapse: Antibiotics were superior to placebo (RR of relapse = 0.62, 95%CI: 0.46-0.84) for preventing relapse in patients with quiescent CD[71]. Nitroimidazoles (metronidazole and ornidazole)have been shown to be effective in preventing post-operative recurrence of CD[148,149]. The evidence on the effect of antibiotics on disease activity and relapse in IBD is limited because a diverse number of antibiotics with different spectra of activity were grouped together making interpretation and generalizability difficult. However, despite the absence of robust data, antibiotics are widely used therapy in IBD.
Oral contraceptives
Risk of disease: Oral contraceptive pills (OCPs) were positively associated with UC and CD in a meta-analysis of 14 case control studies[150]. The pooled RR for women currently taking OCPs was 1.46 (95%CI: 1.26-1.70, P < 0.001, adjusted for smoking) and 1.28 (95%CI: 1.06-1.54, P = 0.011, adjusted for smoking) for CD and UC respectively[150]. The risk of CD was greater with prolonged exposure to OCPs and women who discontinued OCPs were no longer at a significantly increased risk for CD[150]. Similarly, OCPs were shown to increase the risk of UC and CD in another study, however the risk of UC was only increased in patients with a history of smoking, suggesting that smoking was a confounding variable[151]. Hormone replacement therapy (HRT) in post-menopausal women was shown to increase the risk of UC but not CD[152].
Risk of disease progression: A case control study showed that OCPs have no effect on disease activity in CD[153]. In contrast, Kane et al[154] showed that HRT was protective against disease activity in post-menopausal women with IBD (HR = 0.18, 95%CI: 0.04-0.72). A dose-response effect was noted with longer duration of HR, however the results should be interpreted with caution because it was a small single center retrospective study with limited generalizability.
Risk of relapse: A prospective cohort study showed that women who continued to take OCPs were at a threefold increased risk of developing a relapse of CD; this effect was stronger among women who were prescribed OCPs and smoked, suggesting that smoking was a confounding variable[155]. The mechanism by which OCPs increase the risk of IBD is unknown, estrogen enhances humoral immunity and proliferation of macrophages, while progesterone suppresses immune responses[156]. Therefore, it is conceivable that estrogen enhances inflammation and progesterone suppresses inflammation in patients with IBD.
Stress
Stress is defined as a state of disharmony or threatened homeostasis[157]. The hypothalamo-pituitary-adrenal (HPA) axis and the immune system work closely together when the body is confronted with a stressful response. When stimulated by a stress event, the immune system activates the HPA axis by producing cytokines that ultimately result in the production of powerful anti-inflammatory agents such as glucocorticoids[157]. Disruptions of the HPA axis and immune system loop could potentially lead to diseases with an inflammatory and behavioral component due to abnormal responses to stressful stimuli. The loop that connects the immune system to the HPA is complex and disruptions at different levels could lead to different manifestations of disease[158].
Risk of disease: A few studies have shown that stress is associated with increased relapse in patients with UC and CD[159,160]. However, there is no evidence that stress is associated with increased risk of incident IBD (LOE 2b).
Risk of disease progression: An interventional study evaluated the effects of the Breath-Body-Mind Workshop (BBMW) (breathing, movement, and meditation) vs an educational seminar on psychological symptoms, physical symptoms and inflammatory biomarkers of IBD[161]. The BBMW group had significant improvement on Brief Symptom Inventory 18, Beck Anxiety Inventory, Beck Depression Inventory, IBD Questionnaire, and Perceived Stress Questionnaire. Interestingly, median C-reactive protein (CRP) values decreased significantly in the BBMW group but no significant change in CRP values were seen in the educational seminar group[161] (LOE 1b).
Risk of relapse: Low stress was associated with reduced relapse in a cohort study of 101 patients with quiescent CD followed for one year[159]. The INSPIRE study evaluated the role of stress management psychotherapy in active IBD patients with high scores (> 60) on the perceived stress questionnaire[161]. One hundred and fourteen patients were divided into two groups, the first group received usual treatment and the second group received usual treatment and psychotherapy. The intervention did not improve disease or reduce relapse; however, there was a small increase in the IBDQ score (P = 0.009, mean differences 16.3 ± 6.1 in patients with UC[161]. Similarly a Cochrane review of 21 studies and 1745 patients did not show any improvement in IBD relapse or remission rates with psychological interventions aimed at reducing stress[162] (LOE 1b).
Sleep
Sleep disturbances have been strongly associated with IBD and other chronic inflammatory diseases such as Rheumatoid arthritis and Lupus[163]. Sleep disturbances in these diseases are due to cytokines produced by chronic inflammation that are also known to affect sleep. Inflammation plays a role in regulating sleep and the interplay of inflammatory cytokines and the sleep cycle is complex. In animal studies, increased levels of Interleukin-1 (IL-1) and TNF-α were associated with an increase in NREM sleep[164,165]. IL-1 at low levels induces NREM sleep and at higher levels, can cause NREM suppression and sleep fragmentation[166,167]. Interleukin-6 (IL-6) mediates the acute-phase response. IL-6 has been shown to suppress REM sleep and promote wakefulness in patients with IBD[168,169]. Nocturnal diarrhea also disturbs sleep in IBD patients; therefore the high prevalence of sleeping disorders in IBD patients is not surprising (LOE 5).
Risk of disease: It has been hypothesized that sleep disturbances are not merely an outcome, but rather a cause of chronic inflammatory diseases, and there is some evidence to support this hypothesis. Sleep is divided in to two parts, REM and Non REM sleep. NREM sleep accounts for 80% of total sleep time and is broken down into 4 stages. Stages 3 and 4 of NREM sleep are often referred to as slow-wave sleep (SWS) and are considered the most restorative stages of sleep where the greatest impact from immune regulation occurs. The effects of SWS can lead to a decrease in colon contractility, which is considered the “rest period” for the colon, so alterations in this stage of sleep can have direct effects on GI physiology such as diminished mucosal integrity[170,171]. When sleep is disturbed in healthy young volunteers, Interleukin (IL)-1 (beta), TNF- (alpha), and IL-6, the 3 major proinflammatory cytokines that are important in IBD are increased[170,172].
Risk of disease progression: Sleep deprivation was shown to worsen inflammation and delay healing in a murine model of colitis[173]. IBD patients have poor sleep quality, prolonged sleep latency, and increase use of sleeping pills when compared with healthy controls[174,175]. Patients with clinically active IBD have significantly worse sleep than patients with inactive disease[174,176,177] (LOE 2b, 3,5).
Risk of relapse: A cohort study of 3173 subjects showed that poor sleep increases the risk of relapse in patients with inactive CD but not UC[178]. In another cohort study, IBD patients in clinical remission but with abnormal sleep were at increased risk of relapse at six months when compared to patients in clinical remission with good sleep[174]. Currently, the evidence is not strong enough to mandate treating sleep disorders in patients with IBD solely for the purpose of improving IBD outcomes, nevertheless, it is worth noting that sleep disorders can be a significant quality of life issue and all patients with IBD should be screened. In the future screening and treatment of sleep disorders might have therapeutic implications in the treatment of IBD (LOE 2b, 2b).
Vaccinations
The effect of vaccinations on the incidence of IBD is controversial. It was previously thought that vaccinations decreased early childhood infections which in turn may favor the onset of immunologic diseases[179]. Viral or bacterial components and chemical adjuvants (e.g., Aluminum) contained in many vaccines were also considered risk factors for incident IBD because of the potential risk of stimulating the immune system leading to a deregulated inflammatory response[180] (LOE 3b, 5).
Risk of disease: It was first reported in 1995 that the measles vaccine increased the risk of developing IBD. Subsequent studies have not shown any association between measles vaccination and IBD. Epidemiologic studies that investigated other vaccines such as BCG, diphtheria, tetanus, poliomyelitis, smallpox, pertussis, rubella, and mumps, have reported conflicting results[181-183]. A large meta-analysis that included 11 studies (2400 IBD patients and 34000 controls) did not find any significant increased risk of developing IBD after childhood immunization with BCG, diphtheria, tetanus, smallpox, pertussis, measles, mumps, and rubella-containing vaccines[183]. Interestingly, there was an increased risk of IBD after poliomyelitis vaccination, however the studies included in the meta-analysis had significant heterogeneity amongst them, which limits the generalizability of the results[183] (LOE 3b, 3a).
Breast feeding
IBD incidence peaks in early adulthood and therefore early environmental exposures are likely to have a profound effect on future IBD susceptibility. Breastfeeding is an early environmental exposure that affects the development of the immune system and the gut microbiome[184]. Breastfeeding plays a very important role in protecting against early enteric infections[185]. Human milk contains (1) lactoferrin that prevents the multiplication of bacteria by chelating iron; (2) IgA that prevents the binding of bacteria to the epithelium and also neutralizes toxins; and (3) Lactadherin that prevents the binding of rotavirus; the latter is the leading cause of gastroenteritis in infants. Other components of human milk that are protective against infections are lysozyme, MUC1, C3, defensins and fibronectin[186]. The gut microbiome is regulated by breastfeeding as it inhibits the growth of some bacteria due to its anti-bacterial components and promotes the growth of certain bacteria such as Bifidobacterium and Lactobacillus by producing growth factors[184]. Breast milk has anti-inflammatory properties, lactoferrin binds to bacterial toxins such as LPS, PAF-acetyl hydrolase and breaks down inflammatory mediators, IL-10, and TGF-1 modulate inflammatory leucocytes[186]. A meta-analysis of case-control studies with significant heterogeneity found that breastfeeding was protective for both CD and UC[187]. However, other studies have shown that breastfeeding is either a risk factor or has no association with IBD[188,189]. Further research is needed to clarify the direction of the association between breastfeeding and IBD (LOE 2b, 3a, 3b).
Appendectomy
Risk for Crohn’s disease: The association between appendectomy and CD is conflicting and there has been no consistent association between both entities. Some studies have shown that appendectomy is a risk factor for the development of CD[190,191]. In contrast, other studies have shown no association with CD[192]. A meta-analysis showed an increased risk of CD following an appendectomy, however the risk decreased to baseline after 5 years[193] (LOE 3a, 3b).
Risk for ulcerative colitis: Murine models have suggested that removal of the appendix could have an immune modulating effect that protects against UC[194]. Observational studies in humans have shown that appendectomy is inversely related to UC[195,196]. The exact mechanism by which appendectomy protects against UC is unclear. A case-control study showed that patients who had an appendectomy had a significantly lower incidence of UC than matched controls[195]. It is interesting to note that this held true only for patients who had an appendectomy done for appendicitis or mesenteric lymphadenitis[195]. The relationship did not hold true for appendectomies done for non-specific abdominal pain (i.e., appendix is found to be normal on post-operative pathology). Moreover, the inverse relationship of appendectomy to the risk of UC was only seen when the surgeries were done prior to the age of 20 years[195] (LOE 2b, 2b, 5).
