

Abstract
Label-retaining cells, which are characterized by dormancy or slow cycling, may be identified in a number of human normal and cancer tissues, and these cells demonstrate stem cell potential. In glioblastoma, label-retaining assays to enrich glioma stem cells remain to be fully investigated. In the present study, glioblastoma sphere cells cultured in serum-free medium were initially stained with the cell membrane fluorescent marker DiI. The fluorescence intensity during cell proliferation and sphere reformation was observed. At 2 weeks, the DiI-retaining cells were screened by fluorescence-activated cell sorting and compared phenotypically with the DiI-negative cells in terms of in vitro proliferation, clonogenicity and multipotency and for in vivo tumorigenicity, as well as sensitivity to irradiation and temozolomide treatment. It was observed that DiI-retaining cells accounted for a small proportion, <10%, within the glioblastoma spheres and that DiI-retaining cells proliferated significantly more slowly compared with DiI-negative cells (P=0.011, P=0.035 and P=0.023 in the of NCH421k, NCH441 and NCH644 glioblastoma sphere cell lines). Significantly increased clonogenicity (P=0.002, P=0.034 and P=0.016 in the NCH441, NCH644 and NCH421k glioblastoma sphere cell lines) and three-lineage multipotency were observed in DiI-retaining cells in vitro compared with DiI-negative cells. As few as 100 DiI-retaining cells were able to effectively generate tumors in the immunocompromised mouse brain, whereas the same number of DiI-negative cells possessed no such ability, indicating the increased tumorigenicity of DiI-retaining cells compared with DiI-negative cells. Furthermore, DiI-retaining cells demonstrated significant resistance following irradiation (P=0.012, P=0.024 and P=0.036) and temozolomide (P=0.003, P=0.005 and P=0.029) compared with DiI-negative cells in the NCH421k, NCH441 and NCH644 glioblastoma sphere cell lines, respectively. It was concluded that label-retaining cells in glioblastoma spheres manifest clear stem cell features and that the label-retaining assay may be utilized to further enrich glioma stem cells cultured under serum-free conditions for additional study.
Keywords: label-retaining cell, label-retaining assay, glioma, cancer stem cell, DiI
Introduction
Glioblastoma multiforme accounts for up to 60% of all malignant primary brain tumors in adults (1). The average patient survival remains poor despite treatments including surgery, radiotherapy and chemotherapy (1). Numerous studies have focused on glioblastoma ‘stem cells’ or ‘initiating cells’, which constitute a small population of the tumor, but which possess significant tumorigenicity and resistance to conventional radiotherapy and chemotherapy (2).
Due to the marked heterogeneity of glioblastoma tissue, methods for selecting the stem cell population specifically and effectively remain challenging (3). One simple and popular method to isolate cancer stem cells is based on the use of a specific stem cell marker, for example, the widely used cluster of differentiation (CD) 133, a pentaspan transmembrane molecule (4). However, the application of this molecule as a stem cell marker remains debatable. In glioma, CD133-negative tumor cells are able to produce CD133-positive cells; furthermore, CD133-negative cells demonstrate tumorigenicity similar to CD133-positive cells (5,6). The glial progenitor marker A2B5 and the neural stem cell marker CD15 have been used to isolate tumor stem cells, which may also be negative for CD133 (7,8). Additionally, CD133-positive cells are not necessarily positive for A2B5 or CD15 (7,8). Therefore, a more specific cell surface marker for glioma stem cells is required.
One way to circumvent the limitations of the current stem cell markers is to isolate stem cells based on their inherent features, which are distinct from their progenitor or differentiated cells (9–11). Normal stem cells are characterized by dormancy or slow cycling (12,13). During dormancy, no DNA synthesis-associated mutations occur, thus maintaining the homeostasis of the stem cell pool. Upon stress or injury, dormant cells enter into the active proliferation stage and reconstitute the tissue, thus maintaining the homeostasis of the body (12,13). Such a small group of dormant cells exists, for instance, among well-characterized mouse hematopoietic stem cells (12). These cells divide ~5 times in a lifetime, and the division cycle may last as long as 145 days. Upon bone marrow injury or growth factor stimulus treatment, for example granulocyte-colony stimulating factor or interferon α, the dormant cells are activated into fast-cycling cells to reconstitute the blood system (12). Among intestinal stem cells, a similar dormant cell pool has a role similar to that of the cells in the hematopoietic system (13). Therefore, dormant stem cells reside at the top of the cellular hierarchy, and screening dormant cells may be a rational way to enrich stem cells.
Fluorescence label-retaining assays are able to effectively discriminate dormant or slow-cycling cells from fast-cycling cells (14). Following the initial uniform fluorescence labeling of a whole cell population, the fluorescence intensity in cycling cells decreases by half due to cell division (14). Following approximately 6 or 7 rounds of division, the fluorescence intensity is so weak that no label is observable in fast-cycling cells, whereas the fluorescence is retained in dormant or slow-cycling cells; thus, the two cell populations can be effectively discriminated based on their varying proliferative abilities (14).
In the stem cell-permissive culture condition of serum-free medium, suspended glioblastoma spheres are considered rich in glioma stem cells (15). However, the majority of cells in the spheres are progenitors with a distinct proliferative potential compared with stem cells (15). The present study hypothesized that the label-retaining assay may further enrich stem cells in glioblastoma spheres. To address this hypothesis, glioblastoma sphere cells were fluorescently labeled, and the alterations in fluorescence intensity during cultivation were monitored, screened, and used to phenotypically characterize the label-retaining cells.
Materials and methods
Cell lines and cell culture
The NCH421k, NCH441 and NCH644 glioblastoma sphere cell lines were kindly provided by Professor Christel Herold-Mende from the Department of Neurosurgery at Heidelberg University (Heidelberg, Germany) and were cultivated in serum-free medium, which consisted of Dulbecco's modified Eagle's medium (DMEM)/F-12 medium (Sigma-Aldrich, St. Louis, MO, USA) containing 20% BIT serum-free supplement (STEMCELL Technologies, Inc., Vancouver, BC, Canada), 20 ng/ml basic fibroblast growth factor (provitro GmbH, Berlin, Germany) and 20 ng/ml epidermal growth factor (provitro GmbH). To induce differentiation, glioblastoma spheres were grown in DMEM containing 10% fetal calf serum (Biochrom, Ltd., Cambridge, UK).
