

Abstract
Insulin resistance and reduced β-cell glucose sensitivity are present in patients with type 2 diabetes. In the present study, we investigated the changes in β-cell function in patients with type 2 diabetes during a 3-year follow-up study. A total of 48 patients with early-onset type 2 diabetes (EOD) and 55 patients with late-onset type 2 diabetes (LOD) were enrolled. Weight, height, waist circumference, hip circumference, blood pressure and plasma levels of lipids, glucose, fasting serum C-peptide (CPR0) and serum C-peptide 6 min after glucagon stimulation (CPR6) were measured. In addition, islet β-cell secretory activity was detected. Subjects with EOD had lower Systolic blood pressure (SBP), diastolic blood pressure (DBP), body mass index (BMI), fasting CPR0, CPR6 and greater glycated hemoglobin A1c (HbA1c), triglyceride (TG) compared with subjects with LOD. CPR0, CPR6 and TG were decreased in both EOD and LOD groups 3 years later. The ratio of β-cell function failure was 29.17 and 10.91% in the EOD and LOD groups, respectively, and there was significant difference between the two groups. A positive correlation was identified between the CPR0 and waist-hip ratio, HbA1c in the EOD group. A similar positive correlation was observed between CPR0 and BMI in the LOD group. Collectively, islet β-cell function has declined in patients with EOD, and this change may be more evident when compared with those with LOD.
Keywords: early-onset type 2 diabetes, late-onset type 2 diabetes, islet β-cell function, C-peptide response to glucagon test
Introduction
Diabetes is a serious global health issue and has become one of the most common epidemics worldwide. The International Diabetes Federation estimated that ~7% of the world's population aged between 25 and 79 years of age had diabetes in 2010; by 2030 the figure will rise to 8% (1). Non-insulin dependent diabetes mellitus is a leading cause of morbidity worldwide and contributes significantly to premature mortality (2). Early-onset type 2 diabetes (EOD) has emerged as an increasing public health problem, particularly in developing areas such as Asia, which are undergoing rapid socio-economic changes (3,4). A recent national investigation completed in China showed that the prevalence of diabetes and pre-diabetes in individuals aged 18–39 years was ~45% (5).
It is established insulin resistance and reduced β-cell glucose sensitivity are present in patients with type 2 diabetes (6). Previously, the concept that insulin resistance was the primary genetic component of type 2 diabetes became widely believed (7); however, previous studies have indicated that the first-phase insulin secretion is the earliest detectable defect in β-cell function, and impaired β-cell function precedes insulin resistance in the pathogenesis of type 2 diabetes (8,9). Impaired β-cell function is regarded as a key factor in the progression from glucose intolerance to overt type 2 diabetes, and it could be the result of the loss of β-cell mass and/or functional defects (10). Moreover, the relative β-cell volume was decreased from ~20 to 50% in autopsy specimens of patients with type 2 diabetes (11). These results suggested that diabetes is associated with impaired β-cell function; however, the development of β-cell hypofunction is poorly understood with regards to type 2 diabetes.
In the present study, we evaluated the function and deterioration of the islet β-cell in patients with type 2 diabetes by using a 3-year follow-up and compared the difference between late-onset diabetes (LOD) and EOD with the use of contrastive analysis.
Materials and methods
Sample
Patients with type 2 diabetes were recruited from the Department of Endocrinology of Huashan Hospital affiliated to Fudan University (Shanghai, China) between 2003 and 2009. A total of 48 patients (36 men and 12 women) with EOD (age at diagnosis, <40 years) and 55 patients (27 men and 28 women) with LOD (age at diagnosis, ≥40 years) were selected for the present study. Plasma glucose concentrations were measured using an Abbott Bichromatic Analyzer (Abbott Laboratories, Lake Bluff, IL, USA), and diabetes was diagnosed according to the World Health Organization plasma glucose criteria (fasting plasma glucose, ≥126 mg/dl; or 2-h plasma glucose, ≥200 mg/dl (12). In addition, anti-insular cellular antibody (anti-ICA; cat. no. 02068), anti-glutamate decarboxylase antibody (anti-GAD; cat. no. 020107) and anti-insulin antibody (anti-IAA; cat. no. 02114A) all are negative. Patients were excluded if they had type 1 diabetes as defined by acute presentation with diabetic ketoacidosis, heavy ketonuria or continuous requirement of insulin within half a year of diagnosis, or unknown diabetes type. Disease course was no more than 5 years in all subjects and all patients received oral blood glucose lowering drugs based on alimentary control.
