

Abstract
Background
Stroke patients often suffer from a central neuropathic pain syndrome called central post-stroke pain. This syndrome is characterized by evoked pain hypersensitivity as well as spontaneous, on-going pain in the body area affected by the stroke. Clinical evidence strongly suggests a dysfunction in central pain pathways as an important pathophysiological factor in the development of central post-stroke pain, but the exact underlying mechanisms remain poorly understood. To elucidate the underlying pathophysiology of central post-stroke pain, we generated a mouse model that is based on a unilateral stereotactic lesion of the thalamic ventral posterolateral nucleus, which typically causes central post-stroke pain in humans.
Results
Behavioral analysis showed that the sensory changes in our model are comparable to the sensory abnormalities observed in patients suffering from central post-stroke pain. Surprisingly, pharmacological inhibition of spinal and peripheral key components of the pain system had no effect on the induction or maintenance of the evoked hypersensitivity observed in our model. In contrast, microinjection of lidocaine into the thalamic lesion completely reversed injury-induced hypersensitivity.
Conclusions
These results suggest that the evoked hypersensitivity observed in central post-stroke pain is causally linked to on-going neuronal activity in the lateral thalamus.
Keywords: pain, stroke, central post-stroke pain, mouse model
Background
Central post-stroke pain (CPSP) can develop following a variety of lesions, including ischemic and hemorrhagic strokes. The probability of a lesion to cause CPSP depends mainly on its location rather than its size, with lesions in the lower brain stem and thalamus having the greatest potential to cause CPSP.1,2
The prevalence of CPSP in patients with stroke is estimated to be as high as 12%,3–8 and up to 18%3,7 in patients presenting initially with sensory deficits. Typical symptoms of CPSP are reduced temperature or pain sensation, both mediated via the spinothalamic tract (STT),3,9,10 in addition to signs of hypersensitivity in the same area, consisting of allodynia or dysaesthesia to cold and mechanical stimuli.10–12 Interestingly, in humans, thalamic lesions of the ventral posterolateral nucleus (VPL) alone have been shown to be sufficient to cause impaired temperature sensation and CPSP,13 suggesting an important role for the STT and the VPL nucleus in CPSP.
In the current study, we analyze a mouse model of CPSP based on stereotactic lesions in the thalamic VPL nucleus. Lesioning of the VPL nucleus in rats and mice has been shown previously to produce a long-lasting hypersensitivity phenotype.14,15 However, the pathophysiological mechanisms underlying thalamic lesion-induced hypersensitivity remain unclear. Here, we demonstrate that damage to the VPL thalamic nucleus either by hemorraghe or excitotoxicity reliably results in mechanical hypersensitivity and cold allodynia, which are typical features of CPSP. Furthermore, we report the relative contributions of peripheral and spinal components of the pain system and of on-going activity in thalamic neurons to the development and maintenance of this phenotype.
Methods
Behavioral analysis
All animal use procedures were in accordance with ethical guidelines imposed by the local governing body (Regierungspräsidium Karlsruhe, Germany). All behavioral measurements were done in awake, unrestrained, age-matched adult (more than 10 weeks-old) mice of both sexes. The C57BL/6 J mice used in this study were purchased from Janvier (Le Genest Saint Isle, France). Mice were habituated to the experimental setups several times before the analysis. Three baseline measures were obtained for each behavioral test in separate sessions one week prior to the time of the injection (day 0). All behavioral experiments were performed in the hind paw contralateral to the thalamic lesion. The observer was fully blinded to the identity of the groups in all behavioral tests.
Mechanical sensitivity
Animals were placed on an elevated wire grid and mechanical sensitivity was determined upon paw withdrawal to manual application of graded von Frey hairs (0.07–2 g) to the plantar surface of the hind paw. Response frequency was calculated as the mean number of withdrawals out of five applications of the respective filament at 10 s intervals. Withdrawal threshold was determined as the von Frey filament, which elicits at least two paw withdrawal responses out of five applications. To demonstrate all response frequencies to graded von Frey hairs over all forces tested, we constructed a response frequency versus stimulus intensity (applied von Frey force) curve for every group for every time point tested. The integral curve is represented as area under the curve (AUC).
Heat sensitivity
Infrared heat was applied with a Hargreaves apparatus (UgoBasile) to the plantar surface of the hind paw, and the latency to withdrawal of the paw (thermal latency) was measured. The intensity of the infrared lamp was adjusted to obtain baseline latencies in the range of 9–10 s in control mice.
The Cold Plate test was done with a Cold Plate (Bioseb) at −2℃. The latency until the first withdrawal response of the hind paw was recorded, and mice were removed immediately. Cut-off latencies were set at 40 s to avoid tissue damage.
Two choice temperature preference assay
Mice were placed in an apparatus that uses temperature adjustable plates at 30℃ and 18℃ (T2CT, Bioseb). The assay was initiated by placement on the 18℃ plate, and the mouse position was tracked over 10 min.
Intrathalamic lesions
Intracerebral injections were performed under stereotaxic guidance by previously described methods. Injections were made to the VPN of the right thalamus (anterior-posterior to bregma −1.6 mm, lateral to the midline 1.8 mm, ventral to the skull surface −3.2 mm).
Intrathalamic hemorrhage was induced by administration of a 0.3 µl volume containing 0.07 U collagenase Type IV, a protease specifically cleaving bonds between neutral amino acids and glycine in the sequence Pro-Neutral amino acid-Glyc-Pro, which is commonly found in collagen. Excitotoxic lesions were induced by administration of a 0.3 µl volume containing 0.3 µg of the amino acid kainate, a potent glutamate receptor agonist. Control injections consisted of an equal volume of saline.