DiI staining of glioblastoma spheres
Glioblastoma spheres were trypsinized (Thermo Fisher Scientific, Inc., Waltham, MA, USA) into single cell suspensions. DiI cell-labeling solution (5 µl/ml; Thermo Fisher Scientific, Inc.) was added to cell suspensions at a density of 1×106/ml. Following incubation for 15 min at 37°C, the labeled cell suspensions were washed twice with phosphate-buffered saline (PBS) and subsequently suspended in the aforementioned serum-free medium. Following incubation for 10 min at 37°C, the cells were uniformly stained with DiI.
Fluorescence-activated cell sorting (FACS) analysis
Following staining with DiI and culturing in the serum-free medium in a humidified 5% CO2/95% air incubator at 37°C for 2 weeks, the glioblastoma spheres were trypsinized into single cell suspensions. The cells (2×106) were subjected to FACS analysis (FACSAria; BD Biosciences, Franklin Lakes, New Jersey, USA) to separate DiI-retaining and DiI-negative cells. Single cell suspensions without DiI staining were used as the isotype control. The sorted DiI-retaining and DiI-negative cells were expanded in the serum-free mediumn in a humidified 5% CO2/95% air incubator at 37°C for 2–4 weeks. When cell growth reached the exponential phase and clear tumor spheres were observed, the spheres were used for the subsequent experiments.
Cell proliferation assay
Dissociated spheres (100 µl) containing ~5,000 cells were seeded in 5 replicates in a 96-well plate (Thermo Fisher Scientific, Inc.). The proliferative ability of these cells was measured using a cell counting kit-8 (CCK-8; Dojindo Molecular Technologies, Inc., Kumamoto, Japan) according to the manufacturer's protocol.
Clonogenic assay
The glioblastoma spheres were trypsinized into single cell suspensions and plated in 96-well plates in 0.2 ml serum-free medium at a density of 1,000 cells/well, 500 cells/well or 200 cells/well in each row. The cultures were fed 0.02 ml serum-free medium/well every 3 days, until day 14. The glioma spheres with dimensions >75 µm were counted. The sphere-forming frequency was obtained by dividing the number of observed spheres by the number of initially plated cells. For the serial clonogenic assay, the spheres were harvested and cultivated in the serum-free mediumn in a humidified 5% CO2/95% air incubator at 37°C for approximately 1–2 weeks. Subsequently, the spheres were trypsinized into single cell suspensions and replated in 96-well plates as described above. Finally, the sphere-forming frequency was recalculated.
Flow cytometric analysis
The glioblastoma spheres were trypsinized into single cell suspensions for the flow cytometric analysis of CD133-positive cells. Single cell suspensions containing ~5×105 cells were incubated with mouse monoclonal anti-human CD133/1 primary antibody (dilution, 1:10; catalog no., 130-090-422; Miltenyi Biotec GmbH, Bergisch Gladbach, Germany) for 30 min at room temperature; mouse monoclonal anti-human immunoglobulin G1 (dilution, 1:10; catalog no., AM01157PU-N; Acris Antibodies GmbH, Herford, Germany) was used as the isotype control. Following washing 3 times with Flow Cytometry buffer (eBioscience, Inc., San Diego, CA, USA), the single cell suspensions of the NCH421k, NCH441 and NCH644 glioblastoma spheres were incubated with fluorescein isothiocyanate-conjugated goat polyclonal anti-mouse secondary antibody (dilution, 1:64; catalog no., BA1101; Boster Systems, Inc., Pleasanton, CA, USA) in the dark for ~20 min. Following washing 3 times with Flow Cytometry buffer (eBioscience, Inc.), the aforementioned NCH421k, NCH441 and NCH644 glioblastoma sphere single cell suspensions were subjected to flow cytometric analysis (FACSCalibur™; BD Biosciences). Only cells with staining intensities above the maximal level of the isotype control were defined as positive.
Western blot analysis
Following differentiation for 1 week, DiI-retaining and DiI-negative cells were harvested and lysed in radioimmunoprecipitation assay buffer [50 mM Tris (pH 7.4), 0.15 M NaCl, 1% Triton X-100, 1% sodium deoxycholate and 0.1% sodium dodecyl sulfate; Sigma-Aldrich]. The supernatants were recovered, and the total protein concentrations were determined using the Bradford assay (16). In total, 75 µg of protein per well were loaded onto the gel. The proteins were separated by 8% sodium dodecyl sulfate polyacrylamide gel electrophoresis and electrophoretically transferred onto a polyvinylidene difluoride membrane at 4°C overnight. Subsequently, the membrane was blocked at room temperature for 1 h in TST buffer (1.211 g Tris, 8.766 g sodium chloride and 5 ml Tween-20 in 1 l double distilled water at pH 7.4; G-Biosciences, St. Louis, MO, USA) containing 5% non-fat milk (Sigma-Aldrich). The polyvinylidene difluoride membrane was washed with TST buffer (G-Biosciences) and subsequently incubated with the following primary antibodies at 4°C overnight: Mouse monoclonal anti-human glial fibrillary acidic protein (GFAP; dilution, 1:10; catalog no., MA1045; Boster Systems, Inc.), rabbit polyclonal anti-human platelet-derived growth factor receptor α (PDGFRα; dilution, 1:10; catalog no., PA1678; Boster Systems, Inc.) or mouse monoclonal anti-human βIII-tubulin (dilution, 1:10; catalog no., MA1112; Boster Systems, Inc.). The polyvinylidene difluoride membrane was washed again with TST buffer (G-Biosciences) and subsequently incubated with horseradish peroxidase (HRP)-conjugated anti-mouse or anti-rabbit polyclonal secondary antibody (dilution, 1:10,000; catalog nos., NA931 and NA934, respectively; GE Healthcare Life Sciences, Chalfont, UK) and HRP-conjugated mouse monoclonal anti-human β-actin antibody (dilution, 1:25,000; catalog no., ab49900; Abcam, Cambridge, UK) at room temperature for 1 h. Finally, the target proteins on the membrane were detected using an ECL Plus kit (GE Healthcare Life Sciences) according to the manufacturer's protocol. The band intensities of the target proteins were normalized to β-actin using ImageJ 1.46r software (imagej.nih.gov/ij/).