Ethical approval was obtained from Huashan Hospital Clinical Research Ethics Committee. All patients provided written informed consent for data analysis and research purpose at the time of assessment.
Clinical studies
Participants underwent routine physical examinations including the following measurements: Height, weight, waist circumference, hip circumference and resting blood pressure. Height was measured in meters (without shoes), and weight in kilograms (without heavy clothing). Body mass index (BMI) was calculated in kg/m2. Waist circumference was measured using a tape measure while subjects were standing, at the midpoint between the bottom of rip cage and the top of lateral border of iliac crests during minimal respiration. Hip circumference was measured around the maximal circumference of the buttocks, at approximately the level of the pubic symphysis. Waist-hip ratio (WHR) was determined by dividing the mean waist circumference by the mean hip circumference. Systolic blood pressure (SBP) was measured with the subject in the supine position following a 5-min rest.
After an overnight fast, venous blood was sampled for measurement of plasma glucose, glycated hemoglobin A1c (HbA1c), total cholesterol (TC), high-density lipoprotein cholesterol (HDL-C), low-density lipoprotein cholesterol (LDL-C), triglyceride (TG), anti-ICA, anti-GAD and anti-IAA. All kits were purchased from Youbo Biotechnoglgy Co., Ltd. (Shanghai, China).
Islet β-cell secretory activity was measured by means of glucagon stimulation test (Express Technology Co., Ltd., Beijing, China). C-peptide (CPR0) levels were measured at fasting state. Glucagon stimulation test was performed by intravenous loading of 1 mg glucagon. The acute response to glucagon was measured by the C-peptide levels 6 min after glucagon challenge (CPR6).
BMI, HbA1c, CPR0 and CPR6 were measured at the beginning of the experiment, and thereafter, these indexes were examined once every year.
Biochemical assays and laboratory analyses
Blood glucose and lipid levels were analyzed using an automatic biochemical analyzer (AU5800; Beckman Coulter, Inc., Brea, CA, USA) according to the manufacturer's instructions. HbA1c was determined using high-performance liquid chromatography (Shimadzu Corporation, Kyoto, Japan). Blood glucose was measured by a glucose oxidase method using a kit (DiaSys Diagnostic Systems GmbH, Holzheim, Germany). HDL-C was measured by a direct method of immunosuppression using a kit (DiaSys Diagnostic Systems GmbH). LDL-C was measured by a direct measuring method using the a kit (Sekisui Chemical, Co., Ltd., Osaka, Japan). TG and TC measurements were performed using an enzymic kit (Beckman Coulter, Inc.). C-peptide was assayed using a radio-immuno method (Shangsha Yike Biotech Co., Ltd., Changsha, China).
Statistical analysis
Measurement data and non-normally distributed data are expressed as the mean ± standard deviation and interquartile range, respectively. All data were analyzed by use of Kolmogorov-Smirnov normality test and if the data were not normally distributed, a log transformation was performed on these data. χ2 test and two sided t-tests were used for comparison of enumeration and measurement data, respectively. The correlation between islet β-cell function and various indexes were analyzed using Pearson correlation analysis. All statistical analyses were performed using SPSS software, version 17.0 (SPSS, Inc., Chicago, IL, USA).
Results
Baseline general characteristics in the EOD and LOD groups
At the baseline, subjects with EOD had lower levels of SBP, DBP, BMI, CPR0, CPR6 and greater levels of HbA1c and TG compared to subjects with LOD (all P<0.01) (Table I). Compared with the baseline, CPR0, CPR6 and TG were decreased in the EOD and LOD groups at 12, 24 and 36 months (Table II).
Table I.
Baseline general characteristics of study participants.
| Characteristic | EOD | LOD |
|---|---|---|
| Gender (male/female) | 36/12 | 27/28 |
| Age (years) | 35.44±5.11 | 59.96±8.50a |
| SBP (mmHg) | 118.35±15.44 | 136.18±21.21a |
| DBP (mmHg) | 78.33±9.65 | 84.47±10.42a |
| BMI (kg/m2) | 23.31±2.93 | 25.17±2.99a |
| FBS (mmol/l) | 9.63±3.26 | 10.09±1.58 |
| HbA1c (%) | 10.88±2.32 | 9.83±1.46a |
| CPR0 (nmol/l)b | 1.1–2.10 | 1.98–3.46a |
| CPR6 (nmol/l)b | 2.00–4.20 | 5.29–7.48a |
| TC (mmol/l) | 4.53±0.99 | 5.15±1.15 |
| TG (mmol/l)b | 1.31–3.57 | 1.00–2.30a |
| HDL (mmol/l)b | 0.96–1.21 | 1.00–1.24 |
| LDL (mmol/l)b | 1.83–3.46 | 2.22–3.71 |
P<0.01, vs. LOD group.
