

Abstract
Based on the mutational effects on the steady-state kinetics of the electron transfer reaction and our NMR analysis of the interaction site (Sakamoto, K., Kamiya, M., Imai, M., Shinzawa-Itoh, K., Uchida, T., Kawano, K., Yoshikawa, S., and Ishimori, K. (2011) Proc. Natl. Acad. Sci. U.S.A. 108, 12271–12276), we determined the structure of the electron transfer complex between cytochrome c (Cyt c) and cytochrome c oxidase (CcO) under turnover conditions and energetically characterized the interactions essential for complex formation. The complex structures predicted by the protein docking simulation were computationally selected and validated by the experimental kinetic data for mutant Cyt c in the electron transfer reaction to CcO. The interaction analysis using the selected Cyt c-CcO complex structure revealed the electrostatic and hydrophobic contributions of each amino acid residue to the free energy required for complex formation. Several charged residues showed large unfavorable (desolvation) electrostatic interactions that were almost cancelled out by large favorable (Columbic) electrostatic interactions but resulted in the destabilization of the complex. The residual destabilizing free energy is compensated by the van der Waals interactions mediated by hydrophobic amino acid residues to give the stabilized complex. Thus, hydrophobic interactions are the primary factors that promote complex formation between Cyt c and CcO under turnover conditions, whereas the change in the electrostatic destabilization free energy provides the variance of the binding free energy in the mutants. The distribution of favorable and unfavorable electrostatic interactions in the interaction site determines the orientation of the binding of Cyt c on CcO.
Keywords: bioenergetics, cytochrome c, cytochrome c oxidase (complex IV), electron transfer complex, molecular docking
Introduction
The electron transfer (ET)3 reactions in mitochondrial and bacterial respiratory chains are essential processes for energy transduction in cells. A series of ET reactions is terminated at cytochrome c oxidase (CcO), where molecular oxygen is reduced to water. Associated with the reduction of molecular oxygen, CcO functions as a proton pump across the membrane, and the proton gradient is the primary driving force for the generation of ATP (1–5).
In the respiratory chain of mitochondria, the electrons to reduce molecular oxygen at CcO are donated from a small hemoprotein, cytochrome c (Cyt c), and Cyt c is thought to form an ET complex with CcO to promote the ET reaction from the heme iron in Cyt c to the CuA site in CcO (6, 7). Although the reduction of molecular oxygen to water molecules requires four electrons, Cyt c can carry only one electron, implying that Cyt c repetitively associates with and dissociates from CcO and suggesting that the specific interprotein interactions between Cyt c and CcO regulate the binding affinity and the ET rate from Cyt c to CcO.
The amino acid sequence and isoelectric point of Cyt c suggest that many positively charged residues are located on the protein surface (6, 7), as confirmed by solving the three-dimensional structures of Cyt c (8), allowing us to speculate that the electrostatic interactions contribute to the formation of the ET complex between Cyt c and CcO. The dependence of the ET rate on ionic strength supports the significant contribution of the electrostatic interactions to the formation of the ET complex (9). The classical chemical modification of the positively charged Lys residues results in the substantial suppression of oxygen consumption in CcO, also showing that the electrostatic interactions are crucial for the complex formation between Cyt c and CcO (10), although the addition of a bulky side chain might interfere with the specific binding of CcO to Cyt c.
These contributions of electrostatic interactions are supported by a previous docking simulation between Cyt c and CcO (11), but the simulation study on the binding of a PDZ domain, a common structural domain found in signaling proteins, to the target peptide showed that the hydrophobic interactions are the major thermodynamic factors stabilizing the protein complex and that the thermodynamic contribution of the electrostatic interactions to the stability of the protein complex was negligible (12). The thermodynamic contributions of hydrophobic and electrostatic interactions to the formation of the ET complex between Cyt c and CcO therefore remain controversial, and detailed experimental characterization of the interprotein interactions in the ET complex has been quite limited.
Recently, however, we successfully identified the interaction site for CcO on Cyt c using NMR spectroscopy (13). Our NMR analysis using chemical shift perturbations clearly indicated positively charged residues, including several Lys residues (Lys-5, Lys-7, Lys-8, Lys-13, Lys-79, Lys-86, Lys-87, and Lys-88), located in the interaction site for CcO on Cyt c, as we expected (10, 14). In addition to the positively charged residues, several negatively charged residues such as Glu-4, Glu-89, Glu-90, and Asp-93 and some hydrophobic residues such as Ile-9, Ile-11, Met-12, and Ile-81 are also present in the interaction site for CcO, indicating that both electrostatic and hydrophobic interactions would contribute to ET complex formation between CcO and Cyt c, regulating the ET reaction from Cyt c to reduce molecular oxygen and promote proton pumping in CcO (13).
Although we have successfully determined the interaction site of Cyt c for CcO, the NMR measurements can be applied only for ET complexes such as oxidized Cyt c, fully oxidized CcO or reduced Cyt c, and fully reduced CcO complexes where the ET reaction does not occur (15–17). The chemical shift perturbations in the NMR analysis are also often induced at amino acid residues that are not located at the interaction site due to secondary structural perturbation. Furthermore, our previous NMR analysis revealed the CcO interaction site on Cyt c, but no information about the Cyt c interaction site on CcO has been obtained, and a detailed analysis of the interactions such as the energetic components stabilizing the ET complex, particularly ET complexes under turnover conditions where the ET reaction from Cyt c to CcO is induced, has not yet been conducted.
In this study, based on the structural information about the CcO interaction site on Cyt c (13), we constructed the ET complexes between Cyt c and CcO by performing docking simulation. We also mutated the amino acid residues located in the CcO interaction site on Cyt c, and we determined the Michaelis constant, Km, for the ET reaction from Cyt c to CcO by monitoring the oxidation of reduced Cyt c by CcO. The predicted protein complexes were computationally validated by the Km values for wild-type and mutant Cyt c, and a reasonable complex structure under turnover conditions was selected. Based on the mutational effects on the ET reaction and the predicted ET complex, the interactions essential for the formation of the Cyt c-CcO complex under turnover conditions for the ET reaction were energetically characterized.
Experimental Procedures
Protein Expression and Purification
The Escherichia coli strain Rosetta2(DE3)pLysS cells transformed with the plasmids containing the DNA of Cyt c (13, 18) were inoculated in 5 ml of 2×TY medium and grown overnight. This pre-cultured medium was added to 4 liters of 2×TY medium, and the bacteria were further incubated at 37 °C. The expression of human Cyt c was initiated by adding 0.8 mm isopropyl 1-thio-β-d-galactopyranoside to the culture when the cell density reached an absorbance of 0.6 at 600 nm. Then, 0.1 mm δ-aminolevulinic acid was added to promote heme biosynthesis. After incubation for an additional 24 h, the cells were collected by centrifugation.
The cell pellet was resuspended in 50 mm Tris-HCl, pH 7.5, containing 1 g/liter lysozyme, 50 mg/liter DNase I, and 50 mg/liter RNase A and suspended for 3 h to lyse the cell pellet completely. The supernatant of the crude extract was obtained by centrifugation at 18,000 rpm for 5 min and 40,000 rpm for 1 h. This supernatant was purified by HiPrep 16/10 SP XL column (GE Healthcare, Uppsala, Sweden) with a linear salt gradient of 1–300 mm NaCl. The elution sample was concentrated by Amicon ultrafiltration using 5-kDa cutoff membranes. To completely oxidize Cyt c, concentrated Cyt c was stirred for 1 h with 10-fold potassium ferricyanide(III). After being dissolved in 50 mm sodium phosphate buffer, pH 7.0, Cyt c was further purified by Mono S 10/100 GL column (GE Healthcare) with a linear salt gradient. The purified Cyt c fractions were pooled, concentrated, and applied to a HiLoad 16/60 Superdex 75 gel filtration column (GE Healthcare).
Mutagenesis was conducted utilizing the PrimeSTAR mutagenesis basal kit from Takara Bio (Otsu, Japan). DNA oligonucleotides were purchased from Operon Biotechnologies (Tokyo, Japan). The mutated genes were sequenced (Operon Biotechnologies, Tokyo, Japan) to ensure that only the desired mutations were introduced. CcO was purified from bovine heart, as described previously (17), and dissolved in 50 mm sodium phosphate buffer, pH 6.8, containing 0.1% n-decyl-β-d-maltoside.