Intrathalamic drug application
All mice were prepared for drug microinjection into the Nucleus Ventralis Posterolateralis by placing anesthetized (vaporized isoflurane, 4% induction, 2% maintenance) animals in a stereotaxic headholder. For intracranial drug administrations, the skull was exposed and a 26-gauge guide cannula (Plastics One Inc., Roanoke, VA) was directed toward the Nucleus Ventralis Posterolateralis (anterior-posterior to bregma −1.6 mm, lateral to the midline 1.8 mm, ventral to the skull surface −2.2 mm). These coordinates were obtained from the atlas of Paxinos and Watson. The guide cannula was cemented in place. The animals were allowed to recover for at least three days post-surgery before any pharmacological manipulations were made. Collagenase was injected in a volume of 0.3 µl, containing 0.07 U. Lidocaine hydrochloride, a sodium channel blocker, (2%, Sigma–Aldrich) was microinjected in a volume of 5 µl. Injections were made through a 33-gauge injection cannula inserted through the guide cannula and protruding an additional 1 mm into fresh brain tissue to prevent backflow of drug into the guide cannula.
Intrathecal drug application
Intrathecal administration of SSP-SAP, a chemical conjugate of peptidase-resistant [Sar9, Met(O2)11] analog of Substance P and the ribosome-inactivating protein saporin, or saporin alone (SAP), was performed as previously described.16 Briefly, adult mice were anesthetized with fentanyl/medetomidine/midazolam (4:6:16; 0.7 µl/g, i.p.). A small incision was made in the intervertebral membrane, and the cannula was inserted into the sub-arachnoid space, terminating in the L4-5 region. SSP-SAP (10−6 M, n = 9, Advanced Targeting Systems, San Diego, CA) or SAP (10−6 M, n = 10, Advanced Targeting Systems) was injected using an intrathecal cannula attached to a Hamilton syringe and flushed with 5 µl saline. The cannula was withdrawn, haemostasis confirmed and the wound was closed.
Intraplantar drug application
Intraplantar injections of a mixture of QX314 (2%), a membrane impermeable quaternary derivative of lidocaine, and Capsaicin (1µg/µl), a TRPV1-channel agonist, were done in a volume of 20 µl, under light ether anaesthesia lasting for 30 s to 1 min.
Immunohistochemistry
Mice were perfused transcardially with 4% paraformaldehyde (PFA) for light microscopy. Tissue was extracted and postfixed for up to 4 h at 4℃. Immunohistochemistry was performed on Vibratome (50 µm) sections using standard reagents and the following antibodies: anti-NK1 (1:10,000, Chemicon, Temecula, CA), anti-NeuN (1:500, Millipore).
Statistics
All data are expressed as mean ± standard error of the mean (SEM). Two tailed, unpaired student’s t test or ANOVA of repeated measures followed by post hoc Fisher’s test were used. Changes with P < 0.05 were considered to be significant.
Results
Models employed and tested
In an effort to simulate lesions of the thalamus characteristic to CPSP, we employed two different methods. The first involves unilateral microinjection of the excitatory amino acid kainate into the VPL nucleus, which produces neuronal cell death in a dose dependent manner, sparing axons of passage17,18 and thereby creating a confined lesion of the sensory thalamus.
The second model involves a confined hemorrhagic lesion of the sensory thalamus via local injection of collagenase into the VPL nucleus.15
CPSP in humans is characterized by sensory abnormalities including evoked pain or dysaesthesia, elicited by mechanical and cold stimuli.10–12 Therefore, we assessed sensory changes in mice following damage to the VPL in both models and characterized the chronology of their onset. Different somatosensory modalities, such as mechanical touch, pressure, cold and heat, were analyzed progressively for up to four weeks after VPL injury. All behavioral experiments were performed in the hind paw contralateral to the thalamic lesion.
Sensory changes following intrathalamic kainate injection
Within three days after stereotactic injection into the VPL, kainate-injected, but not saline-injected mice, showed a markedly increased frequency of hind paw withdrawal to mechanical stimulation with graded von Frey filaments over 0.07–2 g of applied pressure (Figure 1(a)). While saline-injected control animals showed no significant change of withdrawal threshold throughout the observed course of 12 days, kainate-injected animals demonstrated a significant decrease in mechanical withdrawal threshold by day 3 after VPL injection indicative of mechanical allodynia. This mechanical hypersensitivity was maintained for more than 12 days (Figure 1(a)). To assess cold sensitivity, we used a cold plate at −2℃. No change in response latency of saline-injected mice was observed, while kainate-injected mice showed a transient decrease at four days post-injection that recovered to control values by eight days after injection (Figure 1(b)). In contrast, there was no significant difference in the response latencies to infrared heat applied to the plantar surface of the hind paw between kainate- and saline-injected mice over the entire time course (Figure 1(c)). These results indicate that excitotoxic injury to the VPL induces long-lasting mechanical hypersensitivity that develops three days after injection, while there is only transient cold hypersensitivity and no heat hypersensitivity in these animals.
Figure 1.