Irradiation
Glioblastoma spheres were trypsinized into single cell suspensions and plated at a density of 2×104/ml in 96-well plates. The cells were irradiated at room temperature with a single exposure to photon irradiation at a dose of 10 Gy delivered by a linear accelerator (Elekta Synergy®; Elekta AB, Stockholm, Sweden). Following 72 h of irradiation, the proliferation of the irradiated cells was measured using a CCK-8 according to the manufacturer's protocol.
Chemotherapy
Glioblastoma spheres were trypsinized into single cell suspensions and plated at a density of 2×104/ml in 96-well plates. The cells were treated with 500 µmol/l temozolomide (Sigma-Aldrich). Following 72 h of treatment, the proliferation of the treated cells was measured using a CCK-8 according to the manufacturer's protocol.
In vivo tumorigenicity
In total, 40 female non-obese diabetic/severe combined immunodeficiency (NOD/SCID) mice (6–8 weeks old; Charles River Laboratories, Wilmington, MA, USA) were housed under specific pathogen-free conditions at the temperature of 25±5°C, with 12 h light per day. The mice had free access to sterilized food and water. All the animal experiments were performed according to the China Animal Protection Law following institutional guidelines for animal welfare and experimental conduct. Ethical approval was obtained from the Ethical Committee of Tonji Medical University, Tongji Medical College, Huazhong University of Science and Technology (Wuhan, China). In total, 20 mice were randomly selected to be implanted with the DiI-retaining cells and the remaining mice were implanted with the DiI-negative cells. Cells were implanted stereotactically into the right hemispheres of the NOD/SCID mice. To avoid reflux of cells along the needle tract, small carrier volumes (5 µl) were injected 4 mm parasagittal along the coronary suture at an adequate depth of 3.5 mm. Following pausing for 10 min to allow for diffusion of the carrier fluid into the parenchyma, the injection needle was slowly extracted. Subsequent to the injection, no macroscopic reflux was observed in any of the animals, and the needle tract was sealed with biodegradable bone wax (Ethicon, Inc., Somerville, NJ, USA). The survival and general performance of the mice were monitored daily for 6 months following tumor cell implantation. At this point, the remaining live mice were sacrificed by cervical dislocation, and all the brains were harvested.
Immunohistochemistry
Mouse brains were fixed in 10% neutral-buffered formalin (Sigma-Aldrich) and embedded in paraffin (Leica Biosystems, Wetzlar, Germany). For immunohistochemistry, the dewaxed and hydrated sections were incubated in a warm water bath at 95°C for 20 min in neutral (pH 7.0) antigen retrieval solution (Dako, Glostrup, Denmark). Subsequently, the sections were incubated with the following primary antibodies overnight at 4°C: rabbit anti-human Nestin (dilution, 1:10; Boster Systems, Inc.), mouse anti-human GFAP (dilution, 1:10; Boster Systems, Inc.), mouse anti-human Ki67 (dilution, 1:10; Boster Systems, Inc.) or mouse anti-human CD31 (dilution, 1:10; Boster Systems, Inc.). The subsequent procedures were performed according to the manufacturer's protocol for a mouse- or rabbit-specific HRP/AEC Polymer Detection Immunohistochemistry kit (Abcam). A total of 4 images were randomly acquired under magnification, ×200 using a microscope (Olympus BX52; Olympus, Tokyo, Japan) to calculate the percentages of Nestin-, GFAP- or Ki67-positive cells and the tumor microvessel density according to the method previously reported by Weidner et al (17).
Statistical analysis
Quantitative data are presented as the mean ± standard deviation. The data were analyzed for statistical significance using a two-sided Student's t-test with Excel software 2010 (Microsoft Corporation, Redmond, WA, USA). The survival analysis was performed using a log-rank test. The survival data are presented as a Kaplan-Meier plot using SPSS version 19.0 (IBM SPSS, Armonk, NY, USA). P<0.05 was considered to indicate a statistically significant difference.
Results
Cell populations of DiI fluorescence-retaining cells and DiI-negative cells with varying proliferative potentials may be differentiated within glioblastoma spheres at 2 weeks subsequent to initial DiI labeling
The strong photostable fluorescence, excellent cellular retention and minimal cytotoxicity of DiI make it particularly suitable for long-term labeling and tracking of cells (18). Following the initial uniform staining of DiI in the dissociated glioblastoma sphere cell lines NCH421k, NCH441 and NCH644, the fluorescence intensity was monitored every 2–3 days. With cell proliferation and sphere reformation, the fluorescence intensity of DiI in certain cells decayed in a sustained manner, whereas the fluorescence intensity of DiI in other cells remained strong and constant (Fig. 1A and B). At 2 weeks, two cell populations could be clearly distinguished based on fluorescence intensity, namely, a group of DiI-retaining cells and a group of DiI-negative cells (Fig. 1B). Flow cytometric analysis revealed that DiI-retaining cells accounted for a small population within the glioblastoma spheres at 2 weeks. In the NCH421k cell line, the proportion of DiI-retaining cells was ~8±2%, in NCH441 cells it was ~6±1%, and in NCH644 cells it was ~5±1% (Fig. 1C). FACS analysis was performed to isolate DiI-retaining and DiI-negative cells. The CCK-8 cell proliferation assay revealed that DiI-retaining cells proliferated significantly more slowly compared with DiI-negative cells (P=0.011, P=0.035 and P=0.023 in the NCH421k, NCH441 and NCH644 cell lines, respectively; Fig. 1D). Therefore, based on the fluorescence intensity of DiI in the cells, a group of fast-cycling DiI-negative cells and a group of slow-cycling DiI-retaining cells may be successfully differentiated at 2 weeks subsequent to initial DiI labeling.
Figure 1.