DISCUSSION
The available evidence suggests that environmental exposures have variable effects on individuals with IBD. Advances in genetics and immunology have contributed to our understanding of the pathogenesis of IBD. Environmental exposures or exposomes are believed to be the possible missing link to increasing our understanding of the etiology and increased incidence of IBD in recent years. However, the picture is incomplete. Human studies are extremely limited in their ability to test isolated environmental exposures to demonstrate causation or to assess mechanisms of disease. Given the heterogeneity of environmental factors and the fact that none of the other risk factors (genetic predisposition, immune dysregulation, and dysbiosis) can cause IBD on their own; the challenge is how to effectively translate promising results from animal studies to humans, in order to develop models that incorporate the complex interactions between the environment, genetics, gut microbiota, and the immune system (Figure 1). Recent studies on the incidence of IBD in immigrant populations in western countries further support the role of environmental exposures on the incidence of IBD by suggesting that recent immigrants to western countries from countries with a low risk of IBD acquire the risk of IBD in their adopted country rather than their country of origin[10,26]. However, the impact of modifying specific environmental factors on causation and established disease remain inadequately studied with limited high quality data from interventional studies to guide clinical practice (Tables 3 and 4)[197-210]. Prospective quantification of environmental exposures in patients at risk of IBD (e.g., first degree relatives) at different time points from birth to adulthood may be useful to predict the natural history of IBD. GWAS of SNP-based Gene (G) × Environment (E) analysis using either the case-only test or standard case-control interaction test under different statistical assumptions has been suggested as a means to further study G x G and G x E interactions in IBD[211,212]. Specifically, the case-only analysis is more statistically powerful and tests the association between the environmental exposure and the SNP of interest in the cases[213]. Additionally, omics technologies (genomics, transcriptomics, epigenomics and metabolomics) have provided the opportunity to analyze large population-based databases and their associated biobanks to detect metabolites in blood or urine[213]. Epigenetics is the study of modifications in regulation of gene expression that occur without changes to the DNA sequence at the interface between environment and heritable molecular and cellular phenotypes[214]. Epigenetic studies have identified MicroRNAs (miRNAs) as regulators of autophagy and intracellular bacterial processing in IBD[214]. Quantifying epigenetic changes and further sequencing of the gut microbiome to determine if specific “dysbiotic signatures” are consistent with future development of an IBD phenotype could extend our understanding of the etiopathogenesis of IBD with implications for prevention, diagnosis, and treatment. There is a need for high quality interventional studies that assess the impact of modifying environmental exposures on the natural history and patient outcomes in IBD (LOE 2b, 5).
Figure 1.
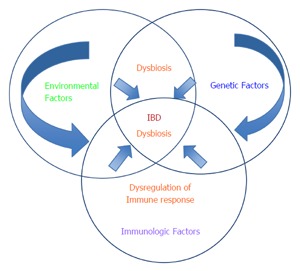
Schematic representation of risk factors contributing to the development of inflammatory bowel diseases. IBD: Inflammatory bowel diseases.
Table 3.
Effect of environmental factors in Crohn’s disease and impact of interventional studies to modify specific environmental factors
| Ref. | Disease onset (Incident CD) | Disease progression | Study population and design | Intervention and comparison group | Outcome |
| Lifestyle | |||||
| Smoking[11,13] (LOE 2b, 2a) | ↑ | ↑ | Cohort study current smokers with CD (n = 474)[17] (LOE 2b) | Smoking cessation counselling | Decreased risk of flares, need for surgery and immunosuppressive therapy[17] |
| Cohort study current smokers with CD (n = 408)[18] (LOE 2b) | Quitters vs non-quitters | Continuing smokers had more disease relapses, and patients who quit smoking had similar relapse incidence compared with non-smokers[18] | |||
| Sleep[177,178] (LOE 2b) | No data | ↑ | None | None | No data |
| Stress[158,159] (LOE 2b) | No data | ↑? | Adult and adolescent patients with IBD Systematic review of RCTs and quasi-RCTs (n = 1745)[162] (LOE 1a) | Multi-modality psychotherapy | No evidence for efficacy of psychological therapy in adult patients with IBD In adolescents, psychological interventions may be beneficial, but the evidence is limited |
| Diet | |||||
| Dietary fat[118] (LOE 3a) | n-6 PUFA↑ | ↓ | CD in remission Systematic review of RCTs (n = 1039)[201] (LOE 1a) | Fish oil n-3 (PUFA) or placebo | Non-significant trend towards lower risk of relapse at 1 yr in fish oil group compared with placebo |
| n-3 PUFA↓ | |||||
| Dietary protein[118,120] (LOE 3a, 2b) | Animal protein( meat and fish)↑ | ↔? | Mild- moderate CD | Restricted diet ( red meat + spelt bread) or control diet (low-fiber, low-fat, and high-carbohydrate ) | Radiologic and endoscopic improvement in restricted diet group (interpret with caution; small study with limited generalizability) |
| Vegetable and diary↓ | RCT (n = 18)[197] (LOE 2b) | ||||
| Dietary fiber[118,120] (LOE 3a, 2b) | Fruit and vegetable fiber↓ | ↓ | Inactive or mildly active CD, RCT (n = 352)[207] (LOE 1b) | High fiber diet vs low fiber | No difference in disease activity, surgery or hospitalizations |
| Food additives [Microparticles (MP)[130,131] (LOE 5)] | High MP-diet↑ | High-MP diet↑ | Active CD RCT (n = 20)[203] (LOE 1b) RCT(n = 83)[202] (LOE 1b) | Low -MP-diet vs control diet | Decrease in CDAI in smaller trial[203] |
| No difference in larger trial[202] | |||||
| Fruits and vegetables[118] (LOE 3a) | ↓ | ↓? | CD in remission | Semi-vegetarian diet or omnivorous diet | Maintenance of remission rates higher on semi-vegetarian diet compared to omnivorous diet |
| RCT (n = 22)[198] (LOE 1b) | |||||
| Food antigens[128] (LOE 4) | No data | ↑ | Active and inactive CD RCT (n = 40)[129] (LOE 2b) Active CD Systematic review RCTs (n = 334)[210] (LOE 1a) | Elimination diet based on IgG positivity to cheese and yeast or sham diet | Daily stool frequency significantly decreased by 11% during a specific diet compared with a sham diet. Abdominal pain reduced and general well-being improved[129] |
| Elemental vs non-elemental diet | No difference in the efficacy between elemental and non-elemental diet[210] | ||||
| Enteral nutrition | No data | ↔ | Active CD Systematic review (n = 192)[210] (LOE 1a) | Enteral nutrition vs corticosteroids | Enteral nutrition less effective than corticosteroids for induction of remission |
| Breastfeeding[187-189] (LOE 3a, 3b, 2b) | ↔ | No data | None | None | No data |
| Pharmacologic agents | |||||
| Nsaids[139-140] (LOE2b) | ↑? | ↑ | Inactive IBD with arthralgia. | Rofecoxib 25 mg or 12.5 mg x 20 d | 41% responded with reduction in arthralgia scores. P < 0.05. No IBD flares |
| Open label trial (n = 32)[144] (LOE 2b) | 9% developed GI side effects | ||||
| Oral contraceptives[150,151,153,155] (LOE 3a, 2b) | ↔ | ↔ | None | None | No data |
| Antibiotics[145-147] (LOE 3b, 3a) | Early exposure↑ | ↓ | Active CD Systematic review of RCTs (n = 1160)[71] (LOE 1a-) | Antibiotic or placebo | Antibiotics superior to placebo at inducing remission |
| Vaccination[183] (LOE 3a) | No effect | No effect | None | None | None |
| Gut microbiome | |||||
| Dysbiosis[80-82] (LOE 4) | ↑ | ↑ | Mild-moderate CD Systematic review of RCTs (n = 746)[199] (LOE 1a) | Probiotics, prebiotics and synbiotics or placebo | Insufficient data to recommend probiotics for use in CD |
| Ecological (Abiotic) | |||||
| Air pollution[33,34] (LOE 2c, 3b) | ↑ | ↑? | None | None | No data |
| Water pollution[36-38] (LOE 5) | ↑? | ↑? | None | None | No data |
| Low Vitamin D[42,44,57,59] (LOE 2b, 3a, 2b) | ↑ | ↑ | CD in remission RCT (n = 94)[51] (LOE 1a) | Vitamin D3 or placebo | Lower relapse rates in patients randomized to vitamin D3 1200 IU/d[51] |
| Mild-moderate CD Cohort study (n = 18)[60] (LOE 2b) | No comparison group | 24 wk of vitamin D3 (up to 5000 IU/d) reduced mean CDAI scores by 112 ± 81 points from 230 ± 74 to 118 ± 66 (P < 0.0001). Quality-of-life scores also improved following vitamin D supplementation[60] | |||
| Surgery | |||||
| Appendectomy[192,193] (LOE 3b, 3a) | ↔ | No data | None | None | No data |
↑ : Increased risk; ↔ : Equivocal risk; ↓ : Decreased risk; ? : Questionable risk. LoE: Levels of Evidence; CD: Crohn's disease; IBD: Inflammatory bowel diseases; PUFA: Polyunsaturated fatty acid.
Table 4.
Effect of environmental factors on ulcerative colitis and impact of interventional studies to modify specific environmental factors
| Ref. | Disease onset (incident UC) | Disease Activity | Study population and design | Intervention and comparison group | Outcome |
| Lifestyle | |||||
| Smoking[11,20,24] (LOE 2b, 3b, 2a) | Current smoking ↓ | ↓ | Mild-moderate UC Systematic review (n = 233)[205] (LOE 1a) (n = 81)[205] (LOE 1a) | Nicotine or placebo | No evidence for efficacy for nicotine preparations in inducing remission in UC |
| Smoking cessation ↑ | Nicotine or corticosteroids | ||||
| Sleep[176,179] (LOE 2b) | No data | ↑ | None | None | No data |
| Stress[158,159] (LOE 5, 2b) | No data | ↑? | Adult and adolescent patients with IBD | Multi-modality psychotherapy | No evidence for efficacy of psychological therapy in adult patients with IBD |
| Systematic review of RCTs and quasi-RCTs (n = 1745)[162] (LOE 1a) | In adolescents, psychological interventions may be beneficial, but the evidence is limited | ||||
| Diet | |||||
| Dietary fat[118] (LOE 3a) | n-3 PUFA ↓ | n-3 PUFA ↓ | UC in remission Systematic review of RCTs (n = 148)[208] (LOE 1a) | fish oil (n-3 PUFA) or placebo | No difference in risk of relapse between n-3 PUFA compared with placebo |
| n-6 PUFA ↑ | |||||
| Dietary milk[116,117] (LOE 5) | ↑ | No data | Active UC | Milk-free diet or sham diet | Fewer relapses on milk-free diet than on sham diet |
| RCT (n = 77)[209] (LOE 2b) | |||||
| Dietary protein[118] (LOE 3a) | ↑ | ↑ | None | None | No data |
| Dietary fiber[118,120] (LOE 2b) | ↔ | ↔ | UC in remission Open label RCT (n = 59)[200](LOE 2b) | Germinated barley food stuff (GBF) + conventional therapy or conventional therapy | Prolonged maintenance of remission in GBF group[200] |
| UC in remission Open label RCT (n = 105)[122] (LOE 2b) | Plantago ovata or Mesalamine | Plantago ovata as effective as Mesalamine in maintenance of remission[122] | |||
| Food antigens[128] (LOE 4) | ↑? | No data | None | None | No data |
| Food additives[131,132] (LOE 5) | ↑? | No data | None | None | No data |
| Breastfeeding[187,189] (LOE 3a, 3b, 2b) | ↔ | No data | None | None | No data |
| Medication | |||||
| Nsaids[139,140] (LOE 2b) | ↑? | ↑ | Quiescent to mild | Rofecoxib 25 mg or 12.5 mg × 20 d | 41% responded with reduction in arthralgia scores. P < 0.05. No IBD flares 9% developed GI side effects |
| UC and CD with arthralgia | |||||
| Prospective Open label trial (n = 32) | |||||
| Oral contraceptives[150,151,153,155] (LOE 3a, 2b) | ↑ | ↔ | None | None | No data |
| Antibiotics[145,147] (LOE 3b, 3a) | Early exposure ↔ | ↓ | Active UC Systematic review of RCTs (n = 9 studies)[71] (LOE 1a) | Antibiotic or placebo | Antibiotics superior to placebo at inducing remission |
| Vaccination[183] (LOE 3a) | No effect | No data | None | None | No data |
| Gut microbiome | |||||
| Dysbiosis[80,83,84] (LOE 4) | ↑ | ↑ | Mild-moderate UC Systematic review of RCTs (n = 650)[199] (LOE 1a) | Probiotics + conventional treatment or placebo | Probiotics effective for induction and maintenance of remission in UC and pouchitis[199] |
| Active UC RCT (n = 70)[204] (LOE 1b) | Fecal microbiota transplant (FMT) or Placebo | FMT induced remission in a significantly greater percentage of patients with active UC than placebo (24% vs 5%)[204] | |||
| Active UC, RCT (n = 100)[206] (LOE1b) | Ciprofloxacin + E-coli Nissle or placebo + E-coli Nissle | No benefit in the use of E. coli Nissle as an add-on treatment to conventional therapies for active UC | |||
| Ecological (Abiotic) | |||||
| Air pollution[33,34] (LOE 2c, 3b) | ↑ | ↑ | None | None | No data |
| Water pollution[36-38] (LOE 5) | ↑ | ↑ | None | None | No data |
| Low Vitamin D[44,57] (LOE 2a, 2b) | ↑ | ↑ | Active UC Cohort study (n = 368)[59] (LOE 2b) | Vitamin D3 or No treatment | Reduction in health-care utilization in the vitamin D treatment group |
| Surgery | |||||
| Appendectomy[195-196] (LOE 2b, 3b) | ↓ | No data | None | None | No data |
↑ : Increased risk; ↔ : Equivocal risk; ↓ : Decreased risk; ? : Questionable risk. LoE: Levels of Evidence; CD: Crohn's disease; IBD: Inflammatory bowel diseases; PUFA: Polyunsaturated fatty acid; UC: Ulcerative colitis.