Sensory changes following unilateral microinjection of kainate into the VPL nucleus of the thalamus. (a) Mechanical threshold to von Frey filaments applied to the plantar surface of the contralateral hind paw after thalamic kainate or saline injection. Area under the curve (AUC) of the mechanical stimulus-response curve is shown on the right. (tP < 0.05 as compared to basal; *P < 0.05 as compared to corresponding control mice; repeated measures ANOVA, post-hoc Fisher’s test; n = 6–10 mice per group.) (b) Withdrawal latencies on a noxious cold plate (−2℃) of kainate-injected and saline-injected mice. AUC of the stimulus-response curves is shown on the right. (tP < 0.05 as compared to basal; *P < 0.05 as compared to corresponding control mice; repeated measures ANOVA, post-hoc Fisher’s test; *P < 0.05, unpaired, two tailed, t test; n = 6–9 mice per group.) (c) Withdrawal latencies to infrared heat applied to the plantar paw surface of kainate-injected and saline-injected mice. AUC of the stimulus-response curves is shown on right. (tP < 0.05 as compared to basal; *P < 0.05 as compared to corresponding control mice; repeated measures ANOVA, post-hoc Fisher’s test; *P < 0.05, unpaired, two tailed, t test; n = 6–9 mice per group.) (d) Immunostaining reveals significant loss of NeuN-immunoreactivity in the right thalamic VPL nucleus seven days after kainate injection. HC, hippocampus; HT, hypothalamus; VPL, ventral posterolateral nucleus of the thalamus; VPM, ventral posteromedial nucleus of the thalamus; IC, internal capsule. Scale bar: 1 mm.
Sensory changes following intrathalamic collagenase injection
CPSP in humans is observed after both ischemic and hemorrhagic insults to the VPL8 suggesting that the development of CPSP in humans is independent of the nature of the injury to the VPL. In order to exclude that the excitatory nature of kainate itself contributed to the observed hypersensitivity, e.g. via exciting thalamic neurons in a manner not characteristic to CPSP, we confirmed our findings using a unilateral microinjection of collagenase IV into the VPL to produce a confined hemorrhagic lesion. Behavioral assessment showed that collagenase-injected mice had a significantly reduced mechanical withdrawal threshold compared to saline injected controls starting at five days post-injection, which was maintained for the entire observation period over 12 days (Figure 2(a)).
Figure 2.

Sensory changes following unilateral microinjection of collagenase into the VPL nucleus of the thalamus. (a) Mechanical threshold to von Frey filaments applied to the plantar surface of the contralateral hind paw after thalamic collagenase or saline injection. Area under the curve (AUC) of the mechanical stimulus-response curve is shown on the right. (tP < 0.05 as compared to basal; *P < 0.05 as compared to corresponding control mice; repeated measures ANOVA, post-hoc Fisher’s test; n = 6–8). (b) Withdrawal latencies to a noxious cold plate (−2℃) of collagenase-injected and saline-injected mice. AUC of the stimulus-response curves is shown on the right. (tP < 0.05 as compared to basal; *P < 0.05 as compared to corresponding control mice; repeated measures ANOVA, post-hoc Fisher’s test; *P < 0.05, unpaired, two tailed, t test; n = 5–10). (c) Percentage of time of a 10 min observation period spent on a plate set at a non-noxious cold temperature (18℃) vs. a plate at 30℃ of collagenase-injected animals vs. controls (*P < 0.05, unpaired two tailed t test; n = 4–11). (d,e) Withdrawal latencies to infrared heat applied to the plantar paw surface of collagenase-injected and control mice. AUC of the stimulus-response curves is shown in (e). (tP < 0.05 as compared to basal; *P < 0.05 as compared to corresponding control mice; repeated measures ANOVA, post-hoc Fisher’s test; *P < 0.05, unpaired, two tailed, t test; n = 6–10.) (f) Hemorrhage in the right thalamus, including the ventral posterolateral nucleus (VPL), three days after collagenase injection. HC, hippocampus; HT, hypothalamus; VPL, ventral posterolateral nucleus of the thalamus; VPM, ventral posteromedial nucleus of the thalamus; IC, internal capsule. Scale bar: 1 mm.
Likewise, collagenase-injected mice, but not saline-injected mice, showed markedly reduced response latencies to noxious cold stimulation applied to the hind paw plantar surface starting at post-operative day 4 and maintained for the entire observation period over 12 days (Figure 2(b)). To investigate whether the observed hypersensitivity to noxious cold stimuli also extends to non-noxious cool temperatures, we performed a thermal place preference test allowing mice to freely choose between two plates set to 30℃ and 18℃, respectively; 14 days after injection, collagenase-injected mice, but not saline-injected mice, showed significantly decreased time at 18℃ compared to non-injected animals, indicating the development of long-lasting cold allodynia (Figure 2(c)). Similar to the kainate model, there was no difference in heat sensitivity between collagenase and saline injected mice (Figure 2(d)).
In summary, these results indicate that a unilateral hemorrhagic lesion in the VPL produces robust and long-lasting mechanical and cold hypersensitivity as well as cold allodynia as predicted from clinical experience. Furthermore, similar phenotypes in both kainate- and collagenase-injected animals imply that the location of the lesion rather than its nature is critical for the development of CPSP. For further analysis, we focused on the collagenase model because of its closer resemblance to a hemorrhagic stroke, typically known to cause CPSP in humans, as well as its more robust phenotype.