Populations of DiI-retaining cells and DiI-negative cells with varying proliferative potentials may be differentiated within glioblastoma spheres at 2 weeks subsequent to initial DiI labeling. (A) Diagram illustrating that the change in the cellular fluorescence intensity in initially labeled cells reflects the proliferative state. (B) Representative images showing the dynamic changes in and distribution of DiI fluorescence (lower panel) during cell proliferation and sphere reformation (upper panel) in the NCH421k cell line at the indicated times (Day 1, Day 3, Day 5, Day 8, Day 11 and Day 14). Scale bars: Day 1 and day 3, 10 µm; day 5, 20 µm; day 8, day 11 and day 14, 50 µm. The cells were initially stained with DiI fluorescence through incubation with DiI cell-labeling solution, as indicated in the Materials and methods. (C) Percentage of DiI-retaining cells in the NCH421k, NCH441 and NCH644 cell lines as analyzed by flow cytometry at 2 weeks subsequent to initial DiI labeling. (D) Proliferative potential of DiI-retaining and DiI-negative cells in the NCH421k, NCH441 and NCH644 cell lines as analyzed by the cell counting kit-8 assay. The data are presented as the mean ± standard deviation from three independent experiments. *P<0.05 compared with the DiI-negative group. OD, optical density.
DiI-retaining cells possess increased self-renewal capacity in vitro compared with DiI-negative cells
The primary DiI-retaining and DiI-negative cells separated by FACS analysis formed glioma spheres after 2–4 weeks. The clonogenicity assay performed on the primary spheres revealed that 2nd passage DiI-retaining cells and DiI-negative cells were able to reform clones. The sphere-forming frequencies of DiI-retaining cells from the NCH441, NCH644 and NCH421k cell lines were 7±2, 10±2 and 6±2%, respectively; these frequencies were lower than the sphere-forming frequencies in DiI-negative cells (8±2, 13±3 and 8±3% for the NCH441, NCH644 and NCH421k cell lines, respectively; P=0.089, P=0.455 and P=0.653, respectively; Fig. 2A, B and C-a). The spheres were harvested again and cultured in serum-free medium for 2 weeks. The secondary clonogenicity assay performed on the secondary spheres revealed that the sphere-forming frequency of the tertiary DiI-retaining cells was significantly increased compared with that of the tertiary DiI-negative cells (P=0.002, P=0.034 and P=0.016, respectively; Fig. 2A, B and C-a). Similarly, the tertiary clonogenicity assay performed on the tertiary spheres revealed that the sphere-forming frequency in the quaternary DiI-retaining cells was significantly increased compared with that of the quaternary DiI-negative cells (P=0.003, P=0.027 and P=0.007, respectively; Fig. 2A, B and C-a). Furthermore, the clonogenicity of the DiI-negative cells decreased significantly with serial passaging (secondary vs. quaternary passages, P=0.013 in NCH441 cells, P=0.003 in NCH644 cells and P=0.028 in NCH421k cells; tertiary vs. quaternary passages, P=0.001 in NCH441 cells, P=0.017 in NCH644 cells and P=0.042 in NCH421k cells), whereas the clonogenicity of the DiI-retaining cells remained stable (Fig. 2A-C). Therefore, DiI-retaining cells possessed significantly increased self-renewal capacity in vitro compared with DiI-negative cells.
Figure 2.

DiI-retaining cells possess greater self-renewal capacity in vitro compared with DiI-negative cells. Sphere-forming frequencies of DiI-retaining and DiI-negative cells in the (A) NCH441, (B) NCH644 (C-a) and NCH421k glioblastoma sphere cell line observed in the serial clonogenicity assay. The data are presented as the mean ± standard deviation from three independent experiments. *P<0.05 compared with DiI-retaining cells at the same passage. **P<0.05 vs. DiI-negative cells at different passages. (C-b) Representative images of the sphere-forming abilities of DiI-retaining and DiI-negative cells in the NCH421k cell line at the indicated passages (secondary, tertiary and quarternary) under phase-contrast microscopy. Scale bar=100 µm.
DiI-retaining cells possess multipotency in vitro
Western blot assays revealed that the glial cell marker GFAP, neural marker βIII-tubulin and oligodendrocyte marker PDGFRα were not expressed in the DiI-retaining group prior to differentiation, as no bands for these three markers were observed (Fig. 3A); following differentiation, GFAP, PDGFRα and βIII-tubulin expression was induced (Fig. 3A). In the DiI-negative group, increased expression of these three lineage markers was additionally observed following differentiation compared with that prior to differentiation (Fig. 3A). Notably, two weaker bands for GFAP and βIII-tubulin were observed prior to differentiation in the DiI-negative group (Fig. 3A). Quantification of the band density on the blots revealed that GFAP, βIII-tubulin and PDGFRα expression increased significantly following differentiation in the DiI-retaining group (P=0.027, P=0.006 and P=0.004, respectively; Fig. 3B-a, B-b and B-c), whereas the increased expression of these three markers was not statistically significant in the DiI-negative group (P=0.789, P=0.658 and P=0.089, respectively; Fig. 3B-a, B-b and B-c).
Figure 3.

DiI-retaining cells possess multipotency in vitro. (A) Expression of GFAP, βIII-tubulin and PDGFRα in DiI-retaining and DiI-negative cells in the NCH421k cell line prior to and following differentiation as assayed by western blot, with β-actin as the internal reference. Quantification of the band densities of (B-a) GFAP, (B-b) βIII-tubulin and (B-c) PDGFRα through normalization to β-actin using ImageJ software. The data are presented as the mean ± standard deviation from three independent experiments. *P<0.05 compared with expression prior to differentiation in the DiI-retaining group. GFAP, glial fibrillary acidic protein; PDGFRα, platelet-derived growth factor receptor α.
DiI-retaining cells exhibit significant resistance to irradiation and temozolomide treatment
The dissociated DiI-retaining and DiI-negative cells were irradiated with a single dose of 10 Gy radiation or were treated with 500 µmol/l temozolomide, a representative first-line drug against malignant glioma. A total of 72 h later, the cell proliferation assay revealed significantly increased proliferation of DiI-retaining cells compared with that of DiI-negative cells (following irradiation, P=0.012 in NCH421k cells, P=0.024 in NCH441 cells and P=0.036 in NCH644 cells; following temozolomide, P=0.003 in NCH421k cells, P=0.005 in NCH441 cells and P=0.029 in NCH644 cells; Fig. 4A and B), indicating that DiI-negative cells were more sensitive to radiation or to temozolomide treatment than DiI-retaining cells. In addition, no significant differences in the proliferation of DiI-retaining cells were observed prior to and following radiation or temozolomide treatment, whereas the proliferation of DiI-negative cells decreased significantly following radiation or temozolomide treatment compared to that prior to radiation or temozolomide treatment (following irradiation, P=0.0002 in NCH421k cells, P=0.005 in NCH441 cells and P=0.003 in NCH644 cells; following temozolomide, P=0.019 in NCH421k cells, P=0.027 in NCH441 cells and P=0.004 in NCH644 cells; Fig. 4A and B). Therefore, DiI-retaining cells exhibited significant resistance to radiotherapy and temozolomide treatment.