Footnotes
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Manuscript source: Invited manuscript
Peer-review started: March 11, 2016
First decision: April 14, 2016
Article in press: June 29, 2016
P- Reviewer: Huang VW S- Editor: Qi Y L- Editor: A E- Editor: Wang CH
References
- 1.Russell RK, Satsangi J. Does IBD run in families? Inflamm Bowel Dis. 2008;14 Suppl 2:S20–S21. doi: 10.1002/ibd.20573. [DOI] [PubMed] [Google Scholar]
- 2.Ek WE, D’Amato M, Halfvarson J. The history of genetics in inflammatory bowel disease. Ann Gastroenterol. 2014;27:294–303. [PMC free article] [PubMed] [Google Scholar]
- 3.Hugot JP, Chamaillard M, Zouali H, Lesage S, Cézard JP, Belaiche J, Almer S, Tysk C, O’Morain CA, Gassull M, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature. 2001;411:599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- 4.Lakatos L, Mester G, Erdelyi Z, Balogh M, Szipocs I, Kamaras G, Lakatos PL. Striking elevation in incidence and prevalence of inflammatory bowel disease in a province of western Hungary between 1977-2001. World J Gastroenterol. 2004;10:404–409. doi: 10.3748/wjg.v10.i3.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wild CP. Complementing the genome with an “exposome”: the outstanding challenge of environmental exposure measurement in molecular epidemiology. Cancer Epidemiol Biomarkers Prev. 2005;14:1847–1850. doi: 10.1158/1055-9965.EPI-05-0456. [DOI] [PubMed] [Google Scholar]
- 6.Wild CP. The exposome: from concept to utility. Int J Epidemiol. 2012;41:24–32. doi: 10.1093/ije/dyr236. [DOI] [PubMed] [Google Scholar]
- 7.Phillips B, Ball C, Sackett D, Badenoch D, Straus S, Haynes B, Dawes M. Oxford Centre for Evidence-based Medicine. Levels of Evidence. Available from: http://www.cebm.net/oxford-centre-evidence-based-medicine-levels-evidence-march-2009/
- 8.Strachan DP. Hay fever, hygiene, and household size. BMJ. 1989;299:1259–1260. doi: 10.1136/bmj.299.6710.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Noverr MC, Huffnagle GB. The ‘microflora hypothesis’ of allergic diseases. Clin Exp Allergy. 2005;35:1511–1520. doi: 10.1111/j.1365-2222.2005.02379.x. [DOI] [PubMed] [Google Scholar]
- 10.Leong RW, Mitrev N, Ko Y. Hygiene Hypothesis: Is the Evidence the Same All Over the World? Dig Dis. 2016;34:35–42. doi: 10.1159/000442922. [DOI] [PubMed] [Google Scholar]
- 11.Higuchi LM, Khalili H, Chan AT, Richter JM, Bousvaros A, Fuchs CS. A prospective study of cigarette smoking and the risk of inflammatory bowel disease in women. Am J Gastroenterol. 2012;107:1399–1406. doi: 10.1038/ajg.2012.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johnson GJ, Cosnes J, Mansfield JC. Review article: smoking cessation as primary therapy to modify the course of Crohn’s disease. Aliment Pharmacol Ther. 2005;21:921–931. doi: 10.1111/j.1365-2036.2005.02424.x. [DOI] [PubMed] [Google Scholar]
- 13.Mahid SS, Minor KS, Soto RE, Hornung CA, Galandiuk S. Smoking and inflammatory bowel disease: a meta-analysis. Mayo Clin Proc. 2006;81:1462–1471. doi: 10.4065/81.11.1462. [DOI] [PubMed] [Google Scholar]
- 14.Mahid SS, Minor KS, Stromberg AJ, Galandiuk S. Active and passive smoking in childhood is related to the development of inflammatory bowel disease. Inflamm Bowel Dis. 2007;13:431–438. doi: 10.1002/ibd.20070. [DOI] [PubMed] [Google Scholar]
- 15.Cosnes J, Nion-Larmurier I, Afchain P, Beaugerie L, Gendre JP. Gender differences in the response of colitis to smoking. Clin Gastroenterol Hepatol. 2004;2:41–48. doi: 10.1016/s1542-3565(03)00290-8. [DOI] [PubMed] [Google Scholar]
- 16.Louis E, Michel V, Hugot JP, Reenaers C, Fontaine F, Delforge M, El Yafi F, Colombel JF, Belaiche J. Early development of stricturing or penetrating pattern in Crohn’s disease is influenced by disease location, number of flares, and smoking but not by NOD2/CARD15 genotype. Gut. 2003;52:552–557. doi: 10.1136/gut.52.4.552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cosnes J, Beaugerie L, Carbonnel F, Gendre JP. Smoking cessation and the course of Crohn’s disease: an intervention study. Gastroenterology. 2001;120:1093–1099. doi: 10.1053/gast.2001.23231. [DOI] [PubMed] [Google Scholar]
- 18.Nunes T, Etchevers MJ, García-Sánchez V, Ginard D, Martí E, Barreiro-de Acosta M, Gomollón F, Arroyo M, Bastida G, Gonzalez B, et al. Impact of Smoking Cessation on the Clinical Course of Crohn’s Disease Under Current Therapeutic Algorithms: A Multicenter Prospective Study. Am J Gastroenterol. 2016;111:411–419. doi: 10.1038/ajg.2015.401. [DOI] [PubMed] [Google Scholar]
- 19.Nunes T, Etchevers MJ, Merino O, Gallego S, García-Sánchez V, Marín-Jiménez I, Menchén L, Barreiro-de Acosta M, Bastida G, García S, et al. High smoking cessation rate in Crohn’s disease patients after physician advice--the TABACROHN Study. J Crohns Colitis. 2013;7:202–207. doi: 10.1016/j.crohns.2012.04.011. [DOI] [PubMed] [Google Scholar]
- 20.Boyko EJ, Koepsell TD, Perera DR, Inui TS. Risk of ulcerative colitis among former and current cigarette smokers. N Engl J Med. 1987;316:707–710. doi: 10.1056/NEJM198703193161202. [DOI] [PubMed] [Google Scholar]
- 21.Mokbel M, Carbonnel F, Beaugerie L, Gendre JP, Cosnes J. [Effect of smoking on the long-term course of ulcerative colitis] Gastroenterol Clin Biol. 1998;22:858–862. [PubMed] [Google Scholar]
- 22.Boyko EJ, Perera DR, Koepsell TD, Keane EM, Inui TS. Effects of cigarette smoking on the clinical course of ulcerative colitis. Scand J Gastroenterol. 1988;23:1147–1152. doi: 10.3109/00365528809090183. [DOI] [PubMed] [Google Scholar]
- 23.Höie O, Wolters F, Riis L, Aamodt G, Solberg C, Bernklev T, Odes S, Mouzas IA, Beltrami M, Langholz E, et al. Ulcerative colitis: patient characteristics may predict 10-yr disease recurrence in a European-wide population-based cohort. Am J Gastroenterol. 2007;102:1692–1701. doi: 10.1111/j.1572-0241.2007.01265.x. [DOI] [PubMed] [Google Scholar]
- 24.Gisbert JP, Marín AC, Chaparro M. Systematic review: factors associated with relapse of inflammatory bowel disease after discontinuation of anti-TNF therapy. Aliment Pharmacol Ther. 2015;42:391–405. doi: 10.1111/apt.13276. [DOI] [PubMed] [Google Scholar]
- 25.Benchimol EI, Mack DR, Guttmann A, Nguyen GC, To T, Mojaverian N, Quach P, Manuel DG. Inflammatory bowel disease in immigrants to Canada and their children: a population-based cohort study. Am J Gastroenterol. 2015;110:553–563. doi: 10.1038/ajg.2015.52. [DOI] [PubMed] [Google Scholar]
- 26.Soon IS, Molodecky NA, Rabi DM, Ghali WA, Barkema HW, Kaplan GG. The relationship between urban environment and the inflammatory bowel diseases: a systematic review and meta-analysis. BMC Gastroenterol. 2012;12:51. doi: 10.1186/1471-230X-12-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bernstein CN, Kraut A, Blanchard JF, Rawsthorne P, Yu N, Walld R. The relationship between inflammatory bowel disease and socioeconomic variables. Am J Gastroenterol. 2001;96:2117–2125. doi: 10.1111/j.1572-0241.2001.03946.x. [DOI] [PubMed] [Google Scholar]
- 28.Declercq C, Gower-Rousseau C, Vernier-Massouille G, Salleron J, Baldé M, Poirier G, Lerebours E, Dupas JL, Merle V, Marti R, et al. Mapping of inflammatory bowel disease in northern France: spatial variations and relation to affluence. Inflamm Bowel Dis. 2010;16:807–812. doi: 10.1002/ibd.21111. [DOI] [PubMed] [Google Scholar]
- 29.Nerich V, Monnet E, Weill A, Vallier N, Vanbockstael V, Auleley GR, Balaire C, Dubost P, Rican S, Allemand H, et al. Fine-scale geographic variations of inflammatory bowel disease in France: correlation with socioeconomic and house equipment variables. Inflamm Bowel Dis. 2010;16:813–821. doi: 10.1002/ibd.21122. [DOI] [PubMed] [Google Scholar]
- 30.Zheng JJ, Zhu XS, Huangfu Z, Gao ZX, Guo ZR, Wang Z. Crohn’s disease in mainland China: a systematic analysis of 50 years of research. Chin J Dig Dis. 2005;6:175–181. doi: 10.1111/j.1443-9573.2005.00227.x. [DOI] [PubMed] [Google Scholar]
- 31.Hu D, Ren J, Wang G, Gu G, Liu S, Wu X, Chen J, Ren H, Hong Z, Li J. Geographic mapping of Crohn’s disease and its relation to affluence in jiangsu province, an eastern coastal province of china. Gastroenterol Res Pract. 2014;2014:590467. doi: 10.1155/2014/590467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaplan GG, Hubbard J, Korzenik J, Sands BE, Panaccione R, Ghosh S, Wheeler AJ, Villeneuve PJ. The inflammatory bowel diseases and ambient air pollution: a novel association. Am J Gastroenterol. 2010;105:2412–2419. doi: 10.1038/ajg.2010.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Salim SY, Kaplan GG, Madsen KL. Air pollution effects on the gut microbiota: a link between exposure and inflammatory disease. Gut Microbes. 2014;5:215–219. doi: 10.4161/gmic.27251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ananthakrishnan AN, McGinley EL, Binion DG, Saeian K. Ambient air pollution correlates with hospitalizations for inflammatory bowel disease: an ecologic analysis. Inflamm Bowel Dis. 2011;17:1138–1145. doi: 10.1002/ibd.21455. [DOI] [PubMed] [Google Scholar]
- 35.Masuyama H, Hiramatsu Y, Kunitomi M, Kudo T, MacDonald PN. Endocrine disrupting chemicals, phthalic acid and nonylphenol, activate Pregnane X receptor-mediated transcription. Mol Endocrinol. 2000;14:421–428. doi: 10.1210/mend.14.3.0424. [DOI] [PubMed] [Google Scholar]