Role of peripheral and spinal elements of the pain system in the induction and maintenance of CPSP
In a first step, we wanted to address the role of an important peripheral component of the pain system, namely TRPV1-expressing nerve fibers, in the maintenance of CPSP. To do so, we used the lidocaine derivate QX-314 to interrupt afferent activity from specific subsets of C and Aδ nociceptors, which selectively express the TRPV1 channel, as described previously.19,20 Coadministration of QX-314 and capsaicin into the contralateral hind paw 13 days after VPL injection of collagenase acutely increased mechanical threshold in saline-injected mice, as described previously in rats;19 however, it failed to reverse mechanical hypersensitivity in collagenase-injected mice (Figure 3(b)). Thus, although combined peripheral application of QX-314 and capsaicin strongly decreased mechanical sensitivity in saline-injected animals, mechanical hypersensitivity associated with thalamic collagenase injection remained unaffected, indicating that activity of TRPV1-expressing afferents does not contribute significantly to this phenomenon.
Figure 3.
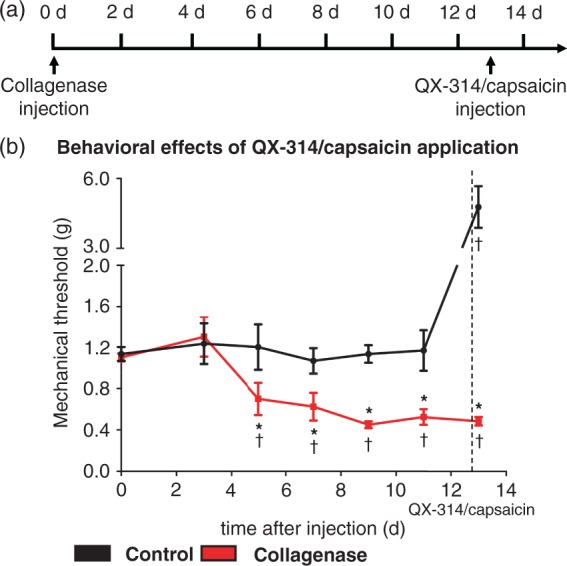
Blocking TRPV1 positive C-fibers does not reverse mechanical hypersensitivity induced by thalamic collagenase injection. (a) Timeline of thalamic collagenase injection and intraplantar QX-314/capsaicin injection. (b) Injection of QX-314/capsaicin significantly increases mechanical threshold in saline-injection mice but fails to do so in collagenase-injected animals (tP < 0.05 as compared to basal; *P < 0.05 as compared to corresponding control mice; repeated measures ANOVA, post-hoc Fisher’s test; n = 6–8).
In a next step, we targeted neurons expressing the neurokinin-1 receptor for substance-P (SP) in lamina I/III of the spinal cord, to elucidate the role of these neurons in the development and maintenance of CPSP. In order to do so, we performed an in vivo ablation of lamina I/III NK1-receptor positive neurons. As described earlier,16 this can be achieved by intrathecal injection of a stabilized Substance P-Saporin (SSP-SAP) construct, which targets lamina I/III NK1-receptor positive neurons in the spinal cord. Selective ablation of lamina I/III NK1-receptor positive neurons with SSP-SAP has been shown to reduce behavioral hyperalgesia in models of inflammation21 and neuropathy,22,23 paralleled by a reduced excitability of dorsal horn neurons in the spinal cord,16,21,22,24 pointing to an important role of this neurons in the maintenance of inflammatory and neuropathic pain states. However, their role in central pain has not been addressed so far. To test contributions of these afferents to the induction or maintenance of VPL injury induced hypersensitivity, we administered SSP-SAP either 28 days prior to or 14 days after intrathalamic injection of collagenase, respectively. As compared to control SAP injections, SSP-SAP injection 28 days prior to intrathalamic collagenase injection neither changed magnitude nor time course of mechanical and cold hypersensitivity (Figure 4(b) and (c)). Similarly, intrathecal SSP-SAP injection 14 days after intrathalamic collagenase injection did not alter mechanical and cold hypersensitivity associated with hemorrhagic injury of the VPL assessed 28 days later (Figure 4(e) and (f)). These results indicate that NK1 receptor positive neurons in the spinal cord do not contribute significantly to the induction or maintenance of the evoked nociceptive hypersensitivity induced by hemorrhagic VPL injury.
Figure 4.

Ablation of lamina I/III NK1 receptor positive neurons in the spinal cord does not affect induction or maintenance of hypersensitivity associated with thalamic collagenase injection. (a) Timeline of spinal NK1 receptor positive neuron ablation by intrathecal SSP-SAP injection and induction of hypersensitivity by thalamic collagenase injection. (b, c) SSP-SAP treatment 28 days prior to thalamic collagenase injection does not prevent development of mechanical (b) or cold (c) hypersensitivity as compared to the corresponding SAP treated control mice. (d) Timeline showing the time points of hypersensitivity induction by thalamic collagenase injection and pharmacological intervention by intrathecal SSP-SAP injection. (e, f) Mechanical (e) and cold (f) hypersensitivity is maintained to a similar degree in SSP-SAP injected animals and controls. (g) Immunohistochemistry with anti-NK1-receptor antibody demonstrating SSP-SAP induced ablation of lamina I NK1 receptor positive neurons in the spinal cord. Scale bar: 300 µm. Data were analyzed via repeated measures ANOVA, post-hoc Fisher’s test in b–f; n = 4–5 mice per group; *P < 0.05 compared to controls.