Indicated that the data were not normally distributed, and these data were expressed as interquartile range (25th and 75th percentiles). EOD, early-onset diabetes; LOD, late-onset diabetes; SBP, systolic blood pressure; DBP, diastolic blood pressure; BMI, body mass index; FBS, fasting blood sugar; HbA1c, glycated hemoglobin A1c; CPR, C-peptide; TC, total cholesterol; TG, triglyceride; HDL, high-density lipoprotein; LDL, low-density lipoprotein.
Table II.
Clinical data of study participants during the 3-year follow-up.
| EOD | LOD | |||||||
|---|---|---|---|---|---|---|---|---|
| Characteristic | Baseline | 12 month | 24 month | 36 month | Baseline | 12 month | 24 month | 36 month |
| BMI (kg/m2) | 23.31±2.93 | 23.72±2.70 | 23.81±2.12 | 23.86±2.63 | 25.17±2.99 | 25.11±2.91 | 25.36±2.98 | 26.03±3.24 |
| HbA1c (%) | 10.88±2.32 | 9.01±2.09 | 8.49±1.67 | 8.39±1.67 | 9.83±1.46 | 8.93±1.32 | 8.38±1.37 | 8.29±1.31a |
| CPR0 (nmol/l) | 1.72±0.76 | 1.68±0.69 | 1.67±078 | 1.45±0.64 | 2.89±1.19 | 2.40±0.95 | 2.18±0.78 | 2.16±0.86a |
| CPR6 (nmol/l) | 2.96±1.67 | 2.84±1.30 | 2.71±0.93 | 2.44±0.84 | 6.66±2.43 | 5.61±1.30 | 4.85±1.22 | 5.20±1.56a |
P<0.01 vs. baseline. EOD, early-onset diabetes; LOD, late-onset diabetes; BMI, body mass index; HbA1c, glycated hemoglobin A1c; CPR, C-peptide.
There was a decreasing trend in the EOD group (Figs. 1–3). HbA1c, CPR0, CPR6 levels were 10.88±2.32, 1.72±0.76 and 2.96±1.67 nmol/l, respectively, when the subjects were recruited, and these were decreased to 8.39±1.67, 1.45±0.64 and 2.44±0.84 nmol/l after 3 years. Compared with the baseline, significant differences were detected in these indexes at 36 months (P<0.01). Furthermore, a decreasing trend was similarly observed in the LOD group (Figs. 1–3). At the baseline, HbA1c, CPR0 and CPR6 levels were 9.83±1.46, 2.89±1.19 and 6.66±2.43 nmol/l, respectively, which decreased to 8.29±1.31, 2.16±0.86 and 5.20±1.56 nmol/l after 3 years. Thus, these indexes showed significant changes during the 3-year follow-up (P<0.01).
Figure 1.
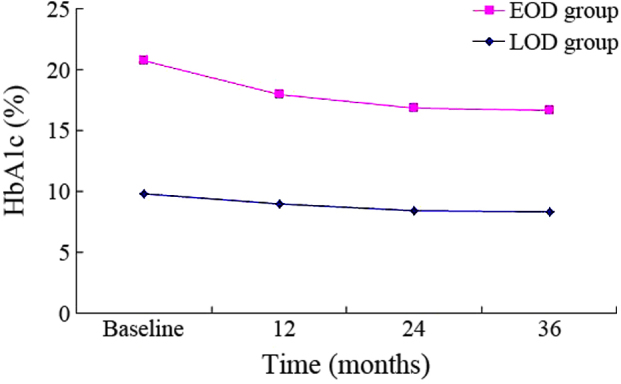
Changes in HbA1c in the 3-year follow-up. EOD, early-onset diabetes; LOD late-onset diabetes; HbA1c, glycated hemoglobin A1c.
Figure 3.
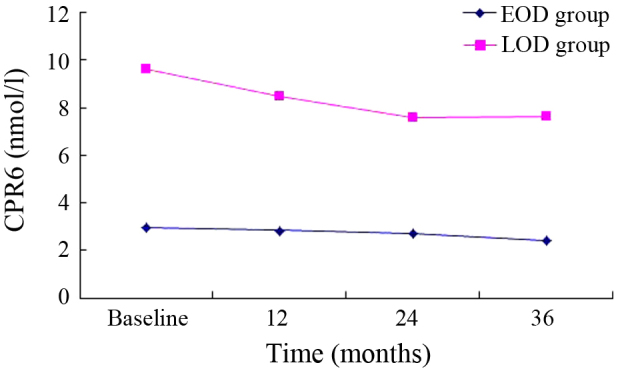
Changes in CPR6 in the 3-year follow-up. EOD, early-onset diabetes; LOD late-onset diabetes; CPR6, C-peptide 6 min after glucagon challenge.