Figure 4.
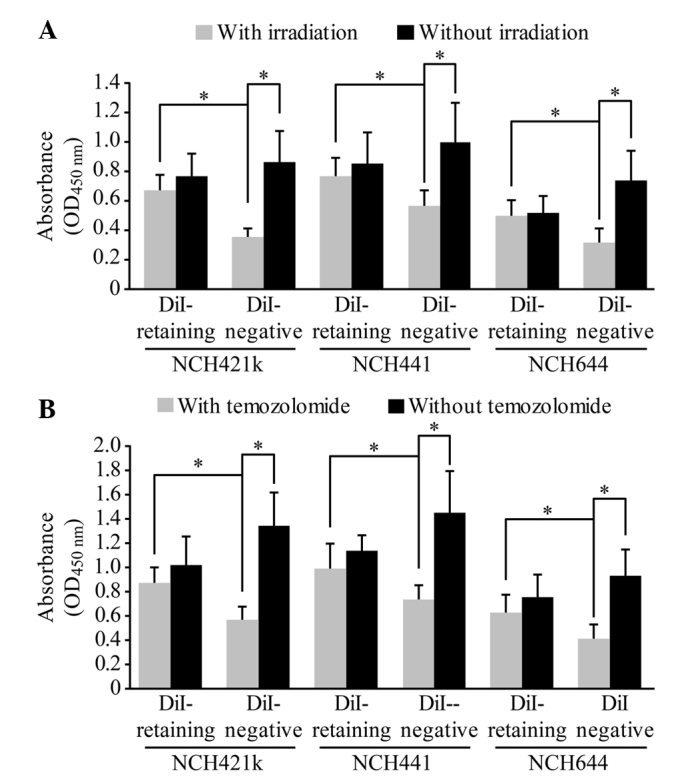
DiI-retaining cells demonstrate significant resistance to irradiation and temozolomide treatment. Proliferative potentials of the DiI-retaining and DiI-negative cells in the NCH421k, NCH441 and NCH644 glioblastoma sphere lines prior to and following (A) irradiation or (B) temozolomide treatment. The data are presented as the mean ± standard deviation from three independent experiments. *P<0.05, DiI-retaining cells vs. DiI-negative cells with irradiation/temozolomide in the respective cell lines, or DiI-negative cells prior to irradiation/temozolomide vs. DiI-negative cells following irradiation/temozolomide. OD, optical density.
The difference in CD133 expression between DiI-retaining cells and DiI-negative cells is not significant
Flow cytometric analysis revealed that CD133-positive cells accounted for ~74±12% of the DiI-retaining cells in the NCH421k cell line, 34±10% of those in the NCH441 cell line and 60±15% of those in the NCH644 cell line. These percentages were slightly higher than the percentages of CD133-positive cells among the DiI-negative populations (64±11% of NCH421k cells, 21±7% of NCH441 cells and 52±13% of NCH644 cells). These differences were not statistically significant (P=0.553, P=0.624 and P=0.867, respectively).
DiI-retaining cells demonstrate significantly increased tumorigenicity in vivo compared with DiI-negative cells
DiI-retaining cells and DiI-negative cells were implanted into the brains of NOD/SCID mice at a serial gradient of 102, 103, 104 and 105. DiI-retaining cells and DiI-negative cells (105 cells/implantation) were able to generate tumors in all treated mice. As the number of implanted cells decreased, higher tumor incidence was observed in the DiI-retaining group compared with the DiI-negative group (Table I). With as few as 102 implanted cells, only DiI-retaining cells were able to effectively generate tumors; at this cell number, no tumors were observed in the DiI-negative group 6 months subsequent to implantation (Table I).
Table I.
Tumor incidence and survival time in non-obese diabetic/severe combined immunodeficiency mice (n=5 in each group) implanted with DiI-retaining cells or DiI-negative cells from the NCH421k cell line at a serial implantation gradient.
| Density of implanted tumor cells | ||||
|---|---|---|---|---|
| Parameter | 102 | 103 | 104 | 105 |
| DiI-retaining | ||||
| Tumor incidence, n | 2 | 3 | 4 | 5 |
| Survival time, daysa | 117±9 | 72±7 | 58±13 | 49±6 |
| DiI-negative | ||||
| Tumor incidence, n | 0 | 1 | 3 | 5 |
| Survival time, daysa | >180 | 125 | 97±13 | 64±5 |
presented as the mean ± standard deviation.
Immunohistochemical analysis revealed strong positive GFAP staining in the two groups and significantly increased Nestin, Ki67 and CD31 in tumors generated from DiI-retaining cells compared with those generated from DiI-negative cells, with the same number of implanted cells (Fig. 5A). These data indicated that the tumors generated from DiI-retaining cells were more enriched in stem cells and exhibited a higher proliferation index and vessel density, thereby more closely resembling the features of human glioblastoma.
Figure 5.

DiI-retaining cells demonstrate significantly increased tumorigenicity in NOD/SCID mice compared with DiI-negative cells. Representative images of (A-a) Nestin (left panel); (A-b) GFAP (left panel); (A-c) Ki67 (left panel) and (A-d) cluster of differentiation 31 (left panel) immunohistochemical staining in the xenografts from NOD/SCID mice implanted with DiI-retaining or DiI-negative cells. Scale bar=100 µm. Quantification of the cells positive for (A-a) Nestin (right panel), (A-b) GFAP (right panel) and (A-c) Ki67 (right panel). *P<0.05. (A-d) MVD in the xenografts from NOD/SCID mice implanted with DiI-retaining or DiI-negative cells. *P<0.05. The data are presented as the mean ± standard deviation. (B) Kaplan-Meier survival analysis of the overall survival of NOD/SCID mice harboring tumors from DiI-retaining or DiI-negative cells. NOD/SCID, non-obese diabetic/severe combined immunodeficiency; GFAP, glial fibrillary acidic protein; MVD, microvessel density.