Steady-state Kinetics
The ET reaction from Cyt c to CcO was measured using a Hitachi U-3310 UV-visible spectrophotometer at room temperature in 50 mm NaPi/NaOH, pH 6.8, 0.1% n-decyl-β-d-maltoside (19, 20). Ferrous Cyt c was prepared by the reduction of ferric Cyt c with dithionite, and excess reductant was removed by PD MiniTrap G-25 chromatography. The concentrations of Cyt c were varied between 0.5 and 40 μm, and the oxidation of Cyt c was followed by measuring absorbance at 550 nm after adding CcO to the reaction solution at a final concentration of 1 nm. The absorption at 550 nm was recorded with intervals of 1 s for 3 min. To determine the end point of the ET reaction from Cyt c to CcO, a small amount of potassium ferricyanide(III) was added to the reaction solution. A first-order oxidation of reduced Cyt c (Equation 1) was observed as reported in previous papers (21, 22):
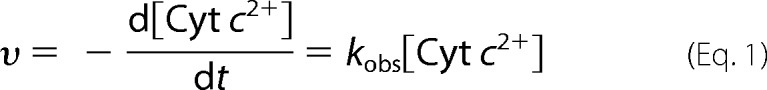 |
where υ is the oxidation rate of reduced Cyt c, and kobs is the apparent rate constant at the various concentrations of Cyt c. Therefore, kobs can be estimated by Equation 2,
where [Cyt c2+]0 denotes the initial concentration of reduced Cyt c.
Consistent with previous reports (21, 22), a single rectangular relationship between υ and [Cyt c2+]0 was obtained under these experimental conditions, as given in Equation 3.
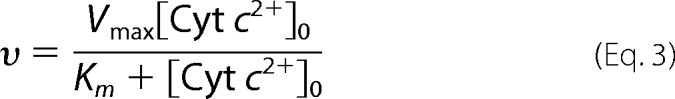 |
We estimated the Michaelis-Menten parameters using [Cyt c2+]0 and kobs as determined by Equation 2. The turnover number, kcat, was calculated by Equation 4,
 |
where [CcO] denotes the total enzyme concentration.
Molecular Docking of Cyt c with CcO
The crystallographic structures of human Cyt c (Protein Data Bank code, 3ZCF, chain A, Fig. 1a) (23) and bovine CcO (3AG2, chains A–M, Fig. 1b) (24) were used as initial geometries for molecular docking simulations because of their high resolution without any amino acid mutations. All hydrogen atoms in the structures were complemented and then energetically minimized using the CHARMm Polar H force field (25) implemented in Discovery Studio 3.5. The heme groups (hemes a, a3, and c) and copper ions (CuA-CuA and CuB) were retained in the structures, whereas the other ions and all crystal water molecules were removed for the following protein-protein docking simulations. The binding interface between Cyt c and CcO was predicted using ZDOCK (26–28), an initial stage rigid-body docking algorithm that utilizes a fast Fourier transform technique for shape complementarity detection. CcO was kept fixed, whereas Cyt c was allowed to translate and rotate around CcO. The top 2000 possible binding interfaces between the two proteins were searched at an angular step size of 15°. The maximum number of clusters, root mean square deviation, and distance cutoffs were set as 100, 10, and 10 Å, respectively. Because Cyt c interacts with CcO in the mitochondrial intermembrane space, amino acid residues that presumably lie at the inner membrane surface of CcO were blocked from interaction in the docking simulations. Thus, the non-membrane region of CcO was used here, as shown in Fig. 1c. This membrane-ignoring method was also used in the previous investigation of the binding interface of the Cyt c-CcO complex (9). The binding site for Cyt c was defined as 21 residues (Glu-4, Lys-5, Lys-7, Lys-8, Ile-9, Ile-11, Met-12, Lys-13, Cys-17, His-18, Gly-24, Asn-31, His-33, Lys-79, Ile-81, Lys-86, Lys-87, Lys-88, Glu-89, Glu-90, and Asp-93) showing a significant chemical shift perturbation, according to our previous work with 1H-15N heteronuclear single quantum coherence spectra for the complex formation between Cyt c and CcO (13). Thus, we used NMR spectral data to exclude unperturbed residues from the possible interaction sites on the surface of Cyt c. A total of 26 poses that passed through the filtering were used with the ZDOCK scoring function, which considers shape complementarity and both electrostatic and desolvation energies. The resulting poses from ZDOCK were re-ranked by ZRANK (29) and clustered. Only seven poses ultimately obtained by this screening process were subjected to further refinement using the RDOCK algorithm (30). The complex structure with the lowest RDOCK energy (E_RDock) was selected as the most probable wild-type complex. The initial geometries of the mutants (I9A, I11A, I81A, K7L/K8L, K13L, K39L, K72L, K79L, and K86L/K87L) at the residues in Cyt c were constructed from the wild-type complex by replacing the side chains of Ile-9, Ile-11, Ile-81, Lys-7/8, Lys-13, Lys-39, Lys-72, Lys-79, and Lys-86/87 in the wild-type with each corresponding residue.
FIGURE 1.

Molecular representation of human Cyt c and bovine CcO. a, structure of human Cyt c (red schematic) with the residues (spheres) targeted by in vitro mutagenesis. In detail, the mutated residues in groups A–C, reflecting the degree of mutational effects (severe, moderate, and unaffected, respectively), are represented by magenta, cyan, and green spheres, respectively. b, structure of bovine CcO with the three important catalytic subunits I–III represented by green, cyan, and magenta, respectively. c, blue region of CcO used for docking includes the part (partial residues in subunits I–IV and VII–XIII) occupying the intermembrane space and the adjacent membrane-embedded region. In CcO, heme groups and copper ions are represented in spheres.
Next, the wild-type and mutant Cyt c-CcO complexes were subjected to a sophisticated minimization. Before the energy minimization, the following preparation of the complexes was performed. Partial charges and parameters for the cofactor sites such as heme a, heme a3, CuA-CuA, and CuB in CcO as well as heme c in Cyt c were taken from the literature (31–34). The total −2 charge of heme c site means that the Cyt c examined here is the reduced form. For standard residues, the default values from the parameters of parm99 (35) were used. The protonation states of His residues at pH 7.0 were determined using the Protein Data Bank code 2PQR webserver (36), and the Arg, Lys, Asp, and Glu residues with ionizable side chains were treated as charged entities. The resulting complexes were solvated in a truncated octahedral box of TIP3P waters extending 12 Å from the surface of the complex. Four, three, and two chloride ions (Cl−) were added to neutralize the total +4 (wild-type, I9A, I11A, and I81A), +3 (K13L, K39L, K72L, and K79L), and +2 (K7L/K8L and K86L/K87L) charges of the systems, respectively. Four-step energy minimizations were then performed as follows: (i) the hydrogen atoms of all residues and the side chain atoms of the mutated residues; (ii) water molecules and ions; (iii) the side chain atoms of all residues, and (iv) all atoms were sequentially relaxed. During the minimizations, the atomic positions of the cofactor sites were constrained with a harmonic potential of 11 kcal mol−1 Å−2. As shown in supplemental Table S1, the central irons of hemes a, a3, and c are ligated to a histidine and/or a methionine from the proteins. Two vinyl groups of heme c are also linked via thioether bonds to two cysteines in Cyt c. These linkages were fixed at crystallographic average bond distance values (see supplemental Table S1) to generate a reasonable complex structure for subsequent binding free energy calculations. All minimizations were conducted using the AMBER10 package (37).
Calculation of Binding Free Energy by MM-PBSA
The MM-PBSA calculation (38) on the wild-type and mutant Cyt c-CcO complexes was used to assess the validity of the binding interface of the resulting Cyt c-CcO optimized complex. In the MM-PBSA approach, the observed overall free energy change (ΔGobs) associated with the complex formation of Cyt c with CcO can be formulated as the sum of two representative free energy terms as shown in Equation 5,
where ΔGobs can be obtained from the Michaelis constant, Km, in this study (ΔGobs = RT ln Km). ΔGbind is the binding free energy of Cyt c with CcO, and ΔGsol is the free energy for the dehydration from the protein surface, corresponding to the solvation free energy change associated with complex formation. MM-PBSA was employed for the calculation of the two energy terms, which were further divided into Equations 6 and 7.
where ΔEMM (sum of the internal, electrostatic, and van der Waals energies) in the right-hand side of Equation 6 consists of ΔEint (ΔEint, internal energy), ΔEcoul (ΔEcoul, electrostatic (Coulombic) energy), and ΔEvdW (ΔEvdW, van der Waals energy), which are internal (bonds, angles, and dihedrals), electrostatic (Coulombic), and van der Waals energies, respectively. Assuming that coordinates of Cyt c and CcO in the complex are identical to the coordinates of the separated forms, ΔEint is likely to be negligibly small due to the lack of structural perturbations in the complex formation, whereas ΔEcoul and ΔEvdW reflect only the non-bonded interaction energies. The contribution of entropic change, −TΔS, was not included, but neglecting ΔEint and −TΔS is considered to be reasonable (39), when only relative mutational effects are of interest and used for validation of the predicted binding interface of the Cyt c-CcO complex.