Comparison of β-cell function failure in the EOD and LOD groups
An >50% reduction in CPR0 or CPR6 was set as the standard of β-cell function failure. A total of 14 subjects (29.17%) in the EOD group and 6 subjects (10.91%) in the LOD group were up to the >50% reduction standard 3 years later, and there was significant difference between the two groups (χ2=5.861, P<0.05) (Fig. 4).
Figure 4.
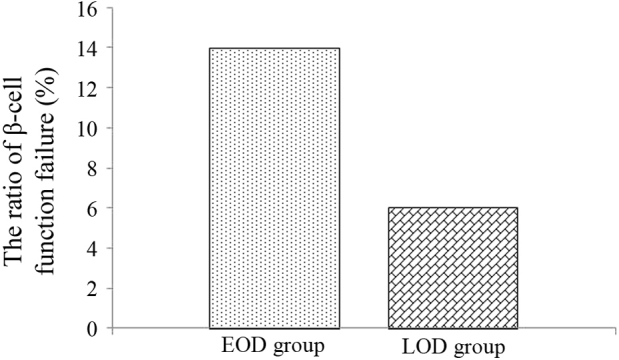
Ratio of β-cell function failure in EOD and LOD groups 3 years later. EOD, early-onset diabetes; LOD late-onset diabetes.
Effect of WHR and BMI on CPR0 and CPR6
Pearson correlation analysis indicated a positive correlation between CPR0 and WHR (P<0.01), HbA1c (P<0.05) in the EOD group after a 3-year follow-up. Furthermore, there was a positive correlation between CPR0 and BMI (P<0.05) in the LOD group.
Discussion
Type 2 diabetes (T2DM) is a complex population, which possesses the characteristics of heterogeneity and disuniformity (13). Previous studies have suggested that following with the progressive aggravation of insulin resistance, the islet β-cell function declines gradually during the pathogenic course of type 2 diabetes (14). However, numerous studies using a variety of techniques have demonstrated that individuals with impaired glucose tolerance or overt type 2 diabetes are characterized by β-cell dysfunction independently of obesity and insulin resistance (15,16). Using a glucose tolerance test, Elbein et al have discovered that first-degree relatives of type 2 diabetic individuals with normal glucose tolerance already possess the characteristic signs of β-cell dysfunction (17). Cnop et al have also reported that in first-degree relatives of type 2 diabetic individuals, the decline in glucose tolerance over time is strongly associated with the loss of β-cell function (18). Thus, early interventions to slow the decline in β-cell function should be considered in high-risk individuals.
Insulin secretion includes two states, the basal (postabsorptive) and stimulated (postprandial) states (19). The postabsorptive state predominates during the interprandial phases and plays a key effect during the period of overnight fast; the postprandial state regulates glucose metabolism as carbohydrate is abundant and must be get rid of (20). Under normal physiological status, postprandial insulin secretion is biphasic and divided into two phases, first-phase and second-phase insulin release (20). Early-phase insulin secretion contains the first phase and part of the second-phase of insulin secretion. Shen et al (21) have found that early-phase insulin secretion is involved in preventing hypernomic postprandial blood glucose and reducing plasma glucose fluctuations.
As the glucagon stimulation test is widely used to evaluate endogenous insulin secretion and shows good reproducibility in the clinic, it is frequently employed to assess β-cell function (22,23). The first measurable sign of β-cell dysfunction that can be observed in type 1 and type 2 diabetes is a defect in the first phase of insulin secretion (24). At 6 min after glucagon stimulation insulin is rapidly secreted, reaching an initial peak value, and this early phase of insulin release lasts no more than 10 min (25). Furthermore, as the 6-min C-peptide level response to 1 mg glucagon is significantly correlated with the area under the curve of C-peptide levels, it is used to assess the first phase of insulin secretion and the residual pancreatic β-cell function (26).
In the present study, glucagon stimulation test (0 and 6 min C-peptide: CPR0 and CPR6) was used to evaluated the β-cell function once a year during the 3-year follow-up in patients with type 2 diabetes. The results suggested that the subjects with EOD had lower BMI, SBP, DBP, CPR0, CPR6 and greater HbA1c and TG compared to subjects with LOD. In the EOD patients, the islet β-cell function declined and could not secrete sufficient insulin, so the plasma glucose was not well controlled and the HbA1c level increased. Hyperglycemia and blood lipid disorder play important roles in the course of islet β-cell deterioration (27). Glucotoxicity and lipotoxicity induced the progressive loss of β-cell function (28).