- 36.Soares A, Guieysse B, Jefferson B, Cartmell E, Lester JN. Nonylphenol in the environment: a critical review on occurrence, fate, toxicity and treatment in wastewaters. Environ Int. 2008;34:1033–1049. doi: 10.1016/j.envint.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 37.Vedani A, Smiesko M, Spreafico M, Peristera O, Dobler M. VirtualToxLab - in silico prediction of the toxic (endocrine-disrupting) potential of drugs, chemicals and natural products. Two years and 2,000 compounds of experience: a progress report. ALTEX. 2009;26:167–176. doi: 10.14573/altex.2009.3.167. [DOI] [PubMed] [Google Scholar]
- 38.Wagner M, Schlüsener MP, Ternes TA, Oehlmann J. Identification of putative steroid receptor antagonists in bottled water: combining bioassays and high-resolution mass spectrometry. PLoS One. 2013;8:e72472. doi: 10.1371/journal.pone.0072472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cantorna MT, Mahon BD. Mounting evidence for vitamin D as an environmental factor affecting autoimmune disease prevalence. Exp Biol Med (Maywood) 2004;229:1136–1142. doi: 10.1177/153537020422901108. [DOI] [PubMed] [Google Scholar]
- 40.Cantorna MT, Mahon BD. D-hormone and the immune system. J Rheumatol Suppl. 2005;76:11–20. [PubMed] [Google Scholar]
- 41.Cantorna MT, Zhu Y, Froicu M, Wittke A. Vitamin D status, 1,25-dihydroxyvitamin D3, and the immune system. Am J Clin Nutr. 2004;80:1717S–1720S. doi: 10.1093/ajcn/80.6.1717S. [DOI] [PubMed] [Google Scholar]
- 42.Ananthakrishnan AN, Khalili H, Higuchi LM, Bao Y, Korzenik JR, Giovannucci EL, Richter JM, Fuchs CS, Chan AT. Higher predicted vitamin D status is associated with reduced risk of Crohn’s disease. Gastroenterology. 2012;142:482–489. doi: 10.1053/j.gastro.2011.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cantorna MT, Munsick C, Bemiss C, Mahon BD. 1,25-Dihydroxycholecalciferol prevents and ameliorates symptoms of experimental murine inflammatory bowel disease. J Nutr. 2000;130:2648–2652. doi: 10.1093/jn/130.11.2648. [DOI] [PubMed] [Google Scholar]
- 44.Del Pinto R, Pietropaoli D, Chandar AK, Ferri C, Cominelli F. Association Between Inflammatory Bowel Disease and Vitamin D Deficiency: A Systematic Review and Meta-analysis. Inflamm Bowel Dis. 2015;21:2708–2717. doi: 10.1097/MIB.0000000000000546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Froicu M, Cantorna MT. Vitamin D and the vitamin D receptor are critical for control of the innate immune response to colonic injury. BMC Immunol. 2007;8:5. doi: 10.1186/1471-2172-8-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Froicu M, Weaver V, Wynn TA, McDowell MA, Welsh JE, Cantorna MT. A crucial role for the vitamin D receptor in experimental inflammatory bowel diseases. Mol Endocrinol. 2003;17:2386–2392. doi: 10.1210/me.2003-0281. [DOI] [PubMed] [Google Scholar]
- 47.Froicu M, Zhu Y, Cantorna MT. Vitamin D receptor is required to control gastrointestinal immunity in IL-10 knockout mice. Immunology. 2006;117:310–318. doi: 10.1111/j.1365-2567.2005.02290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Holick MF. Sunlight and vitamin D for bone health and prevention of autoimmune diseases, cancers, and cardiovascular disease. Am J Clin Nutr. 2004;80:1678S–1688S. doi: 10.1093/ajcn/80.6.1678S. [DOI] [PubMed] [Google Scholar]
- 49.Holmes EA, Xiang F, Lucas RM. Variation in incidence of pediatric Crohn’s disease in relation to latitude and ambient ultraviolet radiation: a systematic review and analysis. Inflamm Bowel Dis. 2015;21:809–817. doi: 10.1097/MIB.0000000000000320. [DOI] [PubMed] [Google Scholar]
- 50.Hughes DJ, McManus R, Neary P, O’morain C, O’sullivan M. Common variation in the vitamin D receptor gene and risk of inflammatory bowel disease in an Irish case-control study. Eur J Gastroenterol Hepatol. 2011;23:807–812. doi: 10.1097/MEG.0b013e328349283e. [DOI] [PubMed] [Google Scholar]
- 51.Jørgensen SP, Agnholt J, Glerup H, Lyhne S, Villadsen GE, Hvas CL, Bartels LE, Kelsen J, Christensen LA, Dahlerup JF. Clinical trial: vitamin D3 treatment in Crohn’s disease - a randomized double-blind placebo-controlled study. Aliment Pharmacol Ther. 2010;32:377–383. doi: 10.1111/j.1365-2036.2010.04355.x. [DOI] [PubMed] [Google Scholar]
- 52.Miheller P, Muzes G, Hritz I, Lakatos G, Pregun I, Lakatos PL, Herszényi L, Tulassay Z. Comparison of the effects of 1,25 dihydroxyvitamin D and 25 hydroxyvitamin D on bone pathology and disease activity in Crohn’s disease patients. Inflamm Bowel Dis. 2009;15:1656–1662. doi: 10.1002/ibd.20947. [DOI] [PubMed] [Google Scholar]
- 53.Naderi N, Farnood A, Habibi M, Derakhshan F, Balaii H, Motahari Z, Agah MR, Firouzi F, Rad MG, Aghazadeh R, et al. Association of vitamin D receptor gene polymorphisms in Iranian patients with inflammatory bowel disease. J Gastroenterol Hepatol. 2008;23:1816–1822. doi: 10.1111/j.1440-1746.2008.05525.x. [DOI] [PubMed] [Google Scholar]
- 54.Narula N, Marshall JK. Management of inflammatory bowel disease with vitamin D: beyond bone health. J Crohns Colitis. 2012;6:397–404. doi: 10.1016/j.crohns.2011.10.015. [DOI] [PubMed] [Google Scholar]
- 55.Nerich V, Jantchou P, Boutron-Ruault MC, Monnet E, Weill A, Vanbockstael V, Auleley GR, Balaire C, Dubost P, Rican S, et al. Low exposure to sunlight is a risk factor for Crohn’s disease. Aliment Pharmacol Ther. 2011;33:940–945. doi: 10.1111/j.1365-2036.2011.04601.x. [DOI] [PubMed] [Google Scholar]
- 56.Pappa HM, Grand RJ, Gordon CM. Report on the vitamin D status of adult and pediatric patients with inflammatory bowel disease and its significance for bone health and disease. Inflamm Bowel Dis. 2006;12:1162–1174. doi: 10.1097/01.mib.0000236929.74040.b0. [DOI] [PubMed] [Google Scholar]
- 57.Raffner Basson A, Swart R, Jordaan E, Mazinu M, Watermeyer G. Vitamin D Deficiency Increases the Risk for Moderate to Severe Disease Activity in Crohn’s Disease Patients in South Africa, Measured by the Harvey Bradshaw Index. J Am Coll Nutr. 2016;35:163–174. doi: 10.1080/07315724.2015.1039665. [DOI] [PubMed] [Google Scholar]
- 58.Simmons JD, Mullighan C, Welsh KI, Jewell DP. Vitamin D receptor gene polymorphism: association with Crohn’s disease susceptibility. Gut. 2000;47:211–214. doi: 10.1136/gut.47.2.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ulitsky A, Ananthakrishnan AN, Naik A, Skaros S, Zadvornova Y, Binion DG, Issa M. Vitamin D deficiency in patients with inflammatory bowel disease: association with disease activity and quality of life. JPEN J Parenter Enteral Nutr. 2011;35:308–316. doi: 10.1177/0148607110381267. [DOI] [PubMed] [Google Scholar]
- 60.Yang L, Weaver V, Smith JP, Bingaman S, Hartman TJ, Cantorna MT. Therapeutic effect of vitamin d supplementation in a pilot study of Crohn’s patients. Clin Transl Gastroenterol. 2013;4:e33. doi: 10.1038/ctg.2013.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhu Y, Mahon BD, Froicu M, Cantorna MT. Calcium and 1 alpha,25-dihydroxyvitamin D3 target the TNF-alpha pathway to suppress experimental inflammatory bowel disease. Eur J Immunol. 2005;35:217–224. doi: 10.1002/eji.200425491. [DOI] [PubMed] [Google Scholar]
- 62.Khalili H, Huang ES, Ananthakrishnan AN, Higuchi L, Richter JM, Fuchs CS, Chan AT. Geographical variation and incidence of inflammatory bowel disease among US women. Gut. 2012;61:1686–1692. doi: 10.1136/gutjnl-2011-301574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Guarner F, Malagelada JR. Gut flora in health and disease. Lancet. 2003;361:512–519. doi: 10.1016/S0140-6736(03)12489-0. [DOI] [PubMed] [Google Scholar]
- 64.Dominguez-Bello MG, Blaser MJ, Ley RE, Knight R. Development of the human gastrointestinal microbiota and insights from high-throughput sequencing. Gastroenterology. 2011;140:1713–1719. doi: 10.1053/j.gastro.2011.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, Knight R. Bacterial community variation in human body habitats across space and time. Science. 2009;326:1694–1697. doi: 10.1126/science.1177486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Penders J, Thijs C, Vink C, Stelma FF, Snijders B, Kummeling I, van den Brandt PA, Stobberingh EE. Factors influencing the composition of the intestinal microbiota in early infancy. Pediatrics. 2006;118:511–521. doi: 10.1542/peds.2005-2824. [DOI] [PubMed] [Google Scholar]
- 67.Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hooper LV, Gordon JI. Commensal host-bacterial relationships in the gut. Science. 2001;292:1115–1118. doi: 10.1126/science.1058709. [DOI] [PubMed] [Google Scholar]
- 69.Sartor RB. Mechanisms of disease: pathogenesis of Crohn’s disease and ulcerative colitis. Nat Clin Pract Gastroenterol Hepatol. 2006;3:390–407. doi: 10.1038/ncpgasthep0528. [DOI] [PubMed] [Google Scholar]
- 70.Chassaing B, Darfeuille-Michaud A. The commensal microbiota and enteropathogens in the pathogenesis of inflammatory bowel diseases. Gastroenterology. 2011;140:1720–1728. doi: 10.1053/j.gastro.2011.01.054. [DOI] [PubMed] [Google Scholar]
- 71.Khan KJ, Ullman TA, Ford AC, Abreu MT, Abadir A, Marshall JK, Talley NJ, Moayyedi P. Antibiotic therapy in inflammatory bowel disease: a systematic review and meta-analysis. Am J Gastroenterol. 2011;106:661–673. doi: 10.1038/ajg.2011.72. [DOI] [PubMed] [Google Scholar]
- 72.Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci USA. 2007;104:13780–13785. doi: 10.1073/pnas.0706625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Harper PH, Lee EC, Kettlewell MG, Bennett MK, Jewell DP. Role of the faecal stream in the maintenance of Crohn’s colitis. Gut. 1985;26:279–284. doi: 10.1136/gut.26.3.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rutgeerts P, Goboes K, Peeters M, Hiele M, Penninckx F, Aerts R, Kerremans R, Vantrappen G. Effect of faecal stream diversion on recurrence of Crohn’s disease in the neoterminal ileum. Lancet. 1991;338:771–774. doi: 10.1016/0140-6736(91)90663-a. [DOI] [PubMed] [Google Scholar]