Moreover, ΔGpolar (ΔGpolar, free energy for polar interactions) and ΔGnonpolar in the right-hand side of Equation 7 are the polar and nonpolar contributions to solvation, respectively. The former term was calculated with the continuum solvation model based on the Poisson-Boltzmann equation (40), whereas the latter term was approximated by a simple linear relation for solvent-accessible surface area change (ΔASA) (41): ΔGnonpolar = γ ΔASA + β, where γ = 0.0072 kcal−1 mol−1 A2 and β = 0 (42). In the Poisson-Boltzmann calculation, the solute and solvent dielectric constants were set to 1 and 78.39, respectively. All the energy terms mentioned above were calculated as the energy difference between the complex (Cyt c-CcO) and the two protein monomers (Cyt c and CcO). Hot spot residues, specific amino acid residues essential for the stabilization of the protein complex, in the binding interface of the Cyt c-CcO complex were identified using the free energy decomposition scheme with the MMPBSA.py script (43) implemented in AmberTools14.
Results
Site-directed Mutagenesis Experiments on Cyt c
To determine hot spot residues in Cyt c in the interaction with CcO, we performed site-directed mutagenesis on 11 residues (Ile-9, Ile-11, Ile-81, Lys-7, Lys-8, Lys-13, Lys-39, Lys-72, Lys-79, Lys-86, and Lys-87) that were predicted to be located at the binding interface of the Cyt c-CcO complex (13). These target isoleucine and lysine were replaced with alanine and leucine, respectively, and a total of nine mutants (I9A, I11A, I81A, K7L/K8L, K13L, K39L, K72L, K79L, and K86L/K87L) and wild-type Cyt c were examined in this study.
The Michaelis-Menten kinetic parameters, Km and kcat, were estimated by the steady-state kinetics for the ET reaction from Cyt c to CcO (19, 20). Based on the Michaelis-Menten kinetic analysis illustrated in Fig. 2, Km and kcat for wild-type Cyt c were determined to be 1.16 ± 0.12 μm and 48.2 ± 0.9 s−1, respectively. The Km value significantly depends on the experimental conditions, including pH and ion strength, and 1.6 μm was previously reported at pH 6.8 in 10 mm bis-Tris (21). Table summarizes the obtained Michaelis-Menten kinetic parameters of the wild-type and mutant proteins, showing that both of Km and kcat were changed by the mutations in Cyt c. As expected, the mutations at lysine residues in the interaction site, Lys-7/8, Lys-13, Lys-72, Lys-79, and Lys-86/87, resulted in substantial mutational effects on the Michaelis-Menten parameters. In particular, the substitution of Leu for Lys-13 induced a severe reduction in the affinity (Km) for CcO, consistent with the result of the chemical modification (10), whereas the mutation at Lys-39, which is located far from the interaction site (13), showed no significant effects on the parameters.
FIGURE 2.
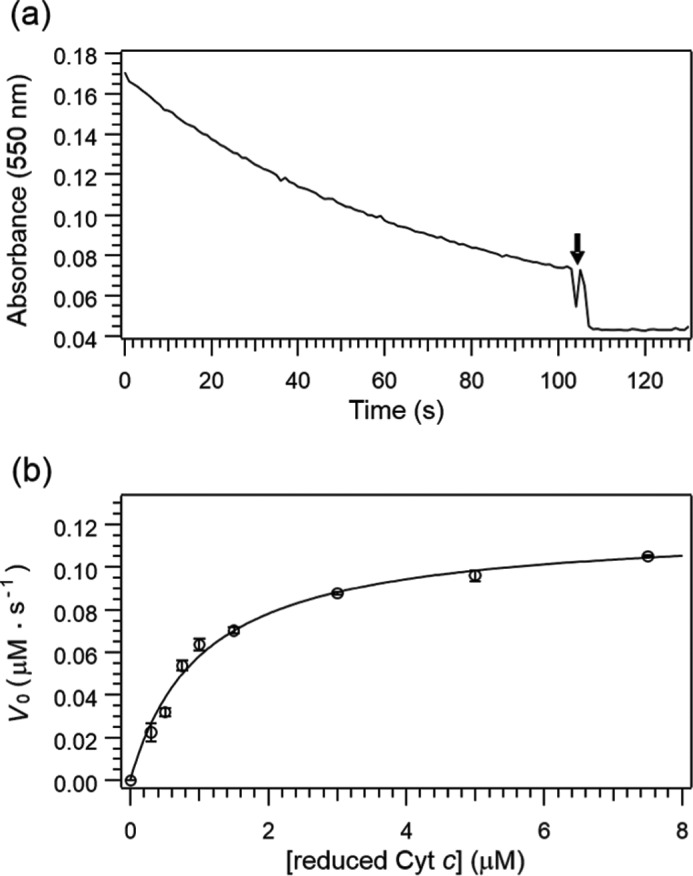
Absorbance change at 550 nm after the addition of CcO (a) and the dependence of the initial rate constants on the concentrations of ferrous Cyt c (b). a, small amount of potassium ferricyanide(III) was added to the reaction mixture (downward arrow) to determine the end point of the ET reaction. The measurements were carried out in the presence of 1 nm CcO dissolved in 50 mm sodium phosphate buffer at pH 6.8 containing 0.1% n-decyl β-d-maltoside. b, solid curve is the best fit to the Michaelis-Menten equation using the least-square analysis.
Moreover, the mutational effects of the hydrophobic residues, Ile-9, Ile-11, and Ile-81, are almost negligible in the Michaelis-Menten parameters as listed in Table 1. These observations are somewhat surprising because our NMR analysis indicated that both electrostatic and hydrophobic interactions contribute to the formation of the ET complex between Cyt c and CcO (13), and a previous simulation study also noted that hydrophobic interactions can significantly contribute to the stabilization of protein-protein complexes (9, 12).
TABLE 1.
Kinetic parameters of the ET reaction from CcO and wild-type and mutant Cyt c
| Groupa | Km | kcat | kcat/Km | |
|---|---|---|---|---|
| μm | s−1 | μm−1s−1 | ||
| K13L | A | 12 ± 1 | 62 ± 2 | 5.3 ± 0.6 |
| K86L/K87L | A | 10 ± 1 | 71 ± 2 | 7.0 ± 0.7 |
| K7L/K8L | B | 7.0 ± 0.7 | 110 ± 10 | 16 ± 2 |
| K72L | B | 5.6 ± 1.1 | 130 ± 10 | 24 ± 5 |
| K79L | B | 5.0 ± 0.8 | 83 ± 4 | 17 ± 3 |
| I9A | C | 0.82 ± 0.15 | 46 ± 1 | 57 ± 10 |
| I11A | C | 1.1 ± 0.2 | 51 ± 1 | 45 ± 7 |
| I81A | C | 1.2 ± 0.1 | 49 ± 1 | 41 ± 2 |
| K39L | C | 1.2 ± 0.1 | 49 ± 1 | 43 ± 2 |
| Wild type | − | 1.2 ± 0.1 | 48 ± 1 | 42 ± 4 |
a Groups A–C represent severe, moderate, and unaffected mutational effects, respectively.
Although these mutational effects are obvious and significant in the kcat of some mutants, the variances of kcat in mutant and wild-type Cyt c were much less than the variances of Km (Table 1), indicating that the ET activity of mutant Cyt c depends mainly on the difference in Km. The nine mutants can therefore be classified into three types, groups A (severe, K13L and K86L/K87L), B (moderate, K7L/K8L, K72L, and K79L), and C (unaffected, I9A, I11A, I81A, and K39L), according to their mutational effects (see Fig. 1a) and based on their Km values.
Determination of the Most Probable Cyt c-CcO Complex
To understand the structural and energetic basis for the interaction between Cyt c and CcO, we predicted the binding interface of the Cyt c-CcO complex with the ZDOCK, ZRANK, and RDOCK algorithms using the NMR information (13) for effective guidance. As described under “Experimental Procedures,” seven refined poses for the wild-type complex were obtained. To select a reasonable complex for the subsequent energy minimization with AMBER, we checked the structural and energetic data of the complexes. Table 2 and supplemental Fig. S1 summarize the characteristics of the seven poses. Pose 1 is considered to be the most probable complex with regard to the lowest RDOCK energy (−33.7 kcal mol−1), whereas poses 6, 24, and 25 are not acceptable, due to their high energies compared with the others. However, pose 1 has a somewhat long distance (15.6 Å) between the vinyl carbon atom of heme c in Cyt c and the aromatic carbon atom of Trp-104 in subunit II of CcO, which is one of the residues that form an exposed hydrophobic cluster near the electron entry site, the CuA site, into CcO (Fig. 3a) (44). Of the energetically reasonable solutions, poses 10 and 13 have relatively short heme c-Trp-104 separations (11.5 and 5.8 Å, (Fig. 3c) respectively), which are comparable with the reported values (11). However, the RDOCK energies of the two poses are ∼10 kcal mol−1 higher than that of pose 1. Given the discussion mentioned above, we finally selected poses 1 and 13 in terms of the RDOCK energy and heme c-Trp-104 distance criteria, respectively. The two probable poses were further examined for energy minimization and detailed binding interaction analysis.