Currently, there are a number of hypotheses regarding the possible mechanism underlying impaired β-cell function (29). First, chronic hyperglycemia has been shown to inhibit β-cell response, which has been demonstrated both in vitro and in vivo (30). Exposure of β-cells to sustained levels of hyperglycemia may deplete the insulin secretory granules from the β-cell, leaving less insulin available for release in response to further hyperglycemia (31). Lowering of glucose levels allows more complete granulation of β-cells and thus improved acute insulin responses (32). In addition, it has been suggested that increased glucose levels activate the hexosamine pathway and contribute to excess generation of reactive oxygen species, resulting in the inhibition of insulin gene transcription and insulin secretion (33).
Another potential cause of β-cell dysfunction is lipotoxicity, accumulated fatty acids and their metabolic products may have a negative effect on the conversion of proinsulin to insulin, resulting in insulin deficiency (34). Additionally, Zhou and Grill has found that continuous sustained exposure to free fatty acids reduces insulin release in response to glucose by inhibiting glucose oxidation in isolated rat islets (35). It has been shown that the accumulated free fatty acids generated by the hydrolysis of TGs in the islets can decrease an elevation in nitric oxide production, inducing β-cell apoptosis (36). Previous epidemiological studies have demonstrated that triglyceride level is an independent determinant of cardiovascular risk, particularly in association with coronary heart disease (37), and reducing the triglyceride level may restrain atherosclerosis progression. The incidence rate of cardiovascular risk in diabetes mellitus is >3 times compared with the healthy population (38), and the lipid disorder displays a significant effect on the occurrence of macrovascular complications in diabetes mellitus (39).
Compared with the baseline, a decline in islet β-cell function was observed in the EOD and LOD groups over a 3-year follow-up, most notably in the EOD group. An >50% reduction in CPR0 or CPR6 was as the threshold β-cell function failure, and the ratio of β-cell function failure in the EOD group was higher compared with the LOD group. These results suggested that islet β-cell function deteriorated more rapidly in patients with EOD compared with LOD. A correlation analysis indicated that during the β-cell function decline, CPR0 showed a positive association with BMI in the EOD group and with WHR and HbA1c in the LOD group. These results suggest that progressive insulin resistance may accelerate β-cell function failure. Furthermore, the loss of insulin sensitivity contributed to adaptive increase in insulin secretion.
The UK prospective diabetes study group (40) has reported that islet β-cell function is reduced by 50% at the time of diagnosis, and a decreasing rate of 5% every year is observed in patients with type 2 diabetes. Along with the increasing age of the normal glucose tolerance population, the islet β-cell function decreases at a rate of 0.5% every year (41). van Haeften et al (42) has reported that the adaptivity of β-cell function to insulin sensitivity already reaches the maximal degree and the β-cell function may decline in patients with abnormal glucose tolerance. During the chronic progression from normal glucose tolerance to overt diabetes, β-cell function failure may be a more important factor than insulin resistance (6).
Previous investigations have shown that lowering the plasma glucose to near normal by intensive insulin therapy may partially recover the β-cell function and insulin secretion (43). Thus, this schema is consistent with therapeutic interventions aimed at preserving β-cell function/insulin secretion and at reducing the burden of insulin resistance to prevent the cardiovascular disease in the patients with EOD.
Figure 2.
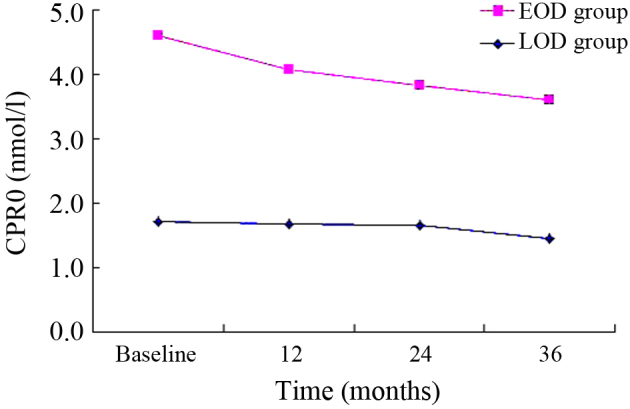
Changes in CPR0 in the 3-year follow-up. EOD, early-onset diabetes; LOD late-onset diabetes; CPR0, C-peptide.