- 75.Lepage P, Seksik P, Sutren M, de la Cochetière MF, Jian R, Marteau P, Doré J. Biodiversity of the mucosa-associated microbiota is stable along the distal digestive tract in healthy individuals and patients with IBD. Inflamm Bowel Dis. 2005;11:473–480. doi: 10.1097/01.mib.0000159662.62651.06. [DOI] [PubMed] [Google Scholar]
- 76.Sartor RB. Microbial influences in inflammatory bowel diseases. Gastroenterology. 2008;134:577–594. doi: 10.1053/j.gastro.2007.11.059. [DOI] [PubMed] [Google Scholar]
- 77.Scanlan PD, Shanahan F, O’Mahony C, Marchesi JR. Culture-independent analyses of temporal variation of the dominant fecal microbiota and targeted bacterial subgroups in Crohn’s disease. J Clin Microbiol. 2006;44:3980–3988. doi: 10.1128/JCM.00312-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bibiloni R, Mangold M, Madsen KL, Fedorak RN, Tannock GW. The bacteriology of biopsies differs between newly diagnosed, untreated, Crohn’s disease and ulcerative colitis patients. J Med Microbiol. 2006;55:1141–1149. doi: 10.1099/jmm.0.46498-0. [DOI] [PubMed] [Google Scholar]
- 79.Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. The human microbiome project. Nature. 2007;449:804–810. doi: 10.1038/nature06244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sokol H, Seksik P, Furet JP, Firmesse O, Nion-Larmurier I, Beaugerie L, Cosnes J, Corthier G, Marteau P, Doré J. Low counts of Faecalibacterium prausnitzii in colitis microbiota. Inflamm Bowel Dis. 2009;15:1183–1189. doi: 10.1002/ibd.20903. [DOI] [PubMed] [Google Scholar]
- 81.Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermúdez-Humarán LG, Gratadoux JJ, Blugeon S, Bridonneau C, Furet JP, Corthier G, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci USA. 2008;105:16731–16736. doi: 10.1073/pnas.0804812105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rajca S, Grondin V, Louis E, Vernier-Massouille G, Grimaud JC, Bouhnik Y, Laharie D, Dupas JL, Pillant H, Picon L, et al. Alterations in the intestinal microbiome (dysbiosis) as a predictor of relapse after infliximab withdrawal in Crohn’s disease. Inflamm Bowel Dis. 2014;20:978–986. doi: 10.1097/MIB.0000000000000036. [DOI] [PubMed] [Google Scholar]
- 83.Bullock NR, Booth JC, Gibson GR. Comparative composition of bacteria in the human intestinal microflora during remission and active ulcerative colitis. Curr Issues Intest Microbiol. 2004;5:59–64. [PubMed] [Google Scholar]
- 84.Varela E, Manichanh C, Gallart M, Torrejón A, Borruel N, Casellas F, Guarner F, Antolin M. Colonisation by Faecalibacterium prausnitzii and maintenance of clinical remission in patients with ulcerative colitis. Aliment Pharmacol Ther. 2013;38:151–161. doi: 10.1111/apt.12365. [DOI] [PubMed] [Google Scholar]
- 85.Zaph C, Du Y, Saenz SA, Nair MG, Perrigoue JG, Taylor BC, Troy AE, Kobuley DE, Kastelein RA, Cua DJ, et al. Commensal-dependent expression of IL-25 regulates the IL-23-IL-17 axis in the intestine. J Exp Med. 2008;205:2191–2198. doi: 10.1084/jem.20080720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sakaguchi S, Ono M, Setoguchi R, Yagi H, Hori S, Fehervari Z, Shimizu J, Takahashi T, Nomura T. Foxp3+ CD25+ CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol Rev. 2006;212:8–27. doi: 10.1111/j.0105-2896.2006.00427.x. [DOI] [PubMed] [Google Scholar]
- 87.Ostman S, Rask C, Wold AE, Hultkrantz S, Telemo E. Impaired regulatory T cell function in germ-free mice. Eur J Immunol. 2006;36:2336–2346. doi: 10.1002/eji.200535244. [DOI] [PubMed] [Google Scholar]
- 88.Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victora GD, Shen Y, Du J, Rubtsov YP, Rudensky AY, et al. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature. 2008;453:236–240. doi: 10.1038/nature06878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Adeeti Chiplunker ANA, Dawn B. Beaulieu, Amar S. Naik, Yelena Zadvornova, Susan Skaros, Kathryn Johnson, Lilani P. Perera, David G. Binion, Mazen Issa. S1145 Long-Term Impact of Clostridium difficile On Inflammatory Bowel Disease. Gastroenterology. 2009;136:Page A–199. [Google Scholar]
- 90.Ananthakrishnan AN, McGinley EL, Binion DG. Excess hospitalisation burden associated with Clostridium difficile in patients with inflammatory bowel disease. Gut. 2008;57:205–210. doi: 10.1136/gut.2007.128231. [DOI] [PubMed] [Google Scholar]
- 91.Gradel KO, Nielsen HL, Schønheyder HC, Ejlertsen T, Kristensen B, Nielsen H. Increased short- and long-term risk of inflammatory bowel disease after salmonella or campylobacter gastroenteritis. Gastroenterology. 2009;137:495–501. doi: 10.1053/j.gastro.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 92.Jess T, Simonsen J, Nielsen NM, Jørgensen KT, Bager P, Ethelberg S, Frisch M. Enteric Salmonella or Campylobacter infections and the risk of inflammatory bowel disease. Gut. 2011;60:318–324. doi: 10.1136/gut.2010.223396. [DOI] [PubMed] [Google Scholar]
- 93.Jodorkovsky D, Young Y, Abreu MT. Clinical outcomes of patients with ulcerative colitis and co-existing Clostridium difficile infection. Dig Dis Sci. 2010;55:415–420. doi: 10.1007/s10620-009-0749-9. [DOI] [PubMed] [Google Scholar]
- 94.Naser SA, Sagramsingh SR, Naser AS, Thanigachalam S. Mycobacterium avium subspecies paratuberculosis causes Crohn’s disease in some inflammatory bowel disease patients. World J Gastroenterol. 2014;20:7403–7415. doi: 10.3748/wjg.v20.i23.7403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rodemann JF, Dubberke ER, Reske KA, Seo DH, Stone CD. Incidence of Clostridium difficile infection in inflammatory bowel disease. Clin Gastroenterol Hepatol. 2007;5:339–344. doi: 10.1016/j.cgh.2006.12.027. [DOI] [PubMed] [Google Scholar]
- 96.Bernstein CN, Rawsthorne P, Blanchard JF. Population-based case-control study of measles, mumps, and rubella and inflammatory bowel disease. Inflamm Bowel Dis. 2007;13:759–762. doi: 10.1002/ibd.20089. [DOI] [PubMed] [Google Scholar]
- 97.Criscuoli V, Rizzuto MR, Cottone M. Cytomegalovirus and inflammatory bowel disease: is there a link? World J Gastroenterol. 2006;12:4813–4818. doi: 10.3748/wjg.v12.i30.4813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Greenberg R, Greenwald B, Roth JS, Ioffe O, Cross R. Squamous dysplasia of the rectum in a patient with ulcerative colitis treated with 6-mercaptopurine. Dig Dis Sci. 2008;53:760–764. doi: 10.1007/s10620-007-9935-9. [DOI] [PubMed] [Google Scholar]
- 99.Kambham N, Vij R, Cartwright CA, Longacre T. Cytomegalovirus infection in steroid-refractory ulcerative colitis: a case-control study. Am J Surg Pathol. 2004;28:365–373. doi: 10.1097/00000478-200403000-00009. [DOI] [PubMed] [Google Scholar]
- 100.Kim JJ, Simpson N, Klipfel N, Debose R, Barr N, Laine L. Cytomegalovirus infection in patients with active inflammatory bowel disease. Dig Dis Sci. 2010;55:1059–1065. doi: 10.1007/s10620-010-1126-4. [DOI] [PubMed] [Google Scholar]
- 101.Kojima T, Watanabe T, Hata K, Shinozaki M, Yokoyama T, Nagawa H. Cytomegalovirus infection in ulcerative colitis. Scand J Gastroenterol. 2006;41:706–711. doi: 10.1080/00365520500408584. [DOI] [PubMed] [Google Scholar]
- 102.Sankaran-Walters S, Ransibrahmanakul K, Grishina I, Hung J, Martinez E, Prindiville T, Dandekar S. Epstein-Barr virus replication linked to B cell proliferation in inflamed areas of colonic mucosa of patients with inflammatory bowel disease. J Clin Virol. 2011;50:31–36. doi: 10.1016/j.jcv.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Schunter MO, Walles T, Fritz P, Meyding-Lamadé U, Thon KP, Fellermann K, Stange EF, Lamadé W. Herpes simplex virus colitis complicating ulcerative colitis: A case report and brief review on superinfections. J Crohns Colitis. 2007;1:41–46. doi: 10.1016/j.crohns.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 104.Spieker T, Herbst H. Distribution and phenotype of Epstein-Barr virus-infected cells in inflammatory bowel disease. Am J Pathol. 2000;157:51–57. doi: 10.1016/S0002-9440(10)64516-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wakefield AJ, Fox JD, Sawyerr AM, Taylor JE, Sweenie CH, Smith M, Emery VC, Hudson M, Tedder RS, Pounder RE. Detection of herpesvirus DNA in the large intestine of patients with ulcerative colitis and Crohn’s disease using the nested polymerase chain reaction. J Med Virol. 1992;38:183–190. doi: 10.1002/jmv.1890380306. [DOI] [PubMed] [Google Scholar]
- 106.Wang W, Jovel J, Halloran B, Wine E, Patterson J, Ford G, OʼKeefe S, Meng B, Song D, Zhang Y, et al. Metagenomic analysis of microbiome in colon tissue from subjects with inflammatory bowel diseases reveals interplay of viruses and bacteria. Inflamm Bowel Dis. 2015;21:1419–1427. doi: 10.1097/MIB.0000000000000344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Yanai H, Shimizu N, Nagasaki S, Mitani N, Okita K. Epstein-Barr virus infection of the colon with inflammatory bowel disease. Am J Gastroenterol. 1999;94:1582–1586. doi: 10.1111/j.1572-0241.1999.01148.x. [DOI] [PubMed] [Google Scholar]
- 108.Weinstock JV, Elliott DE. Helminths and the IBD hygiene hypothesis. Inflamm Bowel Dis. 2009;15:128–133. doi: 10.1002/ibd.20633. [DOI] [PubMed] [Google Scholar]