TABLE 2.
Structural and energetic properties of the seven poses for Cyt c-CcO complex
| Pose | Heme c–Τrp-104 distance | ΔΑSΑ (Å2) |
Ε_Rdock | ||||
|---|---|---|---|---|---|---|---|
| Totala | Ib | IIb | IIIb | IV–XIIIb | |||
| Å | kcal/mol | ||||||
| 1 | 15.6 | 1342/1457 | 252 (19) | 197 (15) | 94 (7) | 799 (60) | −33.7 |
| 6 | 11.2 | 1510/1567 | 418 (28) | 333 (22) | 96 (6) | 663 (44) | −7.13 |
| 10 | 11.5 | 1393/1395 | 306 (22) | 305 (22) | 26 (2) | 756 (54) | −23.3 |
| 11 | 17.2 | 1323/1406 | 183 (14) | 189 (14) | 21 (2) | 930 (70) | −24.0 |
| 13 | 5.8 | 1228/1345 | 201 (16) | 401 (33) | 40 (3) | 585 (48) | −22.2 |
| 24 | 13.9 | 1078/1207 | 173 (16) | 405 (38) | 0 (0) | 500 (46) | −9.90 |
| 25 | 20.9 | 1830/2132 | 457 (25) | 310 (17) | 55 (3) | 1007 (55) | −10.3 |
a Total ΔΑSΑ of Cyt c/CcO is shown.
b Contribution of each subunit of CcO to total ΔΑSΑ is given. Percentage (%) is shown in parentheses.
FIGURE 3.

Predicted binding interfaces of the Cyt c-CcO complexes of poses 1 and 13. a, focused view of the heme c (sphere) in Cyt c and Trp-104 (stick) in CcO, the probable electron entry site obtained from pose 1. b, interactions of the mutated residues in Cyt c with contact residues in CcO from pose 1 are highlighted. The amino acid residues in subunits I, II, and III are represented by green, cyan, and magenta (sticks), respectively. Dotted lines indicate hydrogen bonding between Cyt c and CcO residues. c and d, same representation as in a and b, respectively, but the corresponding CcO is from pose 13.
Prior to the energetic discussion, we examined the solvent-accessible surface area change (ΔASA) at the Cyt c-CcO interface associated with the complex formation. As shown in Table 2 and supplemental Table S2, Cyt c makes contact mainly with the three important catalytic subunits I–III of CcO, indicating that the contribution on average reaches more than 50% of the total ΔASA. The order of ΔASA for the three subunits I–III of CcO upon complex formation is I > II > III for poses 1, 6, 10, and 25 and II > I > III for poses 11, 13, and 24. The other subunits IV–XIII are also involved in the molecular recognition between Cyt c and CcO.
However, the total ΔASA for all the poses is larger than 2200 Å2, which is comparable with the value for protein-protein binding associated with major conformational changes that occur upon association (45). Although the large Cyt c-CcO interface might correspond to major conformational changes associated with the complex formation between Cyt c and CcO during the ET process, the small structural fluctuation of Cyt c (46) and small structural changes associated with the reduction of the CuA site (47) suggest that the binding of Cyt c with CcO under turnover conditions does not necessarily require major conformational changes at the interface. These considerations led to the basic assumption that no major conformational changes occur upon the binding of Cyt c to CcO and that the binding interfaces of the mutant complexes are almost the same as in the wild-type complex. In fact, the K13L Cyt c mutant showed the large increase of the Km value for the ET reaction to CcO, whereas the interaction site of the mutant to CcO is still located near the heme-exposed periphery, which is similar to that of the wild-type Cyt c-CcO complex (supplemental Fig. S2).
Molecular Recognition at the Binding Interface of the Cyt c-CcO Complex
Fig. 3, b and d, shows the binding interface of the Cyt c-CcO complex for the wild-type proteins obtained from poses 1 and 13 along with the location of the residues targeted by in vitro and in silico mutagenesis. Lys-13, Lys-86, and Lys-87 (group A) of Cyt c with severe mutational effects should be involved in the interface because these leucine replacements of the residues (K13L and K86L/K87L) exhibited large increases in Km (Table 1).
In pose 1 (Fig. 3b), the three residues are involved in the hydrogen bonds with Glu-119 (I), Leu-136 (I), Ala-137 (I), Ala-139 (I), and Gln-103 (II) in CcO (where roman numerals in parentheses denote subunit number), suggesting that these mutations affect the Cyt c-CcO interaction. In contrast, in pose 13 (Fig. 3d), the three residues have no partners for the hydrogen bonds in CcO. The severe effects of K13L and K86L/K87L can be qualitatively explained by pose 1 but not by pose 13.
Lys-7, Lys-8, Lys-72, and Lys-79 in group B undergo direct interactions with Tyr-105 (II), Asp-119 (II), Asn-38 (III), Gly-64 (VIII), and Asp-74 (IX) in CcO in pose 1. The corresponding CcO residues in pose 13 are Asp-119 (II) and His-36 (III). The two poses show that Asp-119 (II) is expected to be involved in the binding interactions for Cyt c. Among the residues in group C, Lys-39 is the only basic residue that is likely to interact favorably with acidic residues in CcO. However, as Lys-39 is an unaffected residue, it would be exposed on the opposite side from the Cyt c-CcO interface. In pose 1, Lys-39 actually lies at the solvent-exposed surface of Cyt c, whereas in pose 13, it is located in the vicinity of the interface. Taken together, pose 1 is reasonably consistent with the in vitro site-directed mutagenesis and structural requirements for the binding interactions, reflecting the Cyt c-CcO complex under turnover conditions.
Validation of the Predicted Binding Interface of the Cyt c-CcO Complex
We computationally validate the predicted binding interface of the Cyt c-CcO complex obtained from the docking. For this purpose, we performed correlation analysis on the kinetic data, Michaelis-Menten constant (Km), of wild-type and mutant Cyt c with CcO using the MM-PBSA approach. Table 3 lists the observed and calculated overall free energy changes along with the energetic components obtained from the MM-PBSA calculation on the modeled complexes constructed from pose 1. The observed overall free energy change ΔGobs for the complex formation of wild-type and mutant Cyt c with CcO under turnover conditions were calculated from Km, which is nicely reproduced by MM-PBSA. Fig. 4a shows good agreement between ΔGobs and ΔGcalc (r = 0.926). Notably, the calculation correctly discriminates among the wild-type and the three groups A–C, reflecting the degree of mutational effects. Given that ΔGcalc accounts for almost all of the free energy changes associated with complex formation except for the entropic changes, a significant linear correlation supports the assumption that no major conformational changes occur upon the binding of Cyt c with CcO, as reported previously for the protein-ligand complex formation (48–51).
TABLE 3.
Overall free energy change and representative energy terms
Values are given in kilocalories/mol.
| Cyt c | Group | ΔGobsa | ΔGcalcb | ΔEcoul | ΔEvdW | ΔGpolar | ΔGnonpolar | ΔGelectroc |
|---|---|---|---|---|---|---|---|---|
| K13L | A | −6.72 | −6.71 | −1226.21 | −136.36 | 1276.11 | −19.62 | 49.90 |
| K86L/K87L | A | −6.81 | −6.67 | −1154.71 | −135.77 | 1205.76 | −20.59 | 51.05 |
| K7L/K8L | B | −7.03 | −7.22 | −1261.34 | −136.51 | 1302.64 | −20.27 | 41.31 |
| K72L | B | −7.16 | −7.22 | −1166.92 | −141.46 | 1212.28 | −19.24 | 45.36 |
| K79L | B | −7.23 | −7.73 | −1214.39 | −143.74 | 1253.41 | −19.94 | 39.03 |
| I9A | C | −8.30 | −8.16 | −1415.22 | −138.80 | 1441.66 | −20.16 | 26.44 |
| I11A | C | −8.11 | −7.90 | −1430.76 | −133.97 | 1456.55 | −19.57 | 25.78 |
| I81A | C | −8.08 | −7.76 | −1416.59 | −134.45 | 1445.19 | −19.48 | 28.61 |
| K39L | C | −8.10 | −8.04 | −1351.56 | −137.65 | 1378.76 | −19.92 | 27.20 |
| Wild type | − | −8.09 | −8.25 | −1413.23 | −139.49 | 1438.72 | −20.19 | 25.49 |
| Varianced | 0.35 | 0.30 | 11227.38 | 8.64 | 9352.47 | 0.16 | 98.29 | |
| Correlatione | − | 0.926 | 0.921 | −0.131 | −0.909 | −0.116 | 0.977 |
a ΔGobs = RT lnKm (T = 298 K). Km values are from Table 1.
b Data were calculated from the correlation equation (ΔGcalc = 0.0548 (ΔEcoul + ΔEvdW + ΔGpolar + ΔGnonpolar) − 0.899: n = 10, r = 0.926, s = 0.250, F = 48.1) obtained from a linear regression analysis.
c ΔEcoul + ΔGpolar.
d Values are given in kcal2 mol−2.
e Correlation coefficient of each energy term is with ΔGobs.