References
- 1.Shaw JE, Sicree RA, Zimmet PZ. Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res Clin Pract. 2010;87:4–14. doi: 10.1016/j.diabres.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 2.Kahn CR, Vicent D, Doria A. Type II non-insulin-dependent diabetes mellitus. Annu Rev Med. 1996;47:509–531. doi: 10.1146/annurev.med.47.1.509. [DOI] [PubMed] [Google Scholar]
- 3.Chan JC, Cho NH, Tajima N, Shaw J. Diabetes in the western pacific Region-past, present and future. Diabetes Res Clin Pract. 2014;103:244–255. doi: 10.1016/j.diabres.2013.11.012. [DOI] [PubMed] [Google Scholar]
- 4.Chan JC, Malik V, Jia W, Kadowaki T, Yajnik CS, Yoon KH, Hu FB. Diabetes in Asia: Epidemiology, risk factors and pathophysiology. JAMA. 2009;301:2129–2140. doi: 10.1001/jama.2009.726. [DOI] [PubMed] [Google Scholar]
- 5.Xu Y, Wang L, He J, Bi Y, Li M, Wang T, Wang L, Jiang Y, Dai M, Lu J, et al. Prevalence and control of diabetes in Chinese adults. JAMA. 2013;310:948–958. doi: 10.1001/jama.2013.168118. [DOI] [PubMed] [Google Scholar]
- 6.Ferrannini E, Gastaldelli A, Miyazaki Y, Matsuda M, Mari A, DeFronzo RA. Beta-cell function in subjects spanning the range from normal glucose tolerance to overt diabetes: A new analysis. J Clin Endocrinol Metab. 2005;90:493–500. doi: 10.1210/jc.2004-1133. [DOI] [PubMed] [Google Scholar]
- 7.Warram JH, Martin BC, Krolewski AS, Soeldner JS, Kahn CR. Slow glucose removal rate and hyperinsulinemia precede the development of type II diabetes in the offspring of diabetic parents. Ann Intern Med. 1990;113:909–915. doi: 10.7326/0003-4819-113-12-909. [DOI] [PubMed] [Google Scholar]
- 8.Khan S, Jena G. Valproic acid improves glucose homeostasis by increasing Beta-Cell proliferation, function, and reducing its apoptosis through HDAC inhibition in Juvenile Diabetic Rat. J Biochem Mol Toxicol. 2016 Apr 15; doi: 10.1002/jbt.21807. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 9.John E. Is reduced first-phase insulin release the earliest detectable abnormality in individuals destined to develop type 2 diabetes? Diabetes. 2002;51:117–121. doi: 10.2337/diabetes.51.2007.S117. [DOI] [PubMed] [Google Scholar]
- 10.Seghieri M, Rebelos E, Astiarraga BD, Baldi S, Mari A, Ferrannini E. Impact of a mild decrease in fasting plasma glucose on β-cell function in healthy subjects and patients with type 2 diabetes. Am J Physiol Endocrinol Metab. 2016 Apr 12; doi: 10.1152/ajpendo.00428.2015. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 11.Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. β-cell deficit and increased β-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;1:102–110. doi: 10.2337/diabetes.52.1.102. [DOI] [PubMed] [Google Scholar]
- 12.World Health Organization: WHO Expert Committee on Diabetes Mellitus. Second report. World Health Organ Tech Rep Ser. 1980;646:1–80. [PubMed] [Google Scholar]
- 13.McCoy RG, Van Houten HK, Ross JS, Montori VM, Shah ND. HbA1c overtesting and overtreatment among US adults with controlled type 2 diabetes, 2001–13: Observational population based study. BMJ. 2015;351:h6138. doi: 10.1136/bmj.h6138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao W, Wang Q, Yu S. Efficacy, safety and impact on β-cell function of dipeptidyl peptidase-4 inhibitors plus metformin combination therapy in patients with type 2 diabetes and the difference between Asians and Caucasians: A meta-analysis. J Endocrinol Invest. 2016 Apr 12; doi: 10.1007/s40618-016-0465-1. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 15.Tura A, Mari A, Winzer C, Kautzky-Willer A, Pacini G. Impaired beta-cell function in lean normotolerant former gestational diabetic women. Eur J Clin Invest. 2006;36:22–28. doi: 10.1111/j.1365-2362.2006.01587.x. [DOI] [PubMed] [Google Scholar]