The Kaplan-Meier survival analysis revealed that the overall survival of NOD/SCID mice harboring tumors from DiI-retaining cells was significantly reduced compared with that of mice harboring tumors from DiI-negative cells (P=0.016; Fig. 5B).
Discussion
Once cells are initially labeled with fluorescence or with the DNA synthesis substrate bromodeoxyuridine (BrdU), the proliferative status of these cells may be monitored based on labeling intensity (14). In actively proliferating cells, the labeling intensity decreases by half with each division, whereas in dormant or slow-cycling cells, the labeling intensity remains constant or decays marginally (14). Thus, label-retaining cells correspond to dormant or slow-cycling cells (14).
In normal tissues, the label-retaining assay has already been effectively used to isolate stem cells. In the hematopoietic system, intestinal epithelium, hair follicles, myocardium, prostate epithelium and central nervous system, BrdU label-retaining cells manifest stem cell features (19,20). In a number of human tumors, the label-retaining assay is effective at isolating tumor stem cells (21–24). Through initial carboxyfluorescein succinimidyl ester (CFSE) fluorescence cell labeling and the subsequent tracking of fluorescence intensity, Moore et al successfully confirmed that the CFSE-retaining cells in the HCT116 colorectal cancer cell line and in the MDA-MB-231 breast cancer cell line were tumor stem cells (21). Using the fluorescence dye PKH26, the long-term PKH26-retaining cells in melanoma and ovarian cancer were demonstrated to be rich in cancer stem cells (22,23). Dembinski and Krauss (24) reported that the population of long-term DiI-retaining pancreatic cancer cells exhibited stem cell features.
Similar to the present results, Deleyrolle et al (25) used CFSE fluorescence dye and Richichi et al (26) used PKH26 dye to identify label-retaining stem cells in glioma spheres successfully. These cells constitute only a small population within the glioma spheres. Subsequent to tracking CFSE fluorescence for 1 week, Deleyrolle et al (25) reported that the label-retaining cells with marked characteristics of stem cells accounted for ~5% of the population. Richichi et al (26) reported that the PKH26-retaining cell population ranged between 1 and 3% following 2 weeks of observation. The results of the present study suggested that the DiI-retaining cell population in each of the three glioblastoma sphere cell lines was <10% following 2 weeks of observation. Notably, among the three studies, several fluorescent dyes were employed. PKH26 and DiI are lipophilic carbocyanine dyes that intercalate into the lipid bilayer of the cell membrane, whereas CFSE binds to cytoplasmic components (18). Compared with PKH26, DiI fluorescence is more uniformly distributed in the cell membrane; therefore, the fluorescence decay of DiI more accurately reflects cell division (18). Compared with CFSE, the cell membrane distribution of DiI is less toxic to cells (18). Thus, DiI staining appears to be superior to the other two dyes and more conducive to long-term cell tracking.
Through serial clonogenic assays, the present study demonstrated that the clonogenicity of the label-retaining cells remained relatively constant despite serial passaging, whereas the clonogenicity of the label-negative cells decreased significantly, indicating that label-negative cells possess a limited self-renewal capacity. Approximately 100 label-retaining cells were necessary to generate tumors effectively in immunocompromised mice over 6 months, whereas no tumors were generated with the same number of label-negative cells over the same time interval, highlighting the different degrees of tumorigenicity between the two cell populations. Therefore, the label-retaining cells were greatly enriched in glioma stem cells compared with the label-negative cells.
The present study did not identify any significant differences between label-retaining cells and label-negative cells regarding CD133 expression. Controversies regarding CD133 as a cancer stem cell marker have existed for a long time. A number of studies have demonstrated the tumorigenicity of CD133-negative cells (5,6,27). CD133-positive and CD133-negative stem cells may originate from different cells (6,28). These stem cells are able to coexist in an individual tumor tissue or be differentially distributed in different individuals, reflecting the heterogeneity of glioblastoma (27,29). The label-retaining assay for screening cancer stem cells avoids the diversity and uncertainty of cell surface markers (5,6,27–29). However, certain problems regarding sensitivity and specificity may be encountered in label-retaining assays. Certain dormant cells may enter the proliferative stage and subsequently return to the dormant stage (12), thereby significantly decreasing the labeling intensity and leading to false sorting into the label-negative group. This false sorting may be responsible for the self-renewal capacity of label-negative cells that has been observed in serial clonogenic assays in the present study, which remained significantly weaker compared with that of label-retaining cells. Certain terminally differentiated cells may no longer divide but may retain strong labeling intensity following the initial staining, thus leading to false sorting into the label-retaining group (30). However, as these cells undergo apoptosis and death, their labeling intensities will diminish and finally disappear, whereas the labeling intensity in the dormant stem cells always remains constant during tracking (21–26). Furthermore, the stem cell permissive serum-free condition makes the survival of differentiated cells difficult.
The present study demonstrated that label-retaining cells exhibited significant resistance to radiotherapy and chemotherapy. The dormant cells in numerous human tumors, including ovarian, breast and colorectal cancer, are resistant to conventional radiotherapies or chemotherapies, leading to tumor recurrence or metastasis, sometimes decades later (21,31–33). In glioma, Hussein et al (15) reported that suspended glioma sphere cells were resistant to etoposide treatment, which specifically kills cells at the proliferative stages (S and G2) of the cell cycle. Additional cell cycle analysis revealed that certain cells in glioma spheres reside at the dormant stage of the cell cycle (G0/G1), thus providing a reason for the etoposide resistance (15). Chen et al (34) constructed a Nestin-Δthymidine kinase (ΔTK)-green fluorescence protein (GFP) transgenic mouse model that developed glioma. In this model, the Nestin+/GFP+ progenitor tumor cells were almost Ki67−, indicating that the majority of the progenitor cells were dormant (34). Treating these mice with temozolomide only kills the proliferating Ki67+ tumor cells and therefore only arrests tumor growth (34). As a result, following the conclusion of temozolomide treatment, marked tumor regrowth originating from the dormant Nestin+/GFP+ subpopulation is observed (34). Taken together, the results of these two previous studies and the present study suggest that dormant stem cells may be resistant to radiotherapy and chemotherapy.