Role of lesion-induced thalamic hyperexcitability in the maintenance of CPSP
Lesion-induced thalamic hyperexcitability has been suggested as a possible mechanism underlying CPSP.25–27 To delineate the potential contribution of on-going thalamic hyperexcitability to the maintenance of the nociceptive hypersensitivity induced by thalamic haemorrhage, we selectively inhibited neuronal activity in the injured thalamic area via lidocaine microinjection. Animals were implanted with a guide cannula directed toward the VPL, allowing microinjection into the target area. CPSP was induced by microinjection of collagenase into the VPL and lidocaine (4%, 5µl) or saline was injected 18 days later through the same guide cannula for selective delivery to the injured area. Intrathalamic delivery of saline did not acutely affect the magnitude of hypersensitivity to either mechanical or cold stimuli in collagenase-injected mice. In contrast, microinjection of lidocaine into the VPL completely reversed tactile as well as cold-hypersensitivity associated with hemorrhagic VPL injury (Figure 5(b) and (c)). Notably, mechanical as well as cold hypersensitivity were reversed to baseline levels, but not significantly beyond, arguing against a non-specific effect of lidocaine injection on basal nociception. Thus, on-going neuronal activity in the VPL appears to play an important role in maintenance of CPSP-associated nociceptive hypersensitivity and allodynia.
Figure 5.

Lidocaine microinjection into the injured thalamic VPL nucleus reverses mechanical and cold hypersensitivity induced by thalamic collagenase injection. (a) Timeline of thalamic collagenase injection and intrathalamic lidocaine injection using a guide cannula. (b,c) Lidocaine but not saline microinjection into the injured VPL nucleus reverses mechanical (b) and noxious cold (c) hypersensitivity induced by intrathalamic collagenase injection. (*P < 0.05 compared to controls; repeated measures ANOVA, post-hoc Fisher’s test; n = 4).
Discussion
In this study, we characterize the behavioral phenotype following stereotactical lesioning of the VPL nucleus of the thalamus and delineate the differential contributions of distinct circuits in the somatosensory pathway toward thalamic injury induced hypersensitivity. The thalamus and especially the VPL nucleus are thought to play a key role in the development of CPSP. Central pain is often observed after injuries to the thalamus,1,2 and lesions confined to the VPL have been shown to be sufficient to impair temperature sensation and elicit CPSP in humans.13 Furthermore, there is evidence that thalamic lesions only provoke CPSP if they involve the VPL nucleus,28,29 with an Odds ratio in humans of about 80 to develop CPSP compared to other thalamic lesions.30
Here, we observed that a unilateral stereotactic lesion to the thalamic VPL nucleus is sufficient to cause a pronounced and long-lasting phenotype of tactile and cold hypersensitivity in mice, with a delayed onset of 3 to 5 days after injury. Strikingly, these findings closely resemble the evoked sensory hypersensitivity commonly observed in patients suffering from CPSP. Long-lasting allodynia or dysaesthesia to mechanical or cold stimuli in combination with reduced pain and temperature sensation is a landmark feature of CPSP.10–12 Similarly, a cold environment has been shown to increase spontaneous on-going pain in patients with CPSP,9,10 consistent with our observation of cold hypersensitivity in the mouse model. It is important to note that in recently published similar mouse and rat models increased sensitivity to heat as well as mechanical stimuli was observed.14,15 However, heat hypersensitivity appears to be an uncommon feature of CPSP with the overwhelming majority of patients experiencing warm hypoesthesia in combination with normal heat pain thresholds.31 It is intriguing which factors might account for the different findings in our model compared to the studies of Wasserman and Hanada. This might be due to differences in methodology, for example, controls in the above mentioned studies comprised ipsilateral recordings in collagenase treated animals, while we used saline injected control animals.
Interestingly, symptom onset was delayed by several days after thalamic injury, which mirrors the delayed onset of symptoms in humans.3 Compared to humans, however, where symptom onset can be delayed by weeks to months, this time period seems to be shortened in our model, in a similar fashion as has been shown in rats.15,32
In the current study, we used two different stereotactic lesions of the VPL, an excitotoxic kainate-based lesion as well as a hemorrhagic collagenase-based lesion. Surprisingly, the pattern of sensory abnormalities in both models was comparable, despite a very different injury mechanism. It is known that lesions of diverse etiology can elicit CPSP in humans. Indeed, the distinct location of the lesion, rather than its mechanism, seems to be a strong predictor for the development of CPSP.1,2,30 Our study confirms these observations, and models them in an experimental setting, strongly suggesting that the pathophysiology of CPSP is based on the location of the lesion rather than its etiology. Thus, along the lines described in this article, we propose that animal models of CPSP will be valuable in working out mechanistic contributions as well as testing preclinical interventional strategies.
One key aspect of this study is its focus on addressing contributions of distinct circuits in the somatosensory pathway. Noxious stimuli are processed by well-defined subsets of high-threshold primary sensory neurons, but the exact contribution of different nociceptor subtypes to specific pain states remains unclear. Here, we investigated the role of TRPV1-positive nerve fibers in the maintenance of the evoked nociceptive hypersensitivity observed after collagenase injection. It has previously been shown that the lidocaine derivative QX-314 given in combination with the TRPV1 agonist capsaicin, selectively blocks activity in TRPV1-expressing nociceptive afferents.19 TRPV1-mediated axonal silencing has been shown to lead to deficits in sensitivity to heat and mechanical pressure but not to pinprick or light touch. Additionally, it has been shown to completely reverse inflammation-induced heat, mechanical, and cold hypersensitivity.33 In contrast, in models of nerve injury, it has been shown to reduce mechanical but not cold hypersensitivity. These results point to diverse roles for TRPV1-expressing fibers in the processing of thermal as well as mechanical stimuli in different chronic pain states.33 However, the role of TRPV1-positive fibers in central pain has not been addressed so far. Here, we observed that selective nerve block of TRPV1-positive fibers failed to reverse nociceptive hypersensitivity established in collagenase-injected animals. This is especially striking given that in naive animals TRPV1-mediated axonal silencing causes a substantial increase of von Frey thresholds over baseline. These findings imply that mechanical hypersensitivity in CPSP is not transmitted through TRPV-1 expressing C-fibers, which differs from states of inflammation and peripheral neuropathy.