- 109.Ramanan D, Bowcutt R, Lee SC, Tang MS, Kurtz ZD, Ding Y, Honda K, Gause WC, Blaser MJ, Bonneau RA, et al. Helminth infection promotes colonization resistance via type 2 immunity. Science. 2016;352:608–612. doi: 10.1126/science.aaf3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kabeerdoss J, Pugazhendhi S, Subramanian V, Binder HJ, Ramakrishna BS. Exposure to hookworms in patients with Crohn’s disease: a case-control study. Aliment Pharmacol Ther. 2011;34:923–930. doi: 10.1111/j.1365-2036.2011.04824.x. [DOI] [PubMed] [Google Scholar]
- 111.Setiawan T, Metwali A, Blum AM, Ince MN, Urban JF, Elliott DE, Weinstock JV. Heligmosomoides polygyrus promotes regulatory T-cell cytokine production in the murine normal distal intestine. Infect Immun. 2007;75:4655–4663. doi: 10.1128/IAI.00358-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Weinstock JV, Elliott DE. Helminth infections decrease host susceptibility to immune-mediated diseases. J Immunol. 2014;193:3239–3247. doi: 10.4049/jimmunol.1400927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sandborn WJ, Elliott DE, Weinstock J, Summers RW, Landry-Wheeler A, Silver N, Harnett MD, Hanauer SB. Randomised clinical trial: the safety and tolerability of Trichuris suis ova in patients with Crohn’s disease. Aliment Pharmacol Ther. 2013;38:255–263. doi: 10.1111/apt.12366. [DOI] [PubMed] [Google Scholar]
- 114.Garg SK, Croft AM, Bager P. Helminth therapy (worms) for induction of remission in inflammatory bowel disease. Cochrane Database Syst Rev. 2014;(1):CD009400. doi: 10.1002/14651858.CD009400.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chapman-Kiddell CA, Davies PS, Gillen L, Radford-Smith GL. Role of diet in the development of inflammatory bowel disease. Inflamm Bowel Dis. 2010;16:137–151. doi: 10.1002/ibd.20968. [DOI] [PubMed] [Google Scholar]
- 116.Devkota S, Wang Y, Musch MW, Leone V, Fehlner-Peach H, Nadimpalli A, Antonopoulos DA, Jabri B, Chang EB. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10-/- mice. Nature. 2012;487:104–108. doi: 10.1038/nature11225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ma X, Torbenson M, Hamad AR, Soloski MJ, Li Z. High-fat diet modulates non-CD1d-restricted natural killer T cells and regulatory T cells in mouse colon and exacerbates experimental colitis. Clin Exp Immunol. 2008;151:130–138. doi: 10.1111/j.1365-2249.2007.03530.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hou JK, Abraham B, El-Serag H. Dietary intake and risk of developing inflammatory bowel disease: a systematic review of the literature. Am J Gastroenterol. 2011;106:563–573. doi: 10.1038/ajg.2011.44. [DOI] [PubMed] [Google Scholar]
- 119.Bassaganya-Riera J, DiGuardo M, Viladomiu M, de Horna A, Sanchez S, Einerhand AW, Sanders L, Hontecillas R. Soluble fibers and resistant starch ameliorate disease activity in interleukin-10-deficient mice with inflammatory bowel disease. J Nutr. 2011;141:1318–1325. doi: 10.3945/jn.111.139022. [DOI] [PubMed] [Google Scholar]
- 120.Ananthakrishnan AN, Khalili H, Konijeti GG, Higuchi LM, de Silva P, Korzenik JR, Fuchs CS, Willett WC, Richter JM, Chan AT. A prospective study of long-term intake of dietary fiber and risk of Crohn’s disease and ulcerative colitis. Gastroenterology. 2013;145:970–977. doi: 10.1053/j.gastro.2013.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Rodríguez-Cabezas ME, Gálvez J, Camuesco D, Lorente MD, Concha A, Martinez-Augustin O, Redondo L, Zarzuelo A. Intestinal anti-inflammatory activity of dietary fiber (Plantago ovata seeds) in HLA-B27 transgenic rats. Clin Nutr. 2003;22:463–471. doi: 10.1016/s0261-5614(03)00045-1. [DOI] [PubMed] [Google Scholar]
- 122.Fernández-Bañares F, Hinojosa J, Sánchez-Lombraña JL, Navarro E, Martínez-Salmerón JF, García-Pugés A, González-Huix F, Riera J, González-Lara V, Domínguez-Abascal F, et al. Randomized clinical trial of Plantago ovata seeds (dietary fiber) as compared with mesalamine in maintaining remission in ulcerative colitis. Spanish Group for the Study of Crohn’s Disease and Ulcerative Colitis (GETECCU) Am J Gastroenterol. 1999;94:427–433. doi: 10.1111/j.1572-0241.1999.872_a.x. [DOI] [PubMed] [Google Scholar]
- 123.Heath-Pagliuso S, Rogers WJ, Tullis K, Seidel SD, Cenijn PH, Brouwer A, Denison MS. Activation of the Ah receptor by tryptophan and tryptophan metabolites. Biochemistry. 1998;37:11508–11515. doi: 10.1021/bi980087p. [DOI] [PubMed] [Google Scholar]
- 124.Zelante T, Iannitti RG, Cunha C, De Luca A, Giovannini G, Pieraccini G, Zecchi R, D’Angelo C, Massi-Benedetti C, Fallarino F, et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity. 2013;39:372–385. doi: 10.1016/j.immuni.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 125.Ballegaard M, Bjergstrøm A, Brøndum S, Hylander E, Jensen L, Ladefoged K. Self-reported food intolerance in chronic inflammatory bowel disease. Scand J Gastroenterol. 1997;32:569–571. doi: 10.3109/00365529709025101. [DOI] [PubMed] [Google Scholar]
- 126.Day AS, Whitten KE, Lemberg DA, Clarkson C, Vitug-Sales M, Jackson R, Bohane TD. Exclusive enteral feeding as primary therapy for Crohn’s disease in Australian children and adolescents: a feasible and effective approach. J Gastroenterol Hepatol. 2006;21:1609–1614. doi: 10.1111/j.1440-1746.2006.04294.x. [DOI] [PubMed] [Google Scholar]
- 127.González-Huix F, Fernández-Bañares F, Esteve-Comas M, Abad-Lacruz A, Cabré E, Acero D, Figa M, Guilera M, Humbert P, de León R. Enteral versus parenteral nutrition as adjunct therapy in acute ulcerative colitis. Am J Gastroenterol. 1993;88:227–232. [PubMed] [Google Scholar]
- 128.Takagi S, Utsunomiya K, Kuriyama S, Yokoyama H, Takahashi S, Iwabuchi M, Takahashi H, Takahashi S, Kinouchi Y, Hiwatashi N, et al. Effectiveness of an ‘half elemental diet’ as maintenance therapy for Crohn’s disease: A randomized-controlled trial. Aliment Pharmacol Ther. 2006;24:1333–1340. doi: 10.1111/j.1365-2036.2006.03120.x. [DOI] [PubMed] [Google Scholar]
- 129.Bentz S, Hausmann M, Piberger H, Kellermeier S, Paul S, Held L, Falk W, Obermeier F, Fried M, Schölmerich J, et al. Clinical relevance of IgG antibodies against food antigens in Crohn’s disease: a double-blind cross-over diet intervention study. Digestion. 2010;81:252–264. doi: 10.1159/000264649. [DOI] [PubMed] [Google Scholar]
- 130.Becker HM, Bertschinger MM, Rogler G. Microparticles and their impact on intestinal immunity. Dig Dis. 2012;30 Suppl 3:47–54. doi: 10.1159/000342602. [DOI] [PubMed] [Google Scholar]
- 131.Pineton de Chambrun G, Body-Malapel M, Frey-Wagner I, Djouina M, Deknuydt F, Atrott K, Esquerre N, Altare F, Neut C, Arrieta MC, et al. Aluminum enhances inflammation and decreases mucosal healing in experimental colitis in mice. Mucosal Immunol. 2014;7:589–601. doi: 10.1038/mi.2013.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, Collini S, Pieraccini G, Lionetti P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci USA. 2010;107:14691–14696. doi: 10.1073/pnas.1005963107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS, Schlegel ML, Tucker TA, Schrenzel MD, Knight R, et al. Evolution of mammals and their gut microbes. Science. 2008;320:1647–1651. doi: 10.1126/science.1155725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Muegge BD, Kuczynski J, Knights D, Clemente JC, González A, Fontana L, Henrissat B, Knight R, Gordon JI. Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science. 2011;332:970–974. doi: 10.1126/science.1198719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Walter J, Ley R. The human gut microbiome: ecology and recent evolutionary changes. Annu Rev Microbiol. 2011;65:411–429. doi: 10.1146/annurev-micro-090110-102830. [DOI] [PubMed] [Google Scholar]
- 136.Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, Keilbaugh SA, Bewtra M, Knights D, Walters WA, Knight R, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science. 2011;334:105–108. doi: 10.1126/science.1208344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bonner GF, Fakhri A, Vennamaneni SR. A long-term cohort study of nonsteroidal anti-inflammatory drug use and disease activity in outpatients with inflammatory bowel disease. Inflamm Bowel Dis. 2004;10:751–757. doi: 10.1097/00054725-200411000-00009. [DOI] [PubMed] [Google Scholar]
- 138.Bonner GF, Walczak M, Kitchen L, Bayona M. Tolerance of nonsteroidal antiinflammatory drugs in patients with inflammatory bowel disease. Am J Gastroenterol. 2000;95:1946–1948. doi: 10.1111/j.1572-0241.2000.02263.x. [DOI] [PubMed] [Google Scholar]
- 139.Takeuchi K, Smale S, Premchand P, Maiden L, Sherwood R, Thjodleifsson B, Bjornsson E, Bjarnason I. Prevalence and mechanism of nonsteroidal anti-inflammatory drug-induced clinical relapse in patients with inflammatory bowel disease. Clin Gastroenterol Hepatol. 2006;4:196–202. doi: 10.1016/s1542-3565(05)00980-8. [DOI] [PubMed] [Google Scholar]
- 140.Meyer AM, Ramzan NN, Heigh RI, Leighton JA. Relapse of inflammatory bowel disease associated with use of nonsteroidal anti-inflammatory drugs. Dig Dis Sci. 2006;51:168–172. doi: 10.1007/s10620-006-3103-5. [DOI] [PubMed] [Google Scholar]
- 141.Biancone L, Tosti C, Geremia A, Fina D, Petruzziello C, Emerenziani S, Pallone F. Rofecoxib and early relapse of inflammatory bowel disease: an open-label trial. Aliment Pharmacol Ther. 2004;19:755–764. doi: 10.1111/j.1365-2036.2004.01907.x. [DOI] [PubMed] [Google Scholar]
- 142.Matuk R, Crawford J, Abreu MT, Targan SR, Vasiliauskas EA, Papadakis KA. The spectrum of gastrointestinal toxicity and effect on disease activity of selective cyclooxygenase-2 inhibitors in patients with inflammatory bowel disease. Inflamm Bowel Dis. 2004;10:352–356. doi: 10.1097/00054725-200407000-00005. [DOI] [PubMed] [Google Scholar]