The clarification of the regulation of stem cell proliferation and dormancy enables an improved understanding of the mechanism of tumor cell resistance (35–37). The regulation of the proliferation and dormancy of mouse hematopoietic stem cells has been well characterized (35,36). Proliferative and dormant stem cells reside in distinct niches, including a hypoxic osteoblastic niche that maintains the dormant state and a normoxic vascular endothelial niche that maintains the proliferative state of stem cells (35). Upon bone marrow injury or growth factor stimulus with granulocyte-colony stimulating factor, the dormant cells from the osteoblastic niche migrate into the vascular endothelial niche and enter the proliferative state in response to complex extra- and intracellular signal transduction (35). Numerous molecules participate in this transition, including extracellular factors, for example, thrombopoietin, angiopoietin, transforming growth factor β and Notch1 and intracellular factors, for example, forkhead box O proteins, mammalian target of rapamycin complex 1, F-box and WD repeat domain-containing 7, early growth response protein 1, pre-B-cell leukemia homeobox 1, retinoblastoma protein, c-Cbl, Myc and Bmi1 (36). In glioma, the niche responsible for maintaining the dormant state of glioma stem cells and the dormancy-associated signaling pathways remains to be elucidated. If the dormant stem cells could be induced into the proliferative state, which is sensitive to radiotherapy and chemotherapy, or if the dormant state could be permanently maintained regardless of environmental stimuli (37), then significant progress would be achieved in glioma therapy.
In conclusion, the present study successfully identified a group of dormant or slow-cycling label-retaining cells in glioblastoma spheres cultured in serum-free medium. Compared with label-negative cells, label-retaining cells possessed significant stem cell properties including self-renewal capacity, multipotency in vitro and tumorigenicity in vivo. In addition, these cells demonstrated significant resistance to radiotherapy and chemotherapy. Label-retaining assays may be considered as an effective way to enrich glioma stem cells for additional study.
Acknowledgements
The present study was supported by the National Natural Science Foundation of China (Beijing, China; grant no. 81201681).
References
- 1.Rock K, McArdle O, Forde P, Dunne M, Fitzpatrick D, O'Neill B, Faul C. A clinical review of treatment outcomes in glioblastoma multiforme - the validation in a non-trial population of the results of a randomised Phase III clinical trial: Has a more radical approach improved survival? Br J Radiol. 2012;85:e729–e733. doi: 10.1259/bjr/83796755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jackson M, Hassiotou F, Nowak A. Glioblastoma stem-like cells: At the root of tumor recurrence and a therapeutic target. Carcinogenesis. 2015;36:177–185. doi: 10.1093/carcin/bgu243. [DOI] [PubMed] [Google Scholar]
- 3.Stopschinski BE, Beier CP, Beier D. Glioblastoma cancer stem cells - from concept to clinical application. Cancer Lett. 2013;338:32–40. doi: 10.1016/j.canlet.2012.05.033. [DOI] [PubMed] [Google Scholar]
- 4.Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–5828. [PubMed] [Google Scholar]
- 5.Wang J, Sakariassen PØ, Tsinkalovsky O, Immervoll H, Bøe SO, Svendsen A, Prestegarden L, Røsland G, Thorsen F, Stuhr L, et al. CD133 negative glioma cells form tumors in nude rats and give rise to CD133 positive cells. Int J Cancer. 2008;122:761–768. doi: 10.1002/ijc.23130. [DOI] [PubMed] [Google Scholar]
- 6.Beier D, Hau P, Proescholdt M, Lohmeier A, Wischhusen J, Oefner PJ, Aigner L, Brawanski A, Bogdahn U, Beier CP. CD133(+) and CD133(−) glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res. 2007;67:4010–4015. doi: 10.1158/0008-5472.CAN-06-4180. [DOI] [PubMed] [Google Scholar]
- 7.Ogden AT, Wziri AE, Lochhead RA, Fusco D, Lopez K, Ellis JA, Kang J, Assanah M, McKhann GM, Sisti MB, et al. Identification of A2B5+CD133-tumor-initiating cells in adult human gliomas. Neurosurgery. 2008;62:505–514. doi: 10.1227/01.neu.0000316019.28421.95. [DOI] [PubMed] [Google Scholar]
- 8.Mao XG, Zhang X, Xue XY, Guo G, Wang P, Zhang W, Fei Z, Zhen HN, You SW, Yang H. Brain tumor stem-like cells identified by neural stem cell marker CD15. Transl Oncol. 2009;2:247–257. doi: 10.1593/tlo.09136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma I, Allan AL. The role of human aldehyde dehydrogenase in normal and cancer stem cells. Stem Cell Rev. 2011;7:292–306. doi: 10.1007/s12015-010-9208-4. [DOI] [PubMed] [Google Scholar]
- 10.Meyer M, Reimand J, Lan X, Head R, Zhu X, Kushida M, Bayani J, Pressey JC, Lionel AC, Clarke ID, et al. Single cell-derived clonal analysis of human glioblastoma links functional and genomic heterogeneity. Proc Natl Acad Sci USA. 2015;112:851–856. doi: 10.1073/pnas.1320611111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clément V, Marino D, Cudalbu C, Hamou MF, Mlynarik V, de Tribolet N, Dietrich PY, Gruetter R, Hegi ME, Radovanovic I. Marker-independent identification of glioma-initiating cells. Nat Methods. 2010;7:224–228. doi: 10.1038/nmeth.1430. [DOI] [PubMed] [Google Scholar]
- 12.Wilson A, Laurenti E, Oser G, van der Wath RC, Blanco-Bose W, Jaworski M, Offner S, Dunant CF, Eshkind L, Bockamp E, et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell. 2008;135:1118–1129. doi: 10.1016/j.cell.2008.10.048. [DOI] [PubMed] [Google Scholar]