Another important part of the pain system addressed in this study is lamina I/III NK1-receptor positive neurons in the spinal cord. The neurokinin-1 (NK1) receptor for Substance P (SP) is expressed by the vast majority of neurons in lamina I of the spinal cord, and these neurons have been shown to project to nociceptive centers including the parabrachial nuclei, ventrolateral medulla, periaqueductal grey and thalamus.34 Selective ablation of lamina I NK1-receptor positive neurons with a substance P-saporin (SSP-SAP) conjugate has been shown to reduce behavioral hyperalgesia in rodent models of inflammation21 and peripheral neuropathy,22,23 paralleled by a reduced excitability of dorsal horn neurons in the spinal cord.16,21,22,24 In contrast, acute nociception remains unaffected in SSP-SAP treated animals. This is partially explainable by these neurons being the origin of a spino-bulbo-spinal loob that drives facilitatory serotonergic pathways from the brainstem,23,24 which have been shown to contribute to the maintenance of chronic pain states.35–37 Here, we observed that ablation of NK1-receptor positive neurons in the spinal cord with SSP-SAP was neither sufficient to reverse nociceptive hypersensitivity established in collagenase-injected animals nor did it prevent its development. These results suggest that the hypersensitivity observed in CPSP is independent of serotonergic facilitation driven by NK1-receptor positive neurons. Additionally to their role in serotonergic facilitation, these neurons also constitute important projections to basal forebrain structures involved in limbic circuits. Indirect projections from NK1-receptor positive neurons through the parabrachial area and other reticular centers have been shown to be a major source of nociceptive input to the basal ganglia, amygdala and limbic forebrain areas,38–42 which have been shown to be hyperactive in rodent models of peripheral neuropathy.43–45 Our findings do not support a role for this pathway in the development and maintenance of CPSP. However, it is conceivable that independent projections to cortical areas from the thalamic somatosensory nuclei, sometimes referred to as the lateral pain system, might play a role. Indeed, our results support a role for VPL hyperexcitability in CPSP-associated evoked hypersensitivity. Lesion-induced hyperexcitability has been suggested to play an important role in the pathophysiology of central pain, and there is mounting evidence in support of this idea. For example, an interruption of the STT in rats, primates and humans has been shown to cause hyperactive bursting in the VPL.25–27,46 Additionally, thalamic hyperactivity has been described by use of SPECT and PET in CPSP patients experiencing allodynia.47 Furthermore, many of the drugs effective in CPSP act partly by decreasing neuronal hyperexcitability. In our study, selective inhibition of the injured area of the thalamus by lidocaine microinjection completely reversed the tactile and cold hypersensitivity observed after thalamic collagenase injection, strongly supporting the idea of lesion induced hyperexcitability as the pathophysiological basis for CPSP.
It is equally important to acknowledge here, however, that the present study did not address spontaneous, on-going pain, which, in addition to evoked hypersensitivity is another hallmark feature of CPSP. Thus, the differential contributions of circuits discussed above hold only for evoked hypersensitivity, and spontaneous pain in CPSP might have a different underlying pathophysiology.
Conclusion
In summary, this study establishes a mouse model for CPSP, which is based on a unilateral hemorrhagic lesion in the VPL nucleus of the thalamus and addresses the contributions of distinct circuits in the somatosensory pathway as well as lesion induced hyperexcitability of the lateral thalamus itself as a potential pathophysiological mechanisms for CPSP. Our observations suggest that the evoked nociceptive hypersensitivity component of CPSP is causally linked to an on-going neuronal activity in the lateral thalamus. This mouse model thus contributes to the understanding of the underlying mechanism of CPSP and might represent a useful tool for the development and testing of targeted therapeutic interventions for this severely disabling pain disorder.
Acknowledgments
The authors are grateful to Rose LeFaucheur for secretarial assistance and to Dunja Baumgartl-Ahlert and Hans-Josef Wrede for technical assistance.
Author contributions
SG and KKB performed the experiments and analyzed data; RK, SG and DV designed experiments and wrote the manuscript. All authors read and approved the final manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by ERC Advanced Grants to RK and KAN. DV was supported by a fellowship from the Postdoctoral College financed by the Medical Faculty of Heidelberg University. SG was supported by the Studienstiftung des deutschen Volkes. RK is principal investigator in the Excellence Cluster ‘CellNetworks’ of Heidelberg University and the Molecular Medicine Partnership Unit, Heidelberg.