- 143.Mahadevan U, Loftus EV, Tremaine WJ, Sandborn WJ. Safety of selective cyclooxygenase-2 inhibitors in inflammatory bowel disease. Am J Gastroenterol. 2002;97:910–914. doi: 10.1111/j.1572-0241.2002.05608.x. [DOI] [PubMed] [Google Scholar]
- 144.Reinisch W, Miehsler W, Dejaco C, Harrer M, Waldhoer T, Lichtenberger C, Vogelsang H. An open-label trial of the selective cyclo-oxygenase-2 inhibitor, rofecoxib, in inflammatory bowel disease-associated peripheral arthritis and arthralgia. Aliment Pharmacol Ther. 2003;17:1371–1380. doi: 10.1046/j.1365-2036.2003.01596.x. [DOI] [PubMed] [Google Scholar]
- 145.Hildebrand H, Malmborg P, Askling J, Ekbom A, Montgomery SM. Early-life exposures associated with antibiotic use and risk of subsequent Crohn’s disease. Scand J Gastroenterol. 2008;43:961–966. doi: 10.1080/00365520801971736. [DOI] [PubMed] [Google Scholar]
- 146.Shaw SY, Blanchard JF, Bernstein CN. Association between the use of antibiotics and new diagnoses of Crohn’s disease and ulcerative colitis. Am J Gastroenterol. 2011;106:2133–2142. doi: 10.1038/ajg.2011.304. [DOI] [PubMed] [Google Scholar]
- 147.Ungaro R, Bernstein CN, Gearry R, Hviid A, Kolho KL, Kronman MP, Shaw S, Van Kruiningen H, Colombel JF, Atreja A. Antibiotics associated with increased risk of new-onset Crohn’s disease but not ulcerative colitis: a meta-analysis. Am J Gastroenterol. 2014;109:1728–1738. doi: 10.1038/ajg.2014.246. [DOI] [PubMed] [Google Scholar]
- 148.Rutgeerts P, Hiele M, Geboes K, Peeters M, Penninckx F, Aerts R, Kerremans R. Controlled trial of metronidazole treatment for prevention of Crohn’s recurrence after ileal resection. Gastroenterology. 1995;108:1617–1621. doi: 10.1016/0016-5085(95)90121-3. [DOI] [PubMed] [Google Scholar]
- 149.Rutgeerts P, Van Assche G, Vermeire S, D’Haens G, Baert F, Noman M, Aerden I, De Hertogh G, Geboes K, Hiele M, et al. Ornidazole for prophylaxis of postoperative Crohn’s disease recurrence: a randomized, double-blind, placebo-controlled trial. Gastroenterology. 2005;128:856–861. doi: 10.1053/j.gastro.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 150.Cornish JA, Tan E, Simillis C, Clark SK, Teare J, Tekkis PP. The risk of oral contraceptives in the etiology of inflammatory bowel disease: a meta-analysis. Am J Gastroenterol. 2008;103:2394–2400. doi: 10.1111/j.1572-0241.2008.02064.x. [DOI] [PubMed] [Google Scholar]
- 151.Khalili H, Higuchi LM, Ananthakrishnan AN, Richter JM, Feskanich D, Fuchs CS, Chan AT. Oral contraceptives, reproductive factors and risk of inflammatory bowel disease. Gut. 2013;62:1153–1159. doi: 10.1136/gutjnl-2012-302362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Khalili H, Higuchi LM, Ananthakrishnan AN, Manson JE, Feskanich D, Richter JM, Fuchs CS, Chan AT. Hormone therapy increases risk of ulcerative colitis but not Crohn’s disease. Gastroenterology. 2012;143:1199–1206. doi: 10.1053/j.gastro.2012.07.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Cosnes J, Carbonnel F, Carrat F, Beaugerie L, Gendre JP. Oral contraceptive use and the clinical course of Crohn’s disease: a prospective cohort study. Gut. 1999;45:218–222. doi: 10.1136/gut.45.2.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kane SV, Reddy D. Hormonal replacement therapy after menopause is protective of disease activity in women with inflammatory bowel disease. Am J Gastroenterol. 2008;103:1193–1196. doi: 10.1111/j.1572-0241.2007.01700.x. [DOI] [PubMed] [Google Scholar]
- 155.Timmer A, Sutherland LR, Martin F. Oral contraceptive use and smoking are risk factors for relapse in Crohn’s disease. The Canadian Mesalamine for Remission of Crohn’s Disease Study Group. Gastroenterology. 1998;114:1143–1150. doi: 10.1016/s0016-5085(98)70419-6. [DOI] [PubMed] [Google Scholar]
- 156.Cutolo M, Capellino S, Sulli A, Serioli B, Secchi ME, Villaggio B, Straub RH. Estrogens and autoimmune diseases. Ann N Y Acad Sci. 2006;1089:538–547. doi: 10.1196/annals.1386.043. [DOI] [PubMed] [Google Scholar]
- 157.Sternberg EM, Chrousos GP, Wilder RL, Gold PW. The stress response and the regulation of inflammatory disease. Ann Intern Med. 1992;117:854–866. doi: 10.7326/0003-4819-117-10-854. [DOI] [PubMed] [Google Scholar]
- 158.Bernstein CN, Singh S, Graff LA, Walker JR, Miller N, Cheang M. A prospective population-based study of triggers of symptomatic flares in IBD. Am J Gastroenterol. 2010;105:1994–2002. doi: 10.1038/ajg.2010.140. [DOI] [PubMed] [Google Scholar]
- 159.Bitton A, Dobkin PL, Edwardes MD, Sewitch MJ, Meddings JB, Rawal S, Cohen A, Vermeire S, Dufresne L, Franchimont D, et al. Predicting relapse in Crohn’s disease: a biopsychosocial model. Gut. 2008;57:1386–1392. doi: 10.1136/gut.2007.134817. [DOI] [PubMed] [Google Scholar]
- 160.Gerbarg PL, Jacob VE, Stevens L, Bosworth BP, Chabouni F, DeFilippis EM, Warren R, Trivellas M, Patel PV, Webb CD, et al. The Effect of Breathing, Movement, and Meditation on Psychological and Physical Symptoms and Inflammatory Biomarkers in Inflammatory Bowel Disease: A Randomized Controlled Trial. Inflamm Bowel Dis. 2015;21:2886–2896. doi: 10.1097/MIB.0000000000000568. [DOI] [PubMed] [Google Scholar]
- 161.Boye B, Lundin KE, Jantschek G, Leganger S, Mokleby K, Tangen T, Jantschek I, Pripp AH, Wojniusz S, Dahlstroem A, et al. INSPIRE study: does stress management improve the course of inflammatory bowel disease and disease-specific quality of life in distressed patients with ulcerative colitis or Crohn’s disease? A randomized controlled trial. Inflamm Bowel Dis. 2011;17:1863–1873. doi: 10.1002/ibd.21575. [DOI] [PubMed] [Google Scholar]
- 162.Timmer A, Preiss JC, Motschall E, Rücker G, Jantschek G, Moser G. Psychological interventions for treatment of inflammatory bowel disease. Cochrane Database Syst Rev. 2011;(2):CD006913. doi: 10.1002/14651858.CD006913.pub2. [DOI] [PubMed] [Google Scholar]
- 163.Dickstein JB, Moldofsky H. Sleep, cytokines and immune function. Sleep Med Rev. 1999;3:219–228. doi: 10.1016/s1087-0792(99)90003-5. [DOI] [PubMed] [Google Scholar]
- 164.Opp MR. Cytokines and sleep. Sleep Med Rev. 2005;9:355–364. doi: 10.1016/j.smrv.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 165.Shearer WT, Reuben JM, Mullington JM, Price NJ, Lee BN, Smith EO, Szuba MP, Van Dongen HP, Dinges DF. Soluble TNF-alpha receptor 1 and IL-6 plasma levels in humans subjected to the sleep deprivation model of spaceflight. J Allergy Clin Immunol. 2001;107:165–170. doi: 10.1067/mai.2001.112270. [DOI] [PubMed] [Google Scholar]
- 166.Gamaldo CE, Shaikh AK, McArthur JC. The sleep-immunity relationship. Neurol Clin. 2012;30:1313–1343. doi: 10.1016/j.ncl.2012.08.007. [DOI] [PubMed] [Google Scholar]
- 167.Ranjbaran Z, Keefer L, Stepanski E, Farhadi A, Keshavarzian A. The relevance of sleep abnormalities to chronic inflammatory conditions. Inflamm Res. 2007;56:51–57. doi: 10.1007/s00011-006-6067-1. [DOI] [PubMed] [Google Scholar]
- 168.Vgontzas AN, Papanicolaou DA, Bixler EO, Kales A, Tyson K, Chrousos GP. Elevation of plasma cytokines in disorders of excessive daytime sleepiness: role of sleep disturbance and obesity. J Clin Endocrinol Metab. 1997;82:1313–1316. doi: 10.1210/jcem.82.5.3950. [DOI] [PubMed] [Google Scholar]
- 169.Zisapel N. Sleep and sleep disturbances: biological basis and clinical implications. Cell Mol Life Sci. 2007;64:1174–1186. doi: 10.1007/s00018-007-6529-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Ali T, Orr WC. Sleep disturbances and inflammatory bowel disease. Inflamm Bowel Dis. 2014;20:1986–1995. doi: 10.1097/MIB.0000000000000108. [DOI] [PubMed] [Google Scholar]
- 171.Irwin MR, Wang M, Campomayor CO, Collado-Hidalgo A, Cole S. Sleep deprivation and activation of morning levels of cellular and genomic markers of inflammation. Arch Intern Med. 2006;166:1756–1762. doi: 10.1001/archinte.166.16.1756. [DOI] [PubMed] [Google Scholar]
- 172.Keefer L, Stepanski EJ, Ranjbaran Z, Benson LM, Keshavarzian A. An initial report of sleep disturbance in inactive inflammatory bowel disease. J Clin Sleep Med. 2006;2:409–416. [PubMed] [Google Scholar]
- 173.Tang Y, Preuss F, Turek FW, Jakate S, Keshavarzian A. Sleep deprivation worsens inflammation and delays recovery in a mouse model of colitis. Sleep Med. 2009;10:597–603. doi: 10.1016/j.sleep.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Graff LA, Vincent N, Walker JR, Clara I, Carr R, Ediger J, Miller N, Rogala L, Rawsthorne P, Lix L, et al. A population-based study of fatigue and sleep difficulties in inflammatory bowel disease. Inflamm Bowel Dis. 2011;17:1882–1889. doi: 10.1002/ibd.21580. [DOI] [PubMed] [Google Scholar]
- 175.Ranjbaran Z, Keefer L, Farhadi A, Stepanski E, Sedghi S, Keshavarzian A. Impact of sleep disturbances in inflammatory bowel disease. J Gastroenterol Hepatol. 2007;22:1748–1753. doi: 10.1111/j.1440-1746.2006.04820.x. [DOI] [PubMed] [Google Scholar]
- 176.Ali T, Choe J, Awab A, Wagener TL, Orr WC. Sleep, immunity and inflammation in gastrointestinal disorders. World J Gastroenterol. 2013;19:9231–9239. doi: 10.3748/wjg.v19.i48.9231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Ali T, Madhoun MF, Orr WC, Rubin DT. Assessment of the relationship between quality of sleep and disease activity in inflammatory bowel disease patients. Inflamm Bowel Dis. 2013;19:2440–2443. doi: 10.1097/MIB.0b013e3182a0ea54. [DOI] [PubMed] [Google Scholar]