- 13.Radford IR, Lobachevsky PN. An enteroendocrine cell-based model for a quiescent intestinal stem cell niche. Cell Prolif. 2006;39:403–414. doi: 10.1111/j.1365-2184.2006.00396.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Glauche I, Moore K, Thielecke L, Horn K, Loeffler M, Roeder I. Stem cell proliferation and quiescence - two sides of the same coin. PLoS Comput Biol. 2009;5:e1000447. doi: 10.1371/journal.pcbi.1000447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hussein D, Punjaruk W, Storer LC, Shaw L, Othman R, Peet A, Miller S, Bandopadhyay G, Heath R, Kumari R, et al. Pediatric brain tumor cancer stem cells: Cell cycle dynamics, DNA repair, and etoposide extrusion. Neuro Oncol. 2011;13:70–83. doi: 10.1093/neuonc/noq144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 17.Weidner N, Semple JP, Welch WR, Folkman J. Tumor angiogenesis and metastasis - correlation in invasive breast carcinoma. N Engl J Med. 1991;324:1–8. doi: 10.1056/NEJM199101033240101. [DOI] [PubMed] [Google Scholar]
- 18.Progatzky F, Dallman MJ, Lo Celso C. From seeing to believing: Labelling strategies for in vivo cell-tracking experiments. Interface Focus. 2013;3:20130001. doi: 10.1098/rsfs.2013.0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fernandez-Gonzalez R, Illa-Bochaca I, Shelton DN, Welm BE, Barcellos-Hoff MH, Ortiz-de-Solorzano C. In situ analysis of cell populations: Long-term label-retaining cells. Methods Mol Biol. 2010;621:1–28. doi: 10.1007/978-1-60761-063-2_1. [DOI] [PubMed] [Google Scholar]
- 20.van der Wath RC, Wilson A, Laurenti E, Trumpp A, Liò P. Estimating dormant and active hematopoietic stem cell kinetics through extensive modeling of bromodeoxyuridine label-retaining cell dynamics. PLoS One. 2009;4:e6972. doi: 10.1371/journal.pone.0006972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moore N, Houghton J, Lyle S. Slow-cycling therapy-resistant cancer cells. Stem Cells Dev. 2012;21:1822–1830. doi: 10.1089/scd.2011.0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roesch A, Fukunaga-Kalabis M, Schmidt EC, Zabierowski SE, Brafford PA, Vultur A, Basu D, Gimotty P, Vogt T, Herlyn M. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell. 2010;141:583–594. doi: 10.1016/j.cell.2010.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao MQ, Choi YP, Kang S, Youn JH, Cho NH. CD24+ cells from hierarchically organized ovarian cancer are enriched in cancer stem cells. Oncogene. 2010;29:2672–2680. doi: 10.1038/onc.2010.35. [DOI] [PubMed] [Google Scholar]
- 24.Dembinski JL, Krauss S. Characterization and functional analysis of a slow cycling stem cell-like subpopulation in pancreas adenocarcinoma. Clin Exp Metastasis. 2009;26:611–623. doi: 10.1007/s10585-009-9260-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deleyrolle LP, Harding A, Cato K, Siebzehnrubl FA, Rahman M, Azari H, Olson S, Gabrielli B, Osborne G, Vescovi A, Reynolds BA. Evidence for label-retaining tumour-initiating cells in human glioblastoma. Brain. 2011;134:1331–1343. doi: 10.1093/brain/awr081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Richichi C, Brescia P, Alberizzi V, Fornasari L, Pelicci G. Marker-independent method for isolating slow-dividing cancer stem cells in human glioblastoma. Neoplasia. 2013;15:840–847. doi: 10.1593/neo.13662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen R, Nishimura MC, Bumbaca SM, Kharbanda S, Forrest WF, Kasman IM, Greve JM, Soriano RH, Gilmour LL, Rivers CS, et al. A hierarchy of self-renewing tumor-initiating cell types in glioblastoma. Cancer Cell. 2010;17:362–375. doi: 10.1016/j.ccr.2009.12.049. [DOI] [PubMed] [Google Scholar]
- 28.Lottaz C, Beier D, Meyer K, Kumar P, Hermann A, Schwarz J, Junker M, Oefner PJ, Bogdahn U, Wischhusen J, et al. Transcriptional profiles of CD133+ and CD133- glioblastoma-derived cancer stem cell lines suggest different cells of origin. Cancer Res. 2010;70:2030–2040. doi: 10.1158/0008-5472.CAN-09-1707. [DOI] [PubMed] [Google Scholar]
- 29.Soeda A, Hara A, Kunisada T, Yoshimura S, Iwama T, Park DM. The evidence of glioblastoma heterogeneity. Sci Rep. 2015;5:7979. doi: 10.1038/srep07979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu Q, Nguyen DH, Dong Q, Shitaku P, Chung K, Liu OY, Tso JL, Liu JY, Konkankit V, Cloughesy TF, et al. Molecular properties of CD133+ glioblastoma stem cells derived from treatment-refractory recurrent brain tumors. J Neurooncol. 2009;94:1–19. doi: 10.1007/s11060-009-9919-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fillmore CM, Kuperwasser C. Human breast cancer cell lines contain stem-like cells that self-renew, give rise to phenotypically diverse progeny and survive chemotherapy. Breast Cancer Res. 2008;10:R25. doi: 10.1186/bcr1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Allan AL, Vantyghem SA, Tuck AB, Chambers AF. Tumor dormancy and cancer stem cells: Implications for the biology and treatment of breast cancer metastasis. Breast Dis. 2007;26:87–98. doi: 10.3233/BD-2007-26108. [DOI] [PubMed] [Google Scholar]
- 33.Udagawa T. Tumor dormancy of primary and secondary cancers. APMIS. 2008;116:615–628. doi: 10.1111/j.1600-0463.2008.01077.x. [DOI] [PubMed] [Google Scholar]
- 34.Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG, Parada LF. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012;488:522–526. doi: 10.1038/nature11287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trumpp A, Essers M, Wilson A. Awakening dormant haematopoietic stem cells. Nat Rev Immunol. 2010;10:201–209. doi: 10.1038/nri2726. [DOI] [PubMed] [Google Scholar]
- 36.Wilson A, Laurenti E, Trumpp A. Balancing dormant and self-renewing hematopoietic stem cells. Curr Opin Genet Dev. 2009;19:461–468. doi: 10.1016/j.gde.2009.08.005. [DOI] [PubMed] [Google Scholar]
- 37.Schillert A, Trumpp A, Sprick MR. Label retaining cells in cancer - the dormant root of evil? Cancer Lett. 2013;341:73–79. doi: 10.1016/j.canlet.2013.04.019. [DOI] [PubMed] [Google Scholar]