References
- 1.Bowsher D. Pain after thalamic stroke: right diencephalic predominance and clinical features in 180 patients. Neurology 1998; 51: 927. [DOI] [PubMed] [Google Scholar]
- 2.MacGowan DJ, Janal MN, Clark WC, et al. Central poststroke pain and Wallenberg’s lateral medullary infarction: frequency, character, and determinants in 63 patients. Neurology 1997; 49: 120–125. [DOI] [PubMed] [Google Scholar]
- 3.Andersen G, Vestergaard K, Ingeman-Nielsen M, et al. Incidence of central post-stroke pain. Pain 1995; 61: 187–193. [DOI] [PubMed] [Google Scholar]
- 4.Appelros P. Prevalence and predictors of pain and fatigue after stroke: a population-based study. Int J Rehab Res 2006; 29: 329–333. DOI: 10.1097/MRR.0b013e328010c7b8. [DOI] [PubMed] [Google Scholar]
- 5.Bowsher D. Stroke and central poststroke pain in an elderly population. J Pain 2001; 2: 258–261. DOI: 10.1054/jpai.2001.24549. [DOI] [PubMed] [Google Scholar]
- 6.Jonsson AC, Lindgren I, Hallstrom B, et al. Prevalence and intensity of pain after stroke: a population based study focusing on patients’ perspectives. J Neurol Neurosurg Psychiatry 2006; 77: 590–595. DOI: 10.1136/jnnp.2005.079145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kong KH, Woon VC, Yang SY. Prevalence of chronic pain and its impact on health-related quality of life in stroke survivors. Arch Phys Med Rehabil 2004; 85: 35–40. [DOI] [PubMed] [Google Scholar]
- 8.Weimar C, Kloke M, Schlott M, et al. Central poststroke pain in a consecutive cohort of stroke patients. Cerebrovasc Dis 2002; 14: 261–263. [DOI] [PubMed] [Google Scholar]
- 9.Bowsher D. Central pain: clinical and physiological characteristics. J Neurol Neurosurg Psychiatry 1996; 61: 62–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leijon G, Boivie J. Central post-stroke pain – the effect of high and low frequency TENS. Pain 1989; 38: 187–191. [DOI] [PubMed] [Google Scholar]
- 11.Boivie J, Leijon G, Johansson I. Central post-stroke pain – a study of the mechanisms through analyses of the sensory abnormalities. Pain 1989; 37: 173–185. [DOI] [PubMed] [Google Scholar]
- 12.Vestergaard K, Nielsen J, Andersen G, et al. Sensory abnormalities in consecutive, unselected patients with central post-stroke pain. Pain 1995; 61: 177–186. [DOI] [PubMed] [Google Scholar]
- 13.Kim JS. Pure sensory stroke. Clinical-radiological correlates of 21 cases. Stroke 1992; 23: 983–987. [DOI] [PubMed] [Google Scholar]
- 14.Hanada T, Kurihara T, Tokudome M, et al. Development and pharmacological verification of a new mouse model of central post-stroke pain. Neurosci Res 2014; 78: 72–80. DOI: 10.1016/j.neures.2013.09.005. [DOI] [PubMed] [Google Scholar]
- 15.Wasserman JK, Koeberle PD. Development and characterization of a hemorrhagic rat model of central post-stroke pain. Neuroscience 2009; 161: 173–183. DOI: S0306-4522(09)00329-7 [pii] 10.1016/j.neuroscience.2009.03.042. [DOI] [PubMed] [Google Scholar]
- 16.Suzuki R, Morcuende S, Webber M, et al. Superficial NK1-expressing neurons control spinal excitability through activation of descending pathways. Nat Neurosci 2002; 5: 1319–1326. DOI: 10.1038/nn966. [DOI] [PubMed] [Google Scholar]
- 17.Pisharodi M, Nauta HJ. An animal model for neuron-specific spinal cord lesions by the microinjection of N-methylaspartate, kainic acid, and quisqualic acid. Appl Neurophysiol 1985; 48: 226–233. [DOI] [PubMed] [Google Scholar]
- 18.Schwarcz R, Zaczek R, Coyle JT. Microinjection of kainic acid into the rat hippocampus. Eur J Pharmacol 1978; 50: 209–220. [DOI] [PubMed] [Google Scholar]
- 19.Binshtok AM, Bean BP, Woolf CJ. Inhibition of nociceptors by TRPV1-mediated entry of impermeant sodium channel blockers. Nature 2007; 449: 607–610. DOI: 10.1038/nature06191. [DOI] [PubMed] [Google Scholar]
- 20.Caterina MJ, Schumacher MA, Tominaga M, et al. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 1997; 389: 816–824. DOI: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- 21.Mantyh PW, Rogers SD, Honore P, et al. Inhibition of hyperalgesia by ablation of lamina I spinal neurons expressing the substance P receptor. Science 1997; 278: 275–279. [DOI] [PubMed] [Google Scholar]
- 22.Nichols ML, Allen BJ, Rogers SD, et al. Transmission of chronic nociception by spinal neurons expressing the substance P receptor. Science 1999; 286: 1558–1561. [DOI] [PubMed] [Google Scholar]
- 23.Suzuki R, Rahman W, Rygh LJ, et al. Spinal-supraspinal serotonergic circuits regulating neuropathic pain and its treatment with gabapentin. Pain 2005; 117: 292–303. DOI: 10.1016/j.pain.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 24.Khasabov SG, Rogers SD, Ghilardi JR, et al. Spinal neurons that possess the substance P receptor are required for the development of central sensitization. J Neuroscience 2002; 22: 9086–9098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lenz FA, Gracely RH, Rowland LH, et al. A population of cells in the human thalamic principal sensory nucleus respond to painful mechanical stimuli. Neurosci Lett 1994; 180: 46–50. [DOI] [PubMed] [Google Scholar]