FIGURE 4.

Plots of ΔGobs with ΔGcalc. The calculated values are obtained from the MM-PBSA calculation on the wild-type and mutant complexes constructed from pose 1 (a) and pose 13 (b).The correlation equation (ΔGcalc = 0.0548 (ΔEcoul + ΔEvdW + ΔGpolar + ΔGnonpolar)− 0.899) was used for the calculation.
In general, consistent with these results (Table 3), ΔEcoul is negative because complex formation is driven by favorable Coulombic interactions, whereas ΔGpolar is positive because the desolvation of polarized functional groups is energetically unfavorable. In Table 3, the variances of ΔEcoul and ΔGpolar, which are the electrostatic components of ΔGbind and ΔGsol, respectively, are considerably large, suggesting that the electrostatic interactions have a crucial impact on the variation in the binding of wild-type and mutant Cyt c to CcO. In fact, ΔEcoul and ΔGpolar are well correlated with ΔGobs (r = 0.921 and r = −0.909, respectively), the variance of which corresponds to the variance of Km, suggesting that the electrostatic components are dominant contributors to the variation of ΔGobs. However, as found in most protein-protein as well as protein-ligand systems (48–51), the Coulombic energy for the formation of the electrostatic components is compensated by the desolvation energy for polar residues, and there is actually an anti-correlation between ΔEcoul and ΔGpolar (ΔEcoul = −1.09 ΔGpolar + 163, n = 10, r = 0.999) in this system. Consequently, the variance of the sum of electrostatic contributions (ΔGelectro = ΔEcoul + ΔGpolar) becomes considerably small (98.3 kcal2 mol−2), but the variance of ΔGelectro is still significant compared with the variances of ΔEvdW and ΔGnonpolar, indicating that the electrostatic interactions are the primary factors determining the variation of ΔGobs.
The anti-correlation between ΔEcoul and ΔGpolar in Table 3 also implies that stabilization by ΔEcoul counteracts destabilization by ΔGpolar, and the variance of ΔEcoul overwhelms the variance of ΔGpolar (shown as the linear anti-correlation slope value of 1.09 more than unity). The Coulombic interaction, therefore, dominantly contributes to the variance of ΔGobs; however, the total electrostatic interactions (ΔGelectro) between Cyt c and CcO destabilize the complex formation under turnover conditions because of the large destabilization by the desolvation energy of ΔGpolar.
It should be noted that ΔEvdW and ΔGnonpolar are negative in all of the mutants and wild-type Cyt c, implying that the formation of the van der Waals interactions and the desolvation of nonpolar residues associated with complex formation facilitate the complex formation between Cyt c and CcO. However, the variances of ΔEvdW and ΔGnonpolar in the mutants are quite small (8.64 and 0.16 kcal2 mol−2 for ΔEvdW and ΔGnonpolar, respectively) and show no correlation with the variance of ΔGobs. Therefore, the stabilization by the sum of ΔEvdW and ΔGnonpolar is much larger than the destabilization by ΔGelectro (Table 3), but the contribution is not significant for the variation of ΔGobs.
In contrast to the successful results obtained from pose 1, a worse correlation was achieved when pose 13 was used (r = 0.513, shown in Fig. 4b), due to the incorrect positions of the mutated residues in pose 13, which has little direct interaction with CcO compared with pose 1 (Fig. 3, b and d).
The results mentioned above indicate that the modeled Cyt c-CcO complex structure (pose 1) consistently accounts for the in vitro site-directed mutagenesis experiments characterized by Km, also confirming that the protein-protein interaction site in the Cyt c-CcO complex under turnover conditions is quite similar to the one determined by NMR (13). The computational findings demonstrate that the electrostatic contribution is the most crucial for the variation in the mutational effects and that the van der Waals contribution stabilizes the complex formation of Cyt c with CcO under turnover conditions, as suggested by our NMR results (13).
Discussion
Functional Significance of Predicted Complex Structure of Cyt c-CcO
As clearly shown in Fig. 4a, ΔGobs, the free energies for the Michaelis-Menten complex formation of the Cyt c mutants with CcO in the ET reaction, are well reproduced by using the complex structure, pose 1, indicating that pose 1 is a complex structure under turnover conditions. It is quite interesting that the predicted complex structure based on the interaction analysis using NMR in the equilibrium state reproduced the free energy for the formation of the Michaelis-Menten complex under turnover conditions. This agreement between the free energy for the Cyt c-CcO complex formation in the equilibrium state and that for the formation of the kinetic intermediate indicates that the complex structure under turnover conditions is quite similar to that in the equilibrium state. Such structural similarity between the two states is supported by the rigid dynamic structure of Cyt c (46, 52). The generalized order parameters (s2), the effective correlation time for internal motion (τe), the 15N exchange broadening contributions (Rex) for each residue, and the overall correlation time (τm) clearly showed that the backbone dynamics of Cyt c are highly restricted due to the covalently bound heme that functions as the stable hydrophobic core (46). The residues located on the interaction site did not show detectable Rex at temperatures ranging from 279 to 298 K in the reduced and oxidized forms (52).
It should be noted here that pose 1 shows longer distance for the intermolecular ET reaction between heme c in Cyt c and Trp-104 in subunit II of CcO (15.6 Å for the distance between the vinyl carbon atom of heme c in Cyt c and the aromatic carbon atom of Trp-104 in subunit II of CcO), whereas no correlation between the binding free energy from pose 13, showing shorter distance between heme c and Trp-104, and that from the experimental Km values was observed (Fig. 4b). Although the heme c-Trp-104 distance was used as the criterion for a filtering process in the previous docking studies (9, 11), in which this distance was requested to be less than 15 Å, previous studies (53–55) have revealed that fast electron transfer (kET >104 s−1) will occur any time when the distance between the redox centers of the binding partners is less than 18 Å (55) through efficient electron tunneling across the interface. The quite slow electron transfer rate from Cyt c to CcO (< 400 s−1) (21) indicates the possibility that the redox centers of the two proteins are not so closely located in the electron transfer complex.
Another possibility is that pose 1 is the complex structure under the turnover conditions, but some structural changes would be required for the ET reaction. As reported previously for the interprotein ET reaction system (55), “encounter complexes” are formed before the formation of the protein complex where the ET reaction is induced. Considering that the Michaelis-Menten constants estimated from pose 1 were in good agreement with those from the steady-state kinetics experiments, pose 1 would be one of such encountered complexes essential for the ET reaction between Cyt c and CcO.
Identification of Hot Spot Residues by Free Energy Decomposition
Free energy decomposition on a per residue basis was applied to reveal which residues play important roles in the complex formation. Fig. 5a shows the decomposition of the total free energy change (ΔGtot = ΔEvdW + ΔGelectro + ΔGnonpolar) into the contributions from ΔEvdW, ΔGelectro (= ΔEcoul + ΔGpolar), and ΔGnonpolar for residues with |ΔGtot| ≥ 3.0 kcal mol−1. The primary focus here is to clarify the origin of the major interaction energy for the wild-type Cyt c-CcO complex. As shown in Fig. 5a, the decomposition identified hot spot residues, some of which are the mutated residues (Ile-11, Lys-13, Lys-72, Lys-79, Ile-81, Lys-86, and Lys-87). For a number of residues in Cyt c, ΔEvdW mainly contributes to ΔGtot and stabilizes the complex formation. Ile-11, Met-12, Ile-81, and Val-83, which are hydrophobic residues in the vicinity of the heme c periphery, dominantly contribute to the complex formation. The contribution of some of the hydrophobic residues to van der Waals interactions with CcO, as suggested by the previous docking study (9), has been found in this computational study based on NMR analyses (13). Lys-13, Gln-16, Lys-27, Thr-28, and Lys-86 are also critical for stabilizing complex formation through the van der Waals interactions, despite being polar and charged residues. Among the residues mentioned above, Ile-11, Lys-13, Ile-81, and Lys-86 are the mutated residues. The two mutations to Leu in group A and two of the three mutations to Ala in group C, i.e. K13L, K86L/K87L, I11A, and I81A, exist at the interface and reduce the van der Waals interactions. The counterparts are Leu-136 (I), Ala-139 (I), Gln-103 (II), Glu-157 (II), and Pro-60 (VIII) in CcO, as illustrated in Fig. 5, c–e.
