
Figure 2.
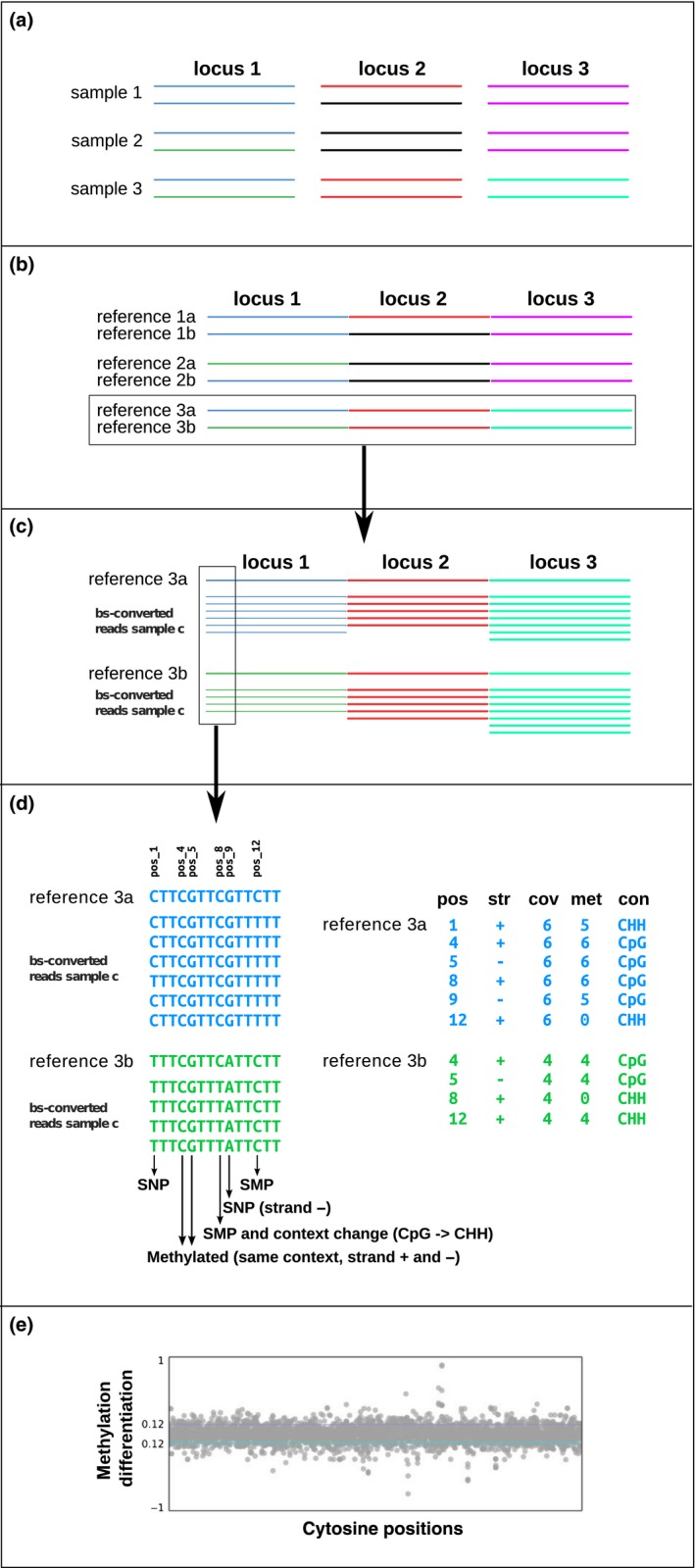
Workflow of the bsRADseq analysis when no reference genome is available. (a) Standard RAD sequencing data set assembled using stacks and unconverted reads (three hypothetical loci, 100 bp long, are exemplified and the different alleles per locus are indicated with different colours. (b) Individual reduced reference genome built using custom python scripts. (c) Mapping the converted reads of each individual sample to its own reduced reference genome using bismark and bowtie. (d) Analysis of the methylation level at each cytosine position separately in each individual sample using bismark. On the left: reads aligned to each reference, 14 base pairs shown; on the right, output of the Bismark_methylation_extractor function for the same base pairs. Pos: position; str: strand; cov: number of reads supporting that position; met: number of reads supporting methylation at that position; cont: context of the cytosine position according to the reference. (e) Results across individual samples are summarized using custom python scripts and statistical tests assessing significance of methylation differentiation between groups of samples can be performed. Here, the average methylation difference between the two Heliosperma species across all CHH positions screened is shown.