- 16.Mari A, Gastaldelli A, Natali A, Ostergard T, Schmitz O, Ferrannini E. Characterization of beta-cell function impairment in first-degree relatives of type 2 diabetic subjects: Modeling analysis of 24-h triple-meal tests. Am J Physiol Endocrinol Metab. 2005;288:E541–E546. doi: 10.1152/ajpendo.00175.2004. [DOI] [PubMed] [Google Scholar]
- 17.Elbein SC, Hasstedt SJ, Wegner K, Kahn SE. Heritability of pancreatic beta-cell function among nondiabetic members of caucasian familial type 2 diabetic kindreds. J Clin Endocrinol Metab. 1999;84:1398–1403. doi: 10.1210/jc.84.4.1398. [DOI] [PubMed] [Google Scholar]
- 18.Cnop M, Vidal J, Hull RL, Utzschneider KM, Carr DB, Schraw T, Scherer PE, Boyko EJ, Fujimoto WY, Kahn SE. Progressive loss of beta-cell function leads to worsening glucose tolerance in first-degree relatives of subjects with type 2 diabetes. Diabetes Care. 2007;30:677–682. doi: 10.2337/dc06-1834. [DOI] [PubMed] [Google Scholar]
- 19.Malin SK, Samat A, Wolski K, Abood B, Pothier CE, Bhatt DL, Nissen S, Brethauer SA, Schauer PR, Kirwan JP, Kashyap SR. Improved acylated ghrelin suppression at 2 years in obese patients with type 2 diabetes: Effects of bariatric surgery vs standard medical therapy. Int J Obes (Lond) 2014;38:364–370. doi: 10.1038/ijo.2013.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Del Prato S, Marchetti P, Bonadonna RC. Phasic insulin release and metabolic regulation in type 2 diabetes. Diabetes. 2002;51(Suppl 1):S109–S116. doi: 10.2337/diabetes.51.2007.S109. [DOI] [PubMed] [Google Scholar]
- 21.Shen J, Chen Z, Chen C, Zhu X, Han Y. Impact of incretin on early-phase insulin secretion and glucose excursion. Endocrine. 2013;44:403–410. doi: 10.1007/s12020-012-9867-9. [DOI] [PubMed] [Google Scholar]
- 22.Scheen AJ, Castillo MJ, Lefèbvre PJ. Assessment of residual insulin secretion in diabetic patients using the intravenous glucagon stimulatory test: Methodological aspects and clinical applications. Diabetes Metab. 1996;22:397–406. [PubMed] [Google Scholar]
- 23.Kondo Y, Satoh S, Nagakura J, Kimura M, Nezu U, Terauchi Y. Defining criteria for the introduction of liraglutide using the glucagon stimulation test in patients with type 2 diabetes. J Diabetes. 2013;4:571–575. doi: 10.1111/jdi.12082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ovalle F, Bell DS. Effect of rosiglitazone versus insulin on the pancreatic beta-cell function of subjects with type 2 diabetes. Diabetes Care. 2004;27:2585–2589. doi: 10.2337/diacare.27.11.2585. [DOI] [PubMed] [Google Scholar]
- 25.Del Prato S. Loss of early insulin secretion leads to postprandial hyperglycaemia. Diabetologia. 2003;46(Suppl 1):M2–M8. doi: 10.1007/s00125-002-0930-6. [DOI] [PubMed] [Google Scholar]
- 26.Bellin MD, Beilman GJ, Dunn TB, Pruett TL, Chinnakotla S, Wilhelm JJ, Ngo A, Radosevich DM, Freeman ML, Schwarzenberg SJ, et al. Islet autotransplantation to preserve beta cell mass in selected patients with chronic pancreatitis and diabetes mellitus undergoing total pancreatectomy. Pancreas. 2013;42:317–321. doi: 10.1097/MPA.0b013e3182681182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Doliba NM, Liu Q, Li C, Chen J, Chen P, Liu C, Frederick DW, Baur JA, Bennett MJ, Naji A, Matschinsky FM. Accumulation of 3-hydroxytetradecenoic acid: Cause or corollary of glucolipotoxic impairment of pancreatic β-cell bioenergetics? Mol Metab. 2015;4:926–939. doi: 10.1016/j.molmet.2015.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Poitout V, Robertson RP. Glucolipotoxicity: Fuel excess and beta-cell dysfunction. Endocr Rev. 2008;29:351–366. doi: 10.1210/er.2007-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Buchanan TA. Pancreatic beta-cell loss and preservation in type 2 diabetes. Clin Ther. 2003;25:B32–B46. doi: 10.1016/S0149-2918(03)80241-2. [DOI] [PubMed] [Google Scholar]