- 26.Lenz FA, Kwan HC, Dostrovsky JO, et al. Characteristics of the bursting pattern of action potentials that occurs in the thalamus of patients with central pain. Brain Res 1989; 496: 357–360. [DOI] [PubMed] [Google Scholar]
- 27.Wang G, Thompson SM. Maladaptive homeostatic plasticity in a rodent model of central pain syndrome: thalamic hyperexcitability after spinothalamic tract lesions. J Neurosci 2008; 28: 11959–11969. DOI: 10.1523/JNEUROSCI.3296-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bogousslavsky J, Regli F, Uske A. Thalamic infarcts: clinical syndromes, etiology, and prognosis. Neurology 1988; 38: 837–848. [DOI] [PubMed] [Google Scholar]
- 29.Hong JH, Bai DS, Jeong JY, et al. Injury of the spino-thalamo-cortical pathway is necessary for central post-stroke pain. Eur Neurol 2010; 64: 163–168. DOI: 10.1159/000319040. [DOI] [PubMed] [Google Scholar]
- 30.Sprenger T, Seifert CL, Valet M, et al. Assessing the risk of central post-stroke pain of thalamic origin by lesion mapping. Brain 2012; 135(Pt 8): 2536–2545. DOI: 10.1093/brain/aws153. [DOI] [PubMed] [Google Scholar]
- 31.Greenspan JD, Ohara S, Sarlani E, et al. Allodynia in patients with post-stroke central pain (CPSP) studied by statistical quantitative sensory testing within individuals. Pain 2004; 109: 357–366. DOI: 10.1016/j.pain.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 32.Yang F, Fu H, Lu YF, et al. Post-stroke pain hypersensitivity induced by experimental thalamic hemorrhage in rats is region-specific and demonstrates limited efficacy of gabapentin. Neurosci Bull 2014; 30: 887–902. DOI: 10.1007/s12264-014-1477-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brenneis C, Kistner K, Puopolo M, et al. Phenotyping the function of TRPV1-expressing sensory neurons by targeted axonal silencing. J Neurosci 2013; 33: 315–326. DOI: 10.1523/JNEUROSCI.2804-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marshall GE, Shehab SA, Spike RC, et al. Neurokinin-1 receptors on lumbar spinothalamic neurons in the rat. Neuroscience 1996; 72: 255–263. [DOI] [PubMed] [Google Scholar]
- 35.Porreca F, Ossipov MH, Gebhart GF. Chronic pain and medullary descending facilitation. Trends Neurosci 2002; 25: 319–325. [DOI] [PubMed] [Google Scholar]
- 36.Ren K, Dubner R. Descending modulation in persistent pain: an update. Pain 2002; 100: 1–6. [DOI] [PubMed] [Google Scholar]
- 37.Urban MO, Gebhart GF. Supraspinal contributions to hyperalgesia. Proc Natl Acad Sci USA 1999; 96: 7687–7692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bernard JF, Huang GF, Besson JM. Nucleus centralis of the amygdala and the globus pallidus ventralis: electrophysiological evidence for an involvement in pain processes. J Neurophysiol 1992; 68: 551–569. [DOI] [PubMed] [Google Scholar]
- 39.Bernard JF, Peschanski M, Besson JM. A possible spino (trigemino)-ponto- amygdaloid pathway for pain. Neurosci Lett 1989; 100: 83–88. [DOI] [PubMed] [Google Scholar]
- 40.Bernard JF, Besson JM. The spino(trigemino)pontoamygdaloid pathway: electrophysiological evidence for an involvement in pain processes. J Neurophysiol 1990; 63: 473–490. [DOI] [PubMed] [Google Scholar]
- 41.Bester H, Menendez L, Besson JM, et al. Spino (trigemino) parabrachiohypothalamic pathway: electrophysiological evidence for an involvement in pain processes. J Neurophysiol 1995; 73: 568–585. [DOI] [PubMed] [Google Scholar]
- 42.Malick A, Strassman RM, Burstein R. Trigeminohypothalamic and reticulohypothalamic tract neurons in the upper cervical spinal cord and caudal medulla of the rat. J Neurophysiol 2000; 84: 2078–2112. [DOI] [PubMed] [Google Scholar]
- 43.Backonja M, Wang B, Miletic V. Responses of neurons in the ventrolateral orbital cortex to noxious cutaneous stimulation in a rat model of peripheral mononeuropathy. Brain Res 1994; 639: 337–340. [DOI] [PubMed] [Google Scholar]
- 44.Paulson PE, Morrow TJ, Casey KL. Bilateral behavioral and regional cerebral blood flow changes during painful peripheral mononeuropathy in the rat. Pain 2000; 84: 233–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Paulson PE, Casey KL, Morrow TJ. Long-term changes in behavior and regional cerebral blood flow associated with painful peripheral mononeuropathy in the rat. Pain 2002; 95: 31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weng HR, Lee JI, Lenz FA, et al. Functional plasticity in primate somatosensory thalamus following chronic lesion of the ventral lateral spinal cord. Neuroscience 2000; 101: 393–401. [DOI] [PubMed] [Google Scholar]
- 47.Peyron R, Garcia-Larrea L, Gregoire MC, et al. Allodynia after lateral-medullary (Wallenberg) infarct. A PET study. Brain 1998; 121(Pt 2): 345–356. [DOI] [PubMed] [Google Scholar]