FIGURE 5.

a, free energy decomposition of ΔGtot on a per residue basis into the contributions from ΔEvdW, ΔGelectro, and ΔGnonpolar (|ΔGtot| ≥ 3.0 kcal mol−1). Three-dimensional views of the residues at the regions of whole interface (b), subunit I (c), subunit II (d), and subunits III–XIII (e) other mainly contributing to the binding of Cyt c to CcO.
Moreover, Lys-79, Lys-87, and Glu-89 are exceptional residues for which ΔGelectro is the largest contribution. The two lysines, which are the mutated residues belonging to groups A and B, exhibit a gain from ΔEcoul in the favorable Coulombic interactions with Glu-119 (I) and Asp-74 (IX) in CcO (Fig. 5, c and e). The replacements of these residues by leucine reduce the favorable interactions and thus the binding affinities (Table 1). In contrast, Glu-89 exhibits the largest loss in ΔGelectro, overcompensated by unfavorable contributions from ΔGpolar, resulting in the destabilization of ΔGtot. The Km value for the E89Q mutant was estimated to be 0.79 ± 0.13 μm, showing the increased affinity to CcO and supporting unfavorable contribution of the interaction mediated with Glu-89 to the complex formation with CcO. This type of unfavorable contribution is also found for Asp-221 (I) and Glu-157 (II) in CcO. We emphasize here that favorable Coulombic interactions (ΔEcoul) are often not strong enough to compensate for unfavorable desolvation effects (ΔGpolar) (Table 3) (48, 56). In fact, the total electrostatic contribution to complex formation is unfavorable (see ΔGelectro in Table 3), implying that the electrostatic interactions do not stabilize the Cyt c-CcO complex under turnover conditions. The distribution of the favorable and unfavorable electrostatic interactions mediated by the charged residues would contribute to fixing the optimum binding orientation of Cyt c on the interaction surface of CcO for the effective ET from Cyt c to CcO.
These results suggest that computational predictions of hot spot residues are in good agreement with previous experimental and docking results (9, 13). The per residue decomposition can identify not only the energetic contribution of the mutated residues believed to lie at the binding interface of the Cyt c-CcO complex but also new critical residues responsible for the binding and for the variation in mutational effects. Moreover, our calculations predict that Glu-89 will unfavorably contribute to the binding of Cyt c.
Cyt c Interaction Site on CcO under Turnover Conditions
Because of the positively charged surface of Cyt c, previous simulation studies focused on the interactions with the negatively charged surface of subunit II of CcO (9, 11). Subunit II is located in the intermembrane space site and induces the CuA site (57), which is supposed to be the entry site for electrons from Cyt c, also suggesting that the electrostatic interaction with negatively charged amino acid residues in subunit II would be the primary interactions to stabilize the ET complex with Cyt c.
As expected from the distribution of the surface charges on Cyt c, the mutational effects of certain lysine residues in the interaction site for CcO, particularly Lys-13, Lys-86, and Lys-87, resulted in a severe decrease in the binding affinity of Cyt c to CcO (Table 3), suggesting the key role of the electrostatic interactions mediated by the lysine residues in complex formation between Cyt c and CcO under turnover conditions. However, the interaction analysis from the docking simulation here revealed that the favorable binding energy associated with the interactions with Lys-13 and Lys-86 comes mainly from ΔEvdW and ΔGnonpolar, whereas the electrostatic interaction mediated by Lys-13 induces destabilization of the Cyt c-CcO complex. This result is rather surprising, as we previously supposed that the interactions mediated by the lysine residues are primarily electrostatic and would stabilize the Cyt c-CcO complex.
Another surprising finding is that the contribution of the interactions mediated by Lys-13, Lys-86, and Lys-87 are not dominant in the stabilization of the Cyt c-CcO complex, as expected from the mutational study. The stabilizing energy for the Cyt c-CcO complex by Lys-13 is substantially less than for hydrophobic residues such as Ile-11, Met-12, and Val-83. Although the contribution of the hydrophobic interactions to the complex formation was suggested by the interaction analysis using NMR (13), such a dominant contribution of the hydrophobic interactions has not been previously reported.
It is quite interesting that van der Waals and nonpolar solvation types of interactions (ΔEvdW and ΔGnonpolar) are mainly attributable to the contribution of Cyt c and subunits IV–XIII of CcO, and not to subunit II (supplemental Table S2 and Fig. 5a), whereas previous simulation studies focused on the interactions between Cyt c and subunit II of CcO (9, 11). The relatively minor contribution of the interactions with subunit II to the stabilization of the Cyt c-CcO complex suggests that the distance between heme c of Cyt c and the CuA site of subunit II of CcO would be longer than we had expected, corresponding to the longer distance between heme c and Trp-104 in subunit II of CcO in the predicted Cyt c-CcO complex (pose 1). The relatively small number of hydrated water molecules (less than 20 water molecules) released in the large interaction site of Cyt c to CcO during the complex formation4 also indicates that the interprotein space between Cyt c and CcO in the complex would still be large enough to accommodate many hydrated water molecules at the interaction site. These specific but not strong interactions between Cyt c and CcO mediated by hydrophobic amino acid residues would promote the rapid dissociation and association of Cyt c to facilitate the four-electron reduction of molecular oxygen in CcO by the one-electron carrier, Cyt c.
In this work, Km is considered to be the dissociation constant, Kd, of the enzyme-substrate complex. However, Km (= (k2 + k3)/k1) is not the dissociation constant, Kd (= k2/k1), in Equation 8,
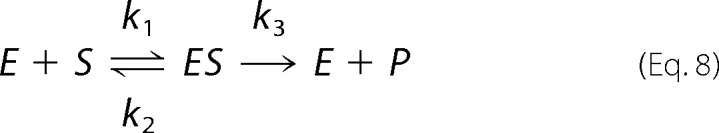 |
The ligand-binding and oxidation states of CcO in the ES complex have not been determined, and the ES complex under these conditions is likely to be a mixture of various intermediate species of CcO. The oxidized CcO under turnover conditions is unlikely to be the fully oxidized state as prepared, which is designated as the resting oxidized state (58, 59). It has been well established that under these experimental conditions for the determination of Km, oxidized Cyt c functions as a competitive inhibitor against reduced Cyt c, as shown in Equation 9,
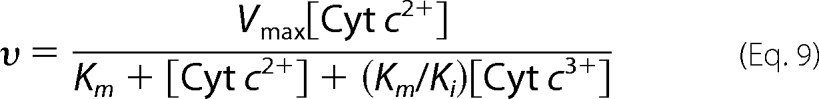 |
Only if Km = Ki, Equation 10 is given,
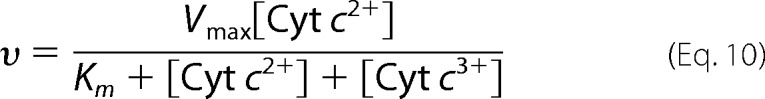 |
This equation indicates that the oxidation process of Cyt c2+ is first order with respect to [Cyt c2+], because [Cyt c2+]+ [Cyt c3+] is constant in each enzyme activity assay system as given above (60). In other words, the first order process indicates that a Ki value is identical to Km. Thus, Km, which shows the stability of the ES complex under turnover conditions, can be evaluated by Ki. Furthermore, these results suggest that the interaction between Cyt c and CcO is independent of the oxidation state of Cyt c as well as the oxidation and ligand binding states of CcO under turnover conditions. Rigorously speaking, the present analysis has been conducted for the interaction between Cyt c and oxidized CcO under turnover conditions, which is different from the resting oxidized CcO, as described above. However, the conclusions are reasonably applicable for the ES complex of CcO.
In summary, we have identified hot spot residues at the binding interface of CcO as well as Cyt c and found that the binding site consists of hydrophobic and charged residues under turnover conditions. The van der Waals interactions mediated by hydrophobic residues are the dominant contributions that stabilize the Cyt c-CcO complex under turnover conditions, as suggested by the NMR measurements (13). Although mutations of the charged residues resulted in severely decreased affinity of Cyt c to CcO and the electrostatic interactions by the charged residues govern the variation in the mutational effects, they do not significantly contribute to the stability of the Cyt c-CcO complex but to the orientation of the binding of Cyt c to CcO to facilitate the ET from Cyt c to CcO under turnover conditions. The energetic contributions to the ET complex formation are more significant in the interactions with subunits IV–XIII than with the electron entry site, subunit II, suggesting a specifically but not tightly packed interface between Cyt c and CcO, which would be essential for the ET reaction via the facilitation of the effective association and dissociation between the two proteins.