- 178.Ananthakrishnan AN, Long MD, Martin CF, Sandler RS, Kappelman MD. Sleep disturbance and risk of active disease in patients with Crohn’s disease and ulcerative colitis. Clin Gastroenterol Hepatol. 2013;11:965–971. doi: 10.1016/j.cgh.2013.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Bach JF. The effect of infections on susceptibility to autoimmune and allergic diseases. N Engl J Med. 2002;347:911–920. doi: 10.1056/NEJMra020100. [DOI] [PubMed] [Google Scholar]
- 180.Baron S, Turck D, Leplat C, Merle V, Gower-Rousseau C, Marti R, Yzet T, Lerebours E, Dupas JL, Debeugny S, et al. Environmental risk factors in paediatric inflammatory bowel diseases: a population based case control study. Gut. 2005;54:357–363. doi: 10.1136/gut.2004.054353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Gilat T, Hacohen D, Lilos P, Langman MJ. Childhood factors in ulcerative colitis and Crohn’s disease. An international cooperative study. Scand J Gastroenterol. 1987;22:1009–1024. doi: 10.3109/00365528708991950. [DOI] [PubMed] [Google Scholar]
- 182.Hansen TS, Jess T, Vind I, Elkjaer M, Nielsen MF, Gamborg M, Munkholm P. Environmental factors in inflammatory bowel disease: a case-control study based on a Danish inception cohort. J Crohns Colitis. 2011;5:577–584. doi: 10.1016/j.crohns.2011.05.010. [DOI] [PubMed] [Google Scholar]
- 183.Hansen TS, Jess T, Vind I, Elkjaer M, Nielsen MF, Gamborg M, Munkholm P. Pineton de Chambrun G. UEG Week 2015 Poster Presentations. United Eur Gastroenterol J. 2015;3(5 Suppl):146–687. [Google Scholar]
- 184.Koloski NA, Bret L, Radford-Smith G. Hygiene hypothesis in inflammatory bowel disease: a critical review of the literature. World J Gastroenterol. 2008;14:165–173. doi: 10.3748/wjg.14.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Goldman AS. The immune system in human milk and the developing infant. Breastfeed Med. 2007;2:195–204. doi: 10.1089/bfm.2007.0024. [DOI] [PubMed] [Google Scholar]
- 186.Brock JH. The physiology of lactoferrin. Biochem Cell Biol. 2002;80:1–6. doi: 10.1139/o01-212. [DOI] [PubMed] [Google Scholar]
- 187.Klement E, Cohen RV, Boxman J, Joseph A, Reif S. Breastfeeding and risk of inflammatory bowel disease: a systematic review with meta-analysis. Am J Clin Nutr. 2004;80:1342–1352. doi: 10.1093/ajcn/80.5.1342. [DOI] [PubMed] [Google Scholar]
- 188.Ekbom A, Adami HO, Helmick CG, Jonzon A, Zack MM. Perinatal risk factors for inflammatory bowel disease: a case-control study. Am J Epidemiol. 1990;132:1111–1119. doi: 10.1093/oxfordjournals.aje.a115754. [DOI] [PubMed] [Google Scholar]
- 189.Thompson NP, Montgomery SM, Wadsworth ME, Pounder RE, Wakefield AJ. Early determinants of inflammatory bowel disease: use of two national longitudinal birth cohorts. Eur J Gastroenterol Hepatol. 2000;12:25–30. doi: 10.1097/00042737-200012010-00006. [DOI] [PubMed] [Google Scholar]
- 190.Andersson RE, Olaison G, Tysk C, Ekbom A. Appendectomy is followed by increased risk of Crohn’s disease. Gastroenterology. 2003;124:40–46. doi: 10.1053/gast.2003.50021. [DOI] [PubMed] [Google Scholar]
- 191.Koutroubakis IE, Vlachonikolis IG, Kapsoritakis A, Spanoudakis S, Roussomoustakaki M, Mouzas IA, Kouroumalis EA, Manousos ON. Appendectomy, tonsillectomy, and risk of inflammatory bowel disease: case-controlled study in Crete. Dis Colon Rectum. 1999;42:225–230. doi: 10.1007/BF02237133. [DOI] [PubMed] [Google Scholar]
- 192.Reif S, Lavy A, Keter D, Broide E, Niv Y, Halak A, Ron Y, Eliakim R, Odes S, Patz J, et al. Appendectomy is more frequent but not a risk factor in Crohn’s disease while being protective in ulcerative colitis: a comparison of surgical procedures in inflammatory bowel disease. Am J Gastroenterol. 2001;96:829–832. doi: 10.1111/j.1572-0241.2001.03529.x. [DOI] [PubMed] [Google Scholar]
- 193.Kaplan GG, Jackson T, Sands BE, Frisch M, Andersson RE, Korzenik J. The risk of developing Crohn’s disease after an appendectomy: a meta-analysis. Am J Gastroenterol. 2008;103:2925–2931. doi: 10.1111/j.1572-0241.2008.02118.x. [DOI] [PubMed] [Google Scholar]
- 194.Mizoguchi A, Mizoguchi E, Chiba C, Bhan AK. Role of appendix in the development of inflammatory bowel disease in TCR-alpha mutant mice. J Exp Med. 1996;184:707–715. doi: 10.1084/jem.184.2.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Andersson RE, Olaison G, Tysk C, Ekbom A. Appendectomy and protection against ulcerative colitis. N Engl J Med. 2001;344:808–814. doi: 10.1056/NEJM200103153441104. [DOI] [PubMed] [Google Scholar]
- 196.Russel MG, Dorant E, Brummer RJ, van de Kruijs MA, Muris JW, Bergers JM, Goedhard J, Stockbrügger RW. Appendectomy and the risk of developing ulcerative colitis or Crohn’s disease: results of a large case-control study. South Limburg Inflammatory Bowel Disease Study Group. Gastroenterology. 1997;113:377–382. doi: 10.1053/gast.1997.v113.pm9247453. [DOI] [PubMed] [Google Scholar]
- 197.Bartel G, Weiss I, Turetschek K, Schima W, Püspök A, Waldhoer T, Gasche C. Ingested matter affects intestinal lesions in Crohn’s disease. Inflamm Bowel Dis. 2008;14:374–382. doi: 10.1002/ibd.20295. [DOI] [PubMed] [Google Scholar]
- 198.Chiba M, Abe T, Tsuda H, Sugawara T, Tsuda S, Tozawa H, Fujiwara K, Imai H. Lifestyle-related disease in Crohn’s disease: relapse prevention by a semi-vegetarian diet. World J Gastroenterol. 2010;16:2484–2495. doi: 10.3748/wjg.v16.i20.2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Ghouri YA, Richards DM, Rahimi EF, Krill JT, Jelinek KA, DuPont AW. Systematic review of randomized controlled trials of probiotics, prebiotics, and synbiotics in inflammatory bowel disease. Clin Exp Gastroenterol. 2014;7:473–487. doi: 10.2147/CEG.S27530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Hanai H, Kanauchi O, Mitsuyama K, Andoh A, Takeuchi K, Takayuki I, Araki Y, Fujiyama Y, Toyonaga A, Sata M, et al. Germinated barley foodstuff prolongs remission in patients with ulcerative colitis. Int J Mol Med. 2004;13:643–647. [PubMed] [Google Scholar]
- 201.Lev-Tzion R, Griffiths AM, Leder O, Turner D. Omega 3 fatty acids (fish oil) for maintenance of remission in Crohn’s disease. Cochrane Database Syst Rev. 2014;(2):CD006320. doi: 10.1002/14651858.CD006320.pub4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Lomer MC, Grainger SL, Ede R, Catterall AP, Greenfield SM, Cowan RE, Vicary FR, Jenkins AP, Fidler H, Harvey RS, et al. Lack of efficacy of a reduced microparticle diet in a multi-centred trial of patients with active Crohn’s disease. Eur J Gastroenterol Hepatol. 2005;17:377–384. doi: 10.1097/00042737-200503000-00019. [DOI] [PubMed] [Google Scholar]
- 203.Lomer MC, Harvey RS, Evans SM, Thompson RP, Powell JJ. Efficacy and tolerability of a low microparticle diet in a double blind, randomized, pilot study in Crohn’s disease. Eur J Gastroenterol Hepatol. 2001;13:101–106. doi: 10.1097/00042737-200102000-00003. [DOI] [PubMed] [Google Scholar]
- 204.Moayyedi P, Surette MG, Kim PT, Libertucci J, Wolfe M, Onischi C, Armstrong D, Marshall JK, Kassam Z, Reinisch W, et al. Fecal Microbiota Transplantation Induces Remission in Patients With Active Ulcerative Colitis in a Randomized Controlled Trial. Gastroenterology. 2015;149:102–109.e6. doi: 10.1053/j.gastro.2015.04.001. [DOI] [PubMed] [Google Scholar]
- 205.Nikfar S, Ehteshami-Ashar S, Rahimi R, Abdollahi M. Systematic review and meta-analysis of the efficacy and tolerability of nicotine preparations in active ulcerative colitis. Clin Ther. 2010;32:2304–2315. doi: 10.1016/j.clinthera.2011.01.004. [DOI] [PubMed] [Google Scholar]
- 206.Petersen AM, Mirsepasi H, Halkjær SI, Mortensen EM, Nordgaard-Lassen I, Krogfelt KA. Ciprofloxacin and probiotic Escherichia coli Nissle add-on treatment in active ulcerative colitis: a double-blind randomized placebo controlled clinical trial. J Crohns Colitis. 2014;8:1498–1505. doi: 10.1016/j.crohns.2014.06.001. [DOI] [PubMed] [Google Scholar]
- 207.Ritchie JK, Wadsworth J, Lennard-Jones JE, Rogers E. Controlled multicentre therapeutic trial of an unrefined carbohydrate, fibre rich diet in Crohn’s disease. Br Med J (Clin Res Ed) 1987;295:517–520. doi: 10.1136/bmj.295.6597.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Turner D, Steinhart AH, Griffiths AM. Omega 3 fatty acids (fish oil) for maintenance of remission in ulcerative colitis. Cochrane Database Syst Rev. 2007;(3):CD006443. doi: 10.1002/14651858.CD006443.pub2. [DOI] [PubMed] [Google Scholar]
- 209.Wright R, Truelove SC. A controlled therapeutic trial of various diets in ulcerative colitis. Br Med J. 1965;2:138–141. doi: 10.1136/bmj.2.5454.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Zachos M, Tondeur M, Griffiths AM. Enteral nutritional therapy for induction of remission in Crohn’s disease. Cochrane Database Syst Rev. 2007;(1):CD000542. doi: 10.1002/14651858.CD000542.pub2. [DOI] [PubMed] [Google Scholar]
- 211.Mukherjee B, Ahn J, Gruber SB, Chatterjee N. Testing gene-environment interaction in large-scale case-control association studies: possible choices and comparisons. Am J Epidemiol. 2012;175:177–190. doi: 10.1093/aje/kwr367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Wang Y, Li D, Wei P. Powerful Tukey’s One Degree-of-Freedom Test for Detecting Gene-Gene and Gene-Environment Interactions. Cancer Inform. 2015;14:209–218. doi: 10.4137/CIN.S17305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Wild CP, Scalbert A, Herceg Z. Measuring the exposome: a powerful basis for evaluating environmental exposures and cancer risk. Environ Mol Mutagen. 2013;54:480–499. doi: 10.1002/em.21777. [DOI] [PubMed] [Google Scholar]
- 214.Mendell JT, Olson EN. MicroRNAs in stress signaling and human disease. Cell. 2012;148:1172–1187. doi: 10.1016/j.cell.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]