- 30.Leahy JL, Bonner-Weir S, Weir GC. Beta-cell dysfunction induced by chronic hyperglycemia. Current ideas on mechanism of impaired glucose-induced insulin secretion. Diabetes Care. 1992;15:4442–4455. doi: 10.2337/diacare.15.3.442. [DOI] [PubMed] [Google Scholar]
- 31.Khattab HA, El-Shitany NA, Abdallah IZ, Yousef FM, Alkreathy HM. Antihyperglycemic potential of grewia asiatica fruit extract against Streptozotocin-induced hyperglycemia in rats: Anti-inflammatory and antioxidant mechanisms. Oxid Med Cell Longev. 2015;2015:549743. doi: 10.1155/2015/549743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van de Bunt M, Manning Fox JE, Dai X, Barrett A, Grey C, Li L, Bennett AJ, Johnson PR, Rajotte RV, Gaulton KJ, et al. Transcript expression data from human islets links regulatory signals from genome-wide association studies for type 2 diabetes and glycemic traits to their downstream effectors. PLoS Genet. 2015;11:e1005694. doi: 10.1371/journal.pgen.1005694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaneto H, Xu G, Song KH, Suzuma K, Bonner-Weir S, Sharma A, Weir GC. Activation of the hexosamine pathway leads to deterioration of pancreatic beta-cell function through the induction of oxidative stress. J Biol Chem. 2001;276:31099–31104. doi: 10.1074/jbc.M104115200. [DOI] [PubMed] [Google Scholar]
- 34.Zhou YP, Grill V. Long-term exposure to fatty acids and ketones inhibits B-cell functions in human pancreatic islets of Langerhans. J Clin Endocrinol Metab. 1995;80:1584–1590. doi: 10.1210/jc.80.5.1584. [DOI] [PubMed] [Google Scholar]
- 35.Zhou YP, Grill VE. Long-term exposure of rat pancreatic islets to fatty acids inhibits glucose-induced insulin secretion and biosynthesis through a glucose fatty acid cycle. J Clin Invest. 1994;93:870–876. doi: 10.1172/JCI117042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jeanne H, Edward S. Beta-cell failure in the pathogenesis of type 2 diabetes mellitus. Curr Diab Rep. 2004;4:169–175. doi: 10.1007/s11892-004-0019-3. [DOI] [PubMed] [Google Scholar]
- 37.Patel A, Barzi F, Jamrozik K, Lam TH, Ueshima H, Whitlock G, Woodward M. Asia Pacific Cohort Studies Collaboration: Serum triglycerides as a risk factor for cardiovascular diseases in the Asia-Pacific region. Circulation. 2004;110:2678–2686. doi: 10.1161/01.CIR.0000145615.33955.83. [DOI] [PubMed] [Google Scholar]
- 38.Taskinen MR. Type 2 diabetes as a lipid disorder. Curr Mol Med. 2005;5:297–308. doi: 10.2174/1566524053766086. [DOI] [PubMed] [Google Scholar]
- 39.Aldebasi YH, Mohieldein AH, Almansour YS, Almutairi BL. Dyslipidemia and lipid peroxidation of Saudi type 2 diabetics with proliferative retinopathy. Saudi Med J. 2013;34:616–622. [PubMed] [Google Scholar]
- 40.No authors listed: U.K. prospective diabetes study 16: Overview of 6 years' therapy of type II diabetes: A progressive disease. U.K. Prospective diabetes study group. Diabetes. 1995;44:1249–1258. doi: 10.2337/diabetes.44.11.1249. [DOI] [PubMed] [Google Scholar]
- 41.Iozzo P, Beck-Nielsen H, Laakso M, Smith U, Yki-Järvinen H, Ferrannini E. Independent influence of age on basal insulin secretion in nondiabetic humans. European group for the study of insulin resistance. J Clin Endocrinol Metab. 1999;84:863–868. doi: 10.1210/jcem.84.3.5542. [DOI] [PubMed] [Google Scholar]
- 42.van Haeften TW, Pimenta W, Mitrakou A, Korytkowski M, Jenssen T, Yki-Jarvinen H, Gerich JE. Relative contributions of beta-cell function and tissue insulin sensitivity to fasting and postglucose-load glycemia. Metabolism. 2000;49:1318–1325. doi: 10.1053/meta.2000.9526. [DOI] [PubMed] [Google Scholar]
- 43.Kishikawa H, Araki E, Wake N, Shichiri M. Intensive insulin therapy to prevent the progression of chronic vascular complications in type 2 diabetes mellitus. Nippon Rinsho. 2002;60(Suppl 9):140–145. (In Japanese) [PubMed] [Google Scholar]