Author Contributions
W. S., S. H., S. Y., K. Y., and K. Ishimori designed the study and wrote the paper. M. I. contributed to purification of the samples. W. S., K. Inoue, and K. S. I. purified the protein samples. W. S., K. Inoue, and T. U. measured kinetics. S. H. performed docking simulation. T. S. measured the NMR spectra. All authors analyzed the results and approved the final version of the manuscript.
Supplementary Material
This work was supported by Grants-in-aid 25288072 and 25109501 (to K. I.), 26234567 (to S. Y.), and 24109014 and 15K13710 (to K. Y.) from the Japan Society for the Promotion of Science, the Global Center of Excellence Program, the Targeted Protein Research Program, CREST from Ministry of Education, Culture, Sports, Science and Technology (MEXT), and MEXT Projects of “Integrated Research on Chemical Synthesis” and “Elements Strategy Initiative to Form Core Research Center.” The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental materials, including the atomic coordinates of the complex determined by the docking simulation.
W. Sato, M. Imai, T. Uchida, K. Shinzawa-Itoh, S. Yoshikawa, and K. Ishimori, submitted for publication.
- ET
- electron transfer
- Cyt c
- cytochrome c
- CcO
- cytochrome c oxidase
- PDZ domain
- post-synaptic density protein (PSD95)-Drosophila disc large tumor suppressor (Dlg1)-zonula occludens-1 protein (zo-1) domain
- bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- MM-PBSA
- molecular mechanics method combined with Poisson-Boltzmann and surface area.
References
- 1. Yoshikawa S., and Shimada A. (2015) Reaction mechanism of cytochrome c oxidase. Chem. Rev. 115, 1936–1989 [DOI] [PubMed] [Google Scholar]
- 2. Malmström B. G. (1979) Cytochrome c oxidase. Structure and catalytic activity. Biochim. Biophys. Acta 549, 281–303 [PubMed] [Google Scholar]
- 3. Papa S. (1982) Molecular mechanism of proton translocation by the cytochrome system and the ATPase of mitochondria. Role of proteins. J. Bioenerg. Biomembr. 14, 69–86 [DOI] [PubMed] [Google Scholar]
- 4. Brunori M., and Wilson M. T. (1995) Electron transfer and proton pumping in cytochrome oxidase. Biochimie 77, 668–676 [DOI] [PubMed] [Google Scholar]
- 5. Kaila V. R., Verkhovsky M. I., and Wikström M. (2010) Proton-coupled electron transfer in cytochrome oxidase. Chem. Rev. 110, 7062–7081 [DOI] [PubMed] [Google Scholar]
- 6. Pettigrew G. W., and Moore G. R. (1987) Cytochrome c–Biological Aspects, pp. 1–28, Springer-Verlag, Berlin [Google Scholar]
- 7. Margoliash E., and Schejter A. (1966) Cytochrome c. Adv. Protein Chem. 21, 113–286 [DOI] [PubMed] [Google Scholar]
- 8. Takano T., Kallai O. B., Swanson R., and Dickerson R. E. (1973) The structure of ferrocytochrome c at 2.45 Å resolution. J. Biol. Chem. 248, 5234–5255 [PubMed] [Google Scholar]
- 9. Roberts V. A., and Pique M. E. (1999) Definition of the interaction domain for cytochrome c on cytochrome c oxidase. III. J. Biol. Chem. 274, 38051–38060 [DOI] [PubMed] [Google Scholar]
- 10. Ferguson-Miller S., Brautigan D. L., and Margoliash E. (1978) Definition of cytochrome c binding domains by chemical modification. III. Kinetics of reaction of carboxydinitrophenyl cytochromes c with cytochrome c oxidase. J. Biol. Chem. 253, 149–159 [PubMed] [Google Scholar]
- 11. Flöck D., and Helms V. (2002) Protein-protein docking of electron transfer complexes: cytochrome c oxidase and cytochrome c. Proteins 47, 75–85 [DOI] [PubMed] [Google Scholar]
- 12. Basdevant N., Weinstein H., and Ceruso M. (2006) Thermodynamic basis for promiscuity and selectivity in protein-protein interactions: PDZ domains, a case study. J. Am. Chem. Soc. 128, 12766–12777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sakamoto K., Kamiya M., Imai M., Shinzawa-Itoh K., Uchida T., Kawano K., Yoshikawa S., and Ishimori K. (2011) NMR basis for interprotein electron transfer gating between cytochrome c and cytochrome c oxidase. Proc. Natl. Acad. Sci. U.S.A. 108, 12271–12276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bisson R., Jacobs B., and Capaldi R. A. (1980) Binding of arylazidocytochrome c derivatives to beef heart cytochrome c oxidase: crosslinking in the high- and low-affinity binding sites. Biochemistry 19, 4173–4178 [DOI] [PubMed] [Google Scholar]
- 15. Banci L., Bertini I., Gray H. B., Luchinat C., Reddig T., Rosato A., and Turano P. (1997) Solution structure of oxidized horse heart cytochrome c. Biochemistry 36, 9867–9877 [DOI] [PubMed] [Google Scholar]
- 16. Banci L., Bertini I., Huber J. G., Spyroulias G. A., and Turano P. (1999) Solution structure of reduced horse heart cytochrome c. J. Bioinorg. Chem. 4, 21–31 [DOI] [PubMed] [Google Scholar]
- 17. Yoshikawa S., Shinzawa-Itoh K., Nakashima R., Yaono R., Yamashita E., Inoue N., Yao M., Fei M. J., Libeu C. P., Mizushima T., Yamaguchi H., Tomizaki T., and Tsukihara T. (1998) Redox-coupled crystal structural changes in bovine heart cytochrome c oxidase. Science 280, 1723–1729 [DOI] [PubMed] [Google Scholar]
- 18. Jeng W.-Y., Chen C.-Y., Chang H.-C., and Chuang W.-J. (2002) Expression and characterization of recombinant human cytochrome c in E. coli. J. Bioenerg. Biomembr. 34, 423–431 [DOI] [PubMed] [Google Scholar]
- 19. Witt H., Malatesta F., Nicoletti F., Brunori M., and Ludwig B. (1998) Cytochrome-c binding site on cytochrome oxidase in Paracoccus denitrificans. Eur. J. Biochem. 251, 367–373 [DOI] [PubMed] [Google Scholar]
- 20. Drosou V., Malatesta F., and Ludwig B. (2002) Mutations in the docking site for cytochrome c on the Paracoccus heme aa3 oxidase. Eur. J. Biochem. 269, 2980–2988 [DOI] [PubMed] [Google Scholar]
- 21. Michel B., and Bosshard H. R. (1989) Oxidation of cytochrome c by cytochrome c oxidase: spectroscopic binding studies and steady-state kinetics support a conformational transition mechanism. Biochemistry 28, 244–252 [DOI] [PubMed] [Google Scholar]
- 22. Speck S. H., Dye D., and Margoliash E. (1984) Single catalytic site model for the oxidation of ferrocytochrome c by mitochondrial cytochrome c oxidase. Proc. Natl. Acad. Sci. U.S.A. 81, 347–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rajagopal B. S., Edzuma A. N., Hough M. A., Blundell K. L., Kagan V. E., Kapralov A. A., Fraser L. A., Butt J. N., Silkstone G. G., Wilson M. T., Svistunenko D. A., and Worrall J. A. (2013) The hydrogen-peroxide-induced radical behaviour in human cytochrome c–phospholipid complexes: implications for the enhanced pro-apoptotic activity of the G41S mutant. Biochem. J. 456, 441–452 [DOI] [PubMed] [Google Scholar]
- 24. Muramoto K., Ohta K., Shinzawa-Itoh K., Kanda K., Taniguchi M., Nabekura H., Yamashita E., Tsukihara T., and Yoshikawa S. (2010) Bovine cytochrome c oxidase structures enable O2 reduction with minimization of reactive oxygens and provide a proton-pumping gate. Proc. Natl. Acad. Sci. U.S.A. 107, 7740–7745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Neria E., Fischer S., and Karplus M. (1996) Simulation of activation free energies in molecular systems. J. Chem. Phys. 105, 1902–1921 [Google Scholar]
- 26. Chen R., and Weng Z. (2002) Docking unbound proteins using shape complementarity, desolvation, and electrostatics. Proteins 47, 281–294 [DOI] [PubMed] [Google Scholar]
- 27. Chen R., and Weng Z. (2003) A novel shape complementarity scoring function for protein-protein docking. Proteins 51, 397–408 [DOI] [PubMed] [Google Scholar]
- 28. Chen R., Li L., and Weng Z. (2003) ZDOCK: An initial-stage protein-docking algorithm. Proteins 52, 80–87 [DOI] [PubMed] [Google Scholar]
- 29. Pierce B., and Weng Z. (2007) ZRANK: Reranking protein docking predictions with an optimized energy function. Proteins 67, 1078–1086 [DOI] [PubMed] [Google Scholar]
- 30. Li L., Chen R., and Weng Z. (2003) RDOCK: Refinement of rigid-body protein docking predictions. Proteins 53, 693–707 [DOI] [PubMed] [Google Scholar]
- 31. Autenrieth F., Tajkhorshid E., Baudry J., and Luthey-Schulten Z. (2004) Classical force field parameters for the heme prosthetic group of cytochrome c. J. Comput. Chem. 25, 1613–1622 [DOI] [PubMed] [Google Scholar]
- 32. Tashiro M., and Stuchebrukhov A. A. (2005) Thermodynamic properties of internal water molecules in the hydrophobic cavity around the catalytic center of cytochrome c oxidase. J. Phys. Chem. B 109, 1015–1022 [DOI] [PubMed] [Google Scholar]
- 33. Seifert A., Tatzel S., Schmid R. D., and Pleiss J. (2006) Multiple molecular dynamics simulations of human p450 monooxygenase CYP2C9: The molecular basis of substrate binding and regioselectivity toward warfarin. Proteins 64, 147–155 [DOI] [PubMed] [Google Scholar]
- 34. Giammona D. A. (1984) An Examination of Conformational Flexibility in Porphyrins and Bulky-Ligand Binding in Myoglobin. Ph.D. thesis, University of California, Davis [Google Scholar]
- 35. Wang J., Cieplak P., and Kollman P. A. (2000) How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 21, 1049–1074 [Google Scholar]
- 36. Dolinsky T. J., Nielsen J. E., McCammon J. A., and Baker N. A. (2004) PDB2PQR: an automated pipeline for the setup of Poisson–Boltzmann electrostatics calculations. Nucleic Acids Res. 32, W665–W667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Case D. A., Cheatham T. E. 3rd., Darden T., Gohlke H., Luo R., Merz K. M. Jr., Onufriev A., Simmerling C., Wang B., and Woods R. J. (2005) The Amber biomolecular simulation programs. J. Comput. Chem. 26, 1668–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kollman P. A., Massova I., Reyes C., Kuhn B., Huo S., Chong L., Lee M., Lee T., Duan Y., Wang W., Donini O., Cieplak P., Srinivasan J., Case D. A., and Cheatham T. E. 3rd. (2000) Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc. Chem. Res. 33, 889–897 [DOI] [PubMed] [Google Scholar]
- 39. Hou T., Wang J., Li Y., and Wang W. (2011) Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 51, 69–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Honig B., and Nicholls A. (1995) Classical electrostatics in biology and chemistry. Science 268, 1144–1149 [DOI] [PubMed] [Google Scholar]
- 41. Lee B., and Richards F. M. (1971) The interpretation of protein structures: estimation of static accessibility. J. Mol. Biol. 55, 379–400 [DOI] [PubMed] [Google Scholar]
- 42. Weiser J., Shenkin P. S., and Still W. C. (1999) Approximate atomic surfaces from linear combinations of pairwise overlaps (LCPO). J. Comput. Chem. 20, 217–230 [Google Scholar]
- 43. Miller B. R. 3rd., McGee T. D. Jr., Swails J. M., Homeyer N., Gohlke H., and Roitberg A. E. (2012) MMPBSA.py:an efficient program for end-state free energy calculations. J. Chem. Theory Comput. 8, 3314–3321 [DOI] [PubMed] [Google Scholar]
- 44. Witt H., Malatesta F., Nicoletti F., Brunori M., and Ludwig B. (1998) Tryptophan 121 of subunit II is the electron entry site to cytochrome-c oxidase in Paracoccus denitrificans. J. Biol. Chem. 273, 5132–5136 [DOI] [PubMed] [Google Scholar]
- 45. Lo Conte L., Chothia C., and Janin J. (1999) The atomic structure of protein-protein recognition sites. J. Mol. Biol. 285, 2177–2198 [DOI] [PubMed] [Google Scholar]
- 46. Sakamoto K., Kamiya M., Uchida T., Kawano K., and Ishimori K. (2010) Redox-controlled backbone dynamics of human cytochrome c revealed by 15N NMR relaxation measurements. Biochem. Biophys. Res. Commun. 398, 231–236 [DOI] [PubMed] [Google Scholar]
- 47. Yoshikawa S., Muramoto K., Shinzawa-Itoh K., Aoyama H., Tsukihara T., Ogura T., Shimokata K., Katayama Y., and Shimada H. (2006) Reaction mechanism of bovine heart cytochrome c oxidase. Biochim. Biophys. Acta 1757, 395–400 [DOI] [PubMed] [Google Scholar]
- 48. Gohlke H., Kiel C., and Case D. A. (2003) Insights into protein–protein binding by binding free energy calculation and free energy decomposition for the Ras-Raf and Ras-RalGDS complexes. J. Mol. Biol. 330, 891–913 [DOI] [PubMed] [Google Scholar]
- 49. Hitaoka S., Matoba H., Harada M., Yoshida T., Tsuji D., Hirokawa T., Itoh K., and Chuman H. (2011) Correlation analyses on binding affinity of sialic acid analogues and anti-influenza drugs with human neuraminidase using ab Initio MO calculations on their complex structures–LERE-QSAR analysis (IV). J. Chem. Inf. Model. 51, 2706–2716 [DOI] [PubMed] [Google Scholar]
- 50. Yoshida T., Hitaoka S., Mashima A., Sugimoto T., Matoba H., and Chuman H. (2012) Combined QM/MM (ONIOM) and QSAR approach to the study of complex formation of matrix metalloproteinase-9 with a series of biphenylsulfonamides–LERE-QSAR analysis (V). J. Phys. Chem. B 116, 10283–10289 [DOI] [PubMed] [Google Scholar]
- 51. Hitaoka S., and Chuman H. (2013) Revisiting the Hansch–Fujita approach and development of a fundamental QSAR. J. Pestic. Sci. 38, 60–67 [Google Scholar]
- 52. Imai M., Saio T., Kumeta H., Uchida T., Inagaki F., and Ishimori K. (2016) Investigation of the redox-dependent modulation of structure and dynamics in human cytochrome c. Biochem. Biophys. Res. Commun. 469, 978–984 [DOI] [PubMed] [Google Scholar]
- 53. Moser C. C., Keske J. M., Warncke K., Farid R. S., and Dutton P. L. (1992) Nature of biological electron transfer. Nature 355, 796–802 [DOI] [PubMed] [Google Scholar]
- 54. Page C. C., Moser C. C., Chen X., and Dutton P. L. (1999) Natural engineering principles of electron tunnelling in biological oxidation-reduction. Nature 402, 47–52 [DOI] [PubMed] [Google Scholar]
- 55. Volkov A. N. (2015) Structure and function of transient encounters of redox proteins. Acc. Chem. Res. 48, 3036–3043 [DOI] [PubMed] [Google Scholar]
- 56. Sheinerman F. B., and Honig B. (2002) On the role of electrostatic interactions in the design of protein–protein interfaces. J. Mol. Biol. 318, 161–177 [DOI] [PubMed] [Google Scholar]
- 57. Tsukihara T., Aoyama H., Yamashita E., Tomizaki T., Yamaguchi H., Shinzawa-Itoh K., Nakashima R., Yaono R., and Yoshikawa S. (1995) Structure of metal sites of oxidized bovine heart cytochrome c oxidase at 2.8 Å. Science 269, 1069–1074 [DOI] [PubMed] [Google Scholar]
- 58. Wilson M. T., Antonini G., Malatesta F., Sarti P., and Brunori M. (1994) Probing the oxygen binding site of cytochrome c oxidase by cyanide. J. Biol. Chem. 269, 24114–24119 [PubMed] [Google Scholar]
- 59. Brunori M., Colosimo A., Sarti P., Antonini E., and Wilson M. T. (1981) ‘Pulsed’ cytochrome oxidase may be produced without the advent of dioxygen. FEBS Lett. 126, 195–198 [DOI] [PubMed] [Google Scholar]
- 60. Minnaert K. (1961) The kinetics of cytochrome c oxidase. I. The system: cytochrome c-cytochrome oxidase-oxygen. Biochim. Biophys. Acta 50, 23